


Abstract
The folate receptor (FR) is overexpressed on a variety of tumor types, whereas its distribution in normal tissues and organs is highly limited. Exploration of the utility of the FR revealed its promising potential for targeting with folate-based radiopharmaceuticals. Herein, we report the principle of the FR-targeting strategy and summarize the development of several folic acid radioconjugates useful for SPECT and PET of cancer diseases. The potential applicability of folate radiopharmaceuticals for FR-targeted radionuclide therapy is also discussed.
Because the availability of efficient and reliable tools for noninvasive diagnosis of diseases is crucial for their management and, thus, for the improvement of a patient's quality of life, identification of targets that are specifically associated with diseased cells is of primary interest. In this respect, the folate receptor (FR) has been intensively studied over almost 2 decades because of its frequent overexpression in cancer cells and its ability to bind and internalize folic acid and conjugates thereof (1).
Folates and folic acid in its oxidized form are water-soluble vitamins of the B-complex group that are exogenously required for optimal health, growth, and development. Folate vitamins act as cofactors for enzymes that are involved in the biosynthesis of DNA and RNA, the amino acid metabolism and epigenetic processes. Thus, folates play a key role for cellular survival and proliferation, whereas impairment of the folate-dependent systems causes several pathophysiologic conditions. Because the hydrophilic nature of folates precludes passive diffusion through the plasma membrane, efficient transport mechanisms are necessary to allow cells the uptake of these essential nutrients. In normal cells, transport is accomplished primarily through the reduced folate carrier (2) and the proton-coupled folate transporter (3). The third uptake system is the high-affinity FR, a glycosyl phosphatidyl inositol–anchored glycoprotein (38–45 kDa) that binds preferentially folic acid (Kd ≈ 10−9 M) and 5-methyltetrahydrofolate and is internalized via endocytosis (4).
In healthy tissues, FR expression is restricted to the lungs, the kidneys, the placenta, and the choroid plexus, where it is confined to the apical surface of polarized epithelia (5). Importantly, the FR is often present in large numbers on epithelial cancers, including tumors of the ovary, cervix, endometrium, lung, kidney, breast, colon, and brain (5,6). Investigations of a variety of FR-positive cancer types revealed that of all the types tested, those of ovarian origin displayed elevated FR levels most frequently (5). The FR is also expressed on hematopoietic malignancies of myeloid origin, including chronic and acute myelogenous leukemias (7). Other tumors, such as sarcomas, lymphomas, pancreatic and testicular cancer, and cancer of the bladder, prostate, and liver, do not commonly upregulate the FR (5).
PRINCIPLE OF FR-TARGETED CANCER RADIOIMAGING
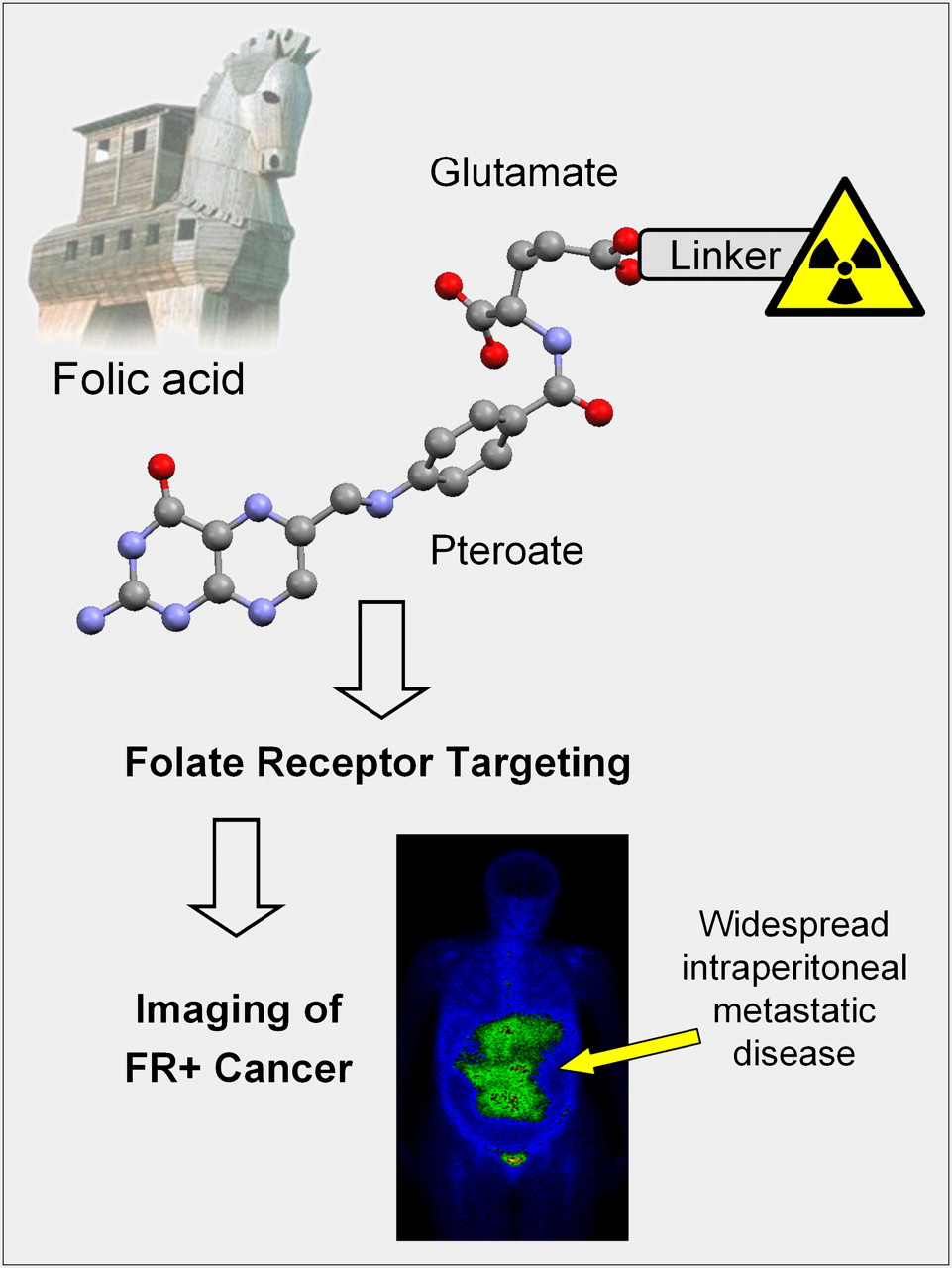
The concept of the FR-targeting strategy makes use of the vitamin folic acid as a molecular Trojan horse for selective delivery of attached probes to FR-expressing cancer cells (Fig. 1). Compared with other targeting agents, such as monoclonal antibodies or peptides, folic acid offers several advantages. It is small (441 Da), stable over a broad range of temperatures and pH values, and thus amenable for site-specific chemical modification. It is inexpensive, nonimmunogenic, and binds to the FR with high affinity even after conjugation to a diagnostic or therapeutic cargo.

Principle of FR-targeted nuclear imaging: radioactive probe is linked to folic acid (pteroylglutamic acid) via glutamate moiety, whereas pteroate moiety is essential for FR binding on cancer cells. SPECT image of cancer patient injected with 99mTc-EC20 (patient scan courtesy of Christopher P. Leamon, Endocyte Inc.).
Because folic acid–targeted imaging agents can serve as noninvasive diagnostic tools to assess the location and severity of FR-positive cancer, a variety of folic acid–conjugated imaging agents have been developed and evaluated in vitro and in vivo. Folic acid conjugates of probes for optical imaging, MRI, and nuclear imaging by SPECT and PET are reported in the literature (Fig. 1) (8,9). Because of the outstanding features of SPECT and PET (e.g., high sensitivity) the overall usage of nuclear medicine procedures is expanding rapidly. Thus, recently, folate-based SPECT and PET tracers have attracted the greatest interest.
FOLIC ACID RADIOCONJUGATES
Radiofolates for SPECT and Potential Therapy
One of the first designs of a folic acid radioconjugate for SPECT used deferoxamine for chelation of the γ-emitting radioisotope 67Ga (10). Tumor targeting was successfully achieved with 67Ga-deferoxamine-folate in mice bearing FR-positive tumor xenografts (∼8.5 percentage injected dose per gram [%ID/g], 4 h after injection). However, significant hepatobiliary excretion of the tracer led to unfavorable abdominal accumulation of radioactivity (11). With the aim of designing a more hydrophilic tracer, a folic acid conjugate with a diethylenetriamine pentaacetate (DTPA) chelator has been developed for radiolabeling with the SPECT isotope 111In (γ-radiation: energy [E] = 171 keV, 245 keV, half-life [t1/2] = 2.8 d). 111In-DTPA-folate was found to clear about 97% through the kidneys, with negligible uptake in the peritoneal cavity (12). On the basis of encouraging preclinical data, 111In-DTPA-folate was tested in clinical trials (13). The tracer was impressively shown to accumulate specifically in FR-positive malignancies of patients with ovarian and endometrial cancer, whereas radioactivity accumulation in normal tissues and organs was observed only in the kidneys and, in some patients, in the liver and spleen.
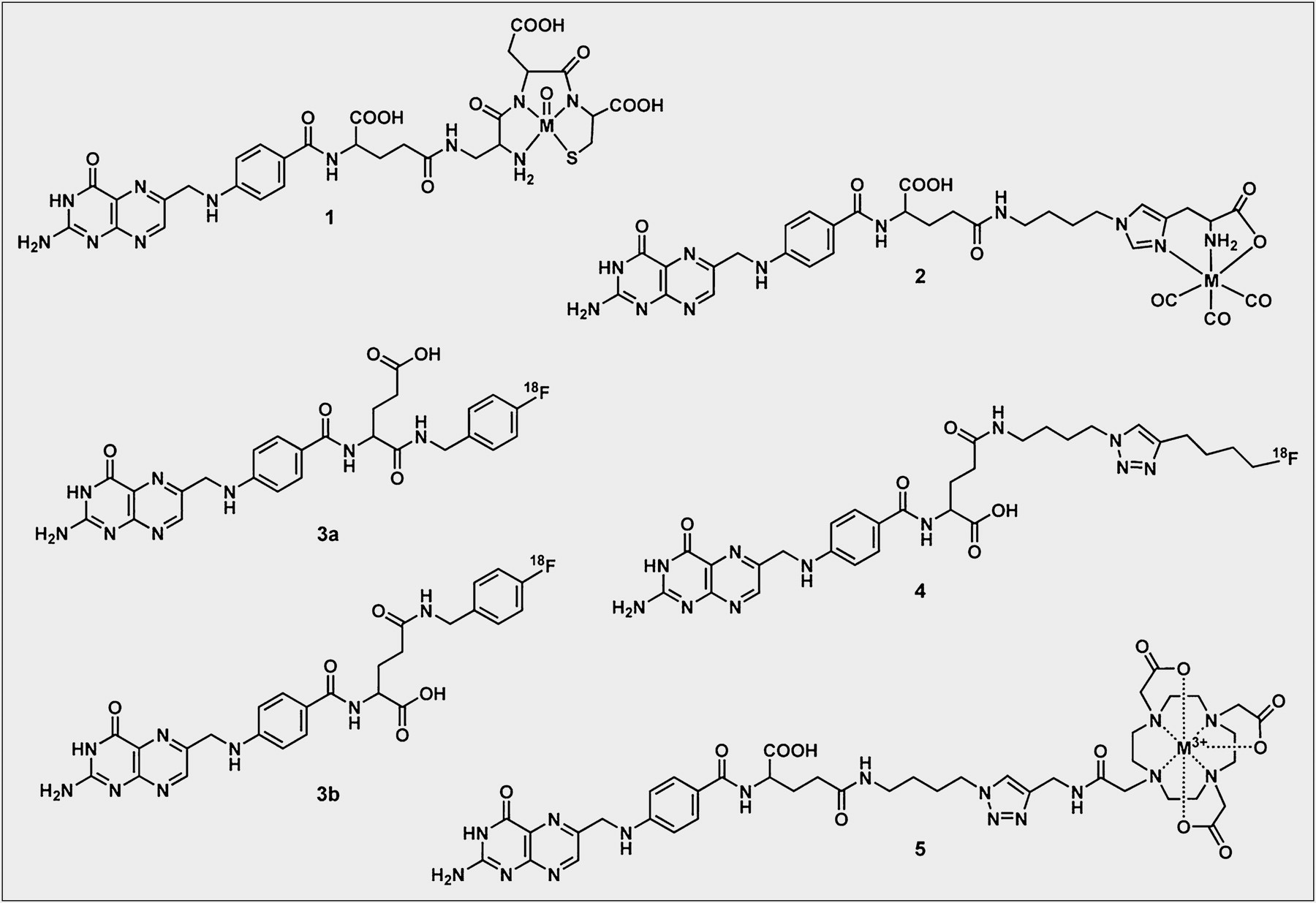
For SPECT purposes with folate-based nuclear imaging agents, 99mTc has emerged as the preferred radionuclide because of its ideal physical decay properties (γ-radiation: E = 140 keV, t1/2 = 6 h), its low cost, and the development of a generator system that makes it readily available on-site in any hospital. Guo et al. reported the synthesis and evaluation of a hydrazinonicotinamide-folate conjugate (14). Hydrazinonicotinamide serves as a monodentate ligand for the octahedral technetium center, whereas the remaining 5 coordination sites about the 99mTc-metal center are occupied by coligands such as tricine and trisodium triphenylphosphine trisulfonate. The preclinical evaluation of the 99mTc-hydrazinonicotinamide-folate was performed with C57BL/6 mice bearing subcutaneous 24JK-FBP tumors (mouse sarcoma cells transfected with the human FR). The 99mTc-folate tracer displayed considerable and FR-specific tumor uptake (∼17.8 %ID/g, 4 h after injection), which declined to background levels if the mice received a coinjection of excess folic acid. Probably the most promising 99mTc-folate candidate is 99mTc-EC20, wherein 99mTc(V) is complexed by a short folate-linked peptide (Cys-Asp-Dap-D-Glu-Pte) (Fig. 2) (15). The in vivo experiments of 99mTc-EC20 were performed on a BALB/c mouse model with syngeneic FR-positive M109 tumors. On the basis of excellent preclinical data that showed a high tumor uptake (∼17.7 %ID/g, 4 h after injection) and elimination primarily via the kidneys, 99mTc-EC20 was introduced into the clinic, where it has been used to image several hundred patients to date. In one clinical trial, imaging with 99mTc-EC20 was performed on 155 patients with a variety of solid tumors (16). The results demonstrated that 68% of the patients showed uptake of 99mTc-EC20 in their tumors. Accumulation of 99mTc-EC20 was also noted in the kidneys and bladder of almost all patients, consistent with the expression of FRs on the proximal tubules of the kidney and the urinary excretion of the tracer. Mild to marked uptake of radioactivity in the liver was blockable by inhibitors of organic anion transport, suggesting mechanisms other than FR binding (16). The authors concluded that 99mTc-EC20 imaging is a safe, noninvasive procedure that may identify FRs in recurrent or metastatic diseases without the need for biopsy to identify patients who may benefit from treatment with FR-targeted therapy.

A completely different strategy was approached while developing organometallic 99mTc-folates (17). The water- and air-stable organometallic tricarbonyl complex [99mTc(H2O)3(CO)3]+ was previously proved to be a versatile reagent for labeling a variety of bioconjugates with 99mTc. Importantly, this strategy potentially allows the synthesis of isostructural compounds for diagnosis and therapy while using the “matched” pair 99mTc/188Re (188Re: β−-decay, average E [Eav] = 763 keV, γ-radiation, E = 155 keV, t1/2 = 17 h) (18). A series of organometallic 99mTc-folate derivatives has been developed and evaluated. In vivo, these tracers exhibited specific uptake in FR-positive tumors (∼2–4 %ID/g, 4 h after injection) and kidneys. Undesirably, 99mTc(CO)3-radiofolates accumulated to a significant extent in the intestinal tract as a consequence of the hydrophobic character of the tricarbonyl-moiety. The most promising organometallic folate tracer, 99mTc(CO)3-histidine-folate (Fig. 2), was used for preclinical imaging studies of tumor-bearing mice with a small-animal multipinhole SPECT/CT scanner (19). This study allowed precise determination of radioactivity uptake and distribution in FR-expressing tissues and organs such as KB tumor xenografts, kidneys, salivary glands, and the choroid plexus in the brain.
More recently, a folate conjugate with a DOTA chelator has been synthesized while using a “Click”-chemistry approach (Fig. 2) (20). This radioconjugate was successfully radiolabeled with 111In for SPECT and with 177Lu (177Lu: β−-decay, Eav = 134 keV, γ-radiation, E = 113 keV, 208 keV, t1/2 = 6.7 d) for potential therapeutic application. In preclinical studies with tumor-bearing mice, the new folate radiotracer displayed an excellent overall tissue distribution with specific accumulation in FR-positive KB tumor xenografts (111In, ∼5.8 %ID/g, and 177Lu, 7.5 %ID/g, 4 h after injection) but with minimal radioactivity retention in nontargeted organs and tissues (21). Significant uptake was, however, observed in the kidneys, resulting in the same low tumor-to-kidney ratios (<0.15) that were observed with previously evaluated radiofolates.
Radiofolates for PET
The development of a folate-based PET agent is an attractive concept because PET would be the most accurate method for noninvasive diagnosis of cancer, particularly of small metastases. Mathias et al. reported the radiosynthesis of the first PET folate tracers, 66Ga- and 68Ga-deferoxamine-folate (22). 68Ga is a generator isotope with a short half-life (89% β+-decay, Eav = 830 keV, t1/2 = 68 min), whereas 66Ga has a relatively long half-life but an unfavorably high positron energy (56% β+-decay, Eav = 1,740 keV, t1/2 = 9.5 h). In this study, FR-positive tumors and kidneys were clearly visualized on small-animal PET images of a KB tumor–bearing mouse 25 h after injection of 66Ga-deferoxamine-folate. However, the same drawback of a high intestinal accumulation of radioactivity that was reported for 67Ga-deferoxamine-folate also hampered a further development of these PET folates.
The design of a 18F-radiolabeled folate tracer is a promising approach because, compared with other radionuclides, 18F (97% β+-decay, Eav = 250 keV, t1/2 = 110 min) displays excellent decay characteristics for PET. The first 18F-folate tracer reported in the literature was a folic acid conjugate with 4-fluorbenzylamine as a prosthetic group, referred to as 18F-fluorobenzylamine-folate (Fig. 2) (23). 18F-fluorobenzylamine was coupled with ester-activated folic acid to obtain γ- and α-18F-fluorobenzylamine-folate isomers in a ratio of 4:1. The last reaction step yielded 15%–44% 18F-fluorobenzylamine-folate tracer after purification via high-performance liquid chromatography. PET studies performed with 18F-fluorobenzylamine-folate in tumor-bearing mice were successful in visualizing KB tumor xenografts (∼6.5 %ID/g, 2 h after injection). Beside uptake in FR-expressing kidneys, massive radioactivity uptake was observed in the gallbladder (>250 %ID/g, 2 h after injection) and the intestinal tract. To address the drawback of a low radiochemical yield experienced with 18F-fluorobenzylamine-folate, a more versatile radiosynthetic strategy was approached that used a Click-chemistry reaction (24). The folate precursor, folic acid-γ-(4-azido)-butylamide, was prepared according to a previously described method (25). The radiosynthesis of the 18F-Click-folate (Fig. 2) comprised 2 main reaction steps. First, the prosthetic group, 6-18F-fluoro-1-hexyne, was produced from the corresponding p-tosylate precursor with an excellent radiochemical yield (70%–85%) and purity (>95%). The second reaction step comprised the 1,4-triazole formation by Cu(I)-catalyzed cycloaddition of the 6-18F-fluoro-1-hexyne and folic acid γ-(4-azido)-butylamide. This Click reaction succeeded without the need for protection groups and directly provided the final 18F-Click-folate (20). In vivo studies performed with KB tumor–bearing mice revealed a relatively high and FR-specific tumor uptake (∼3 %ID/g, 45 min after injection) and a reasonable tumor-to-kidney ratio. However, the strongly lipophilic character of the 18F-Click-folate resulted again in high accumulation of radioactivity in the bile (>600 %ID/g, 45 min after injection) and in the intestinal tract. Because both of these 18F-PET tracers provided suboptimal results, further investment in the design of 18F-PET folates will be necessary for optimization of both the radiosynthesis and the in vivo properties of the tracer.
PERSPECTIVE
A critical aspect of the FR-targeting strategy is the physiologic expression of FRs in the kidneys. Reabsorption of folates from primary urine via FRs is a physiologic process that prevents constant loss of these important vitamins (26). Not surprisingly, small-molecular-weight folic acid radioconjugates undergo the same fate, which results in a significant renal uptake of radioactivity. Thus, folate-based radionuclide therapy with particle-emitting isotopes has not been envisaged so far because low tumor-to-kidney ratios (<0.15) of radiofolates would present a high risk for damage to the kidneys.
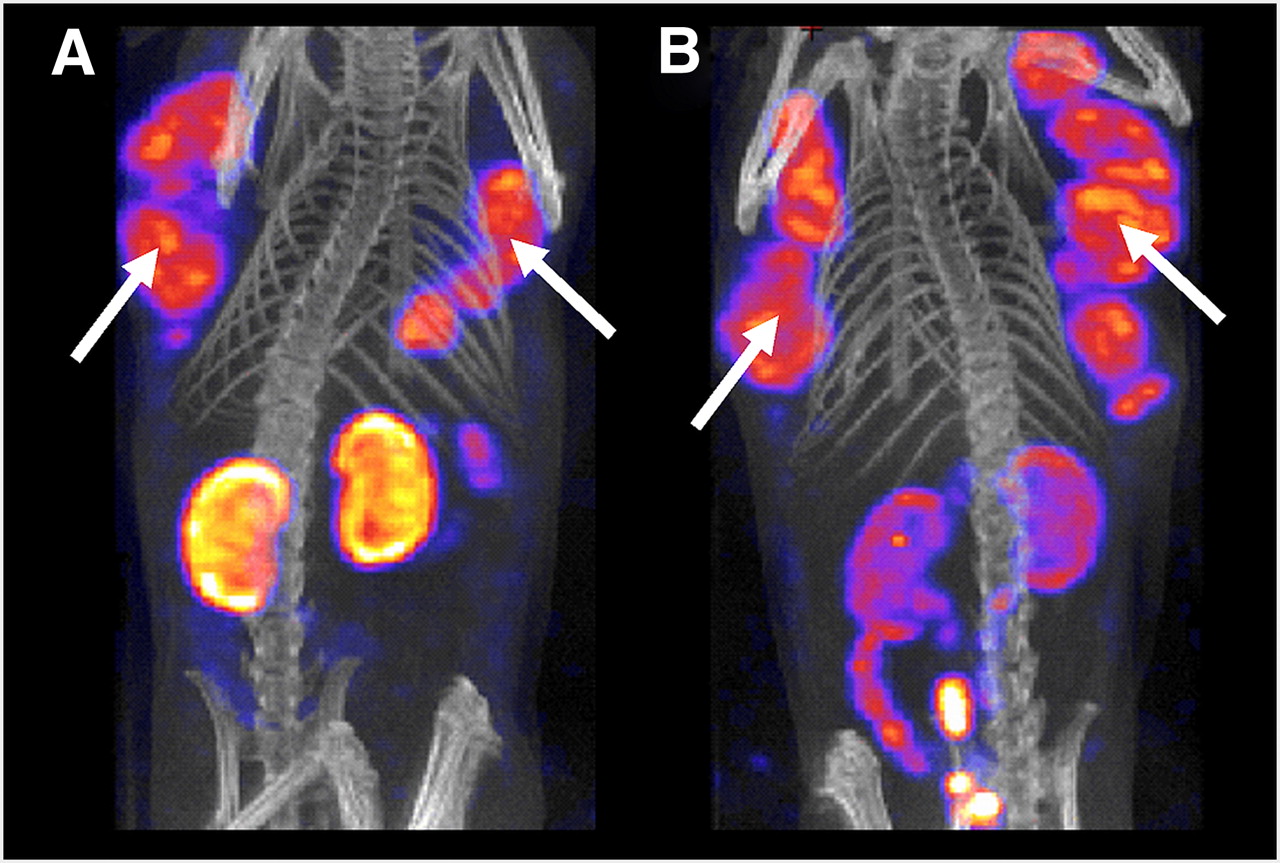
Recent data suggest, however, that predosing with the antifolate pemetrexed significantly reduces kidney uptake of radiofolates while retaining the desired radiotracer accumulation in the tumor (Fig. 3) (21,27,28). The exact underlying mechanism of this observation is not yet completely understood. However, the accessibility of reasonable tumor-to-kidney ratios allows a therapeutic application of radiofolates to be taken into consideration now. But the use of pemetrexed—a chemotherapeutic agent with potential side effects—only for the sake of kidney protection could be problematic. On the other hand, a potential synergistic anticancer effect of therapeutic radiofolates and pemetrexed would be a strong argument to justify the use of this combination for tumor treatment.

SPECT/CT images of female mice bearing human ovarian IGROV-1 tumor xenografts (arrows), 4 h after injection of 111In-DTPA-folate alone (A) and in combination with predosed pemetrexed (B) (28).
CONCLUSION
Using folic acid radioconjugates for SPECT and PET of cancer has proven to be a versatile strategy in preclinical and clinical studies. Although the SPECT tracer 99mTc-EC20 is currently used in the clinic, a suitable PET folate tracer is still lacking. The question of whether FR-targeted radionuclide therapy will be used in the future depends on an appropriate folate tracer design and on combination with substances that potentiate the therapeutic antitumor effect or protect individuals from the risk of nephropathy.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication June 23, 2010.
- Accepted for publication September 8, 2010.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- GEP-NETS UPDATE: Radionuclide therapy in neuroendocrine tumors
- DOTA Conjugate with an Albumin-Binding Entity Enables the First Folic Acid-Targeted 177Lu-Radionuclide Tumor Therapy in Mice
- A Unique Matched Quadruplet of Terbium Radioisotopes for PET and SPECT and for {alpha}- and {beta}--Radionuclide Therapy: An In Vivo Proof-of-Concept Study with a New Receptor-Targeted Folate Derivative