


Abstract
Deoxycytidine kinase (dCK) is a rate-limiting enzyme in the deoxyribonucleoside salvage pathway and a critical determinant of therapeutic activity for several nucleoside analog prodrugs. We have previously reported the development of 1-(2′-deoxy-2′-18F-fluoro-β-d-arabinofuranosyl)cytosine (18F-FAC), a new probe for PET of dCK activity in immune disorders and certain cancers. The objective of the current study was to develop PET probes with improved metabolic stability and specificity for dCK. Toward this goal, several candidate PET probes were synthesized and evaluated in vitro and in vivo. Methods: High-pressure liquid chromatography was used to analyze the metabolic stability of 18F-FAC and several newly synthesized analogs with the natural d-enantiomeric sugar configuration or the corresponding unnatural l-configuration. In vitro kinase and uptake assays were used to determine the affinity of the 18F-FAC l-nucleoside analogs for dCK. The biodistribution of selected l-analogs in mice was determined by small-animal PET/CT. Results: Candidate PET probes were selected using the following criteria: low susceptibility to deamination, high affinity for purified recombinant dCK, high uptake in dCK-expressing cell lines, and biodistribution in mice reflective of the tissue-expression pattern of dCK. Among the 10 newly developed candidate probes, 1-(2′-deoxy-2′-18F-fluoro-β-l-arabinofuranosyl)cytosine (l-18F-FAC) and 1-(2′-deoxy-2′-18F-fluoro-β-l-arabinofuranosyl)-5-methylcytosine (l-18F-FMAC) most closely matched the selection criteria. The selection of l-18F-FAC and l-18F-FMAC was validated by showing that these two PET probes could be used to image animal models of leukemia and autoimmunity. Conclusion: Promising in vitro and in vivo data warrant biodistribution and dosimetry studies of l-18F-FAC and l-18F-FMAC in humans.
- deoxycytidine kinase (dCK)
- cytidine deaminase (CDA)
- PET probes
- deoxyribonucleoside salvage
- autoimmune
- malignant lymphoproliferation
Deoxycytidine kinase (dCK) catalyzes a rate-limiting phosphorylation step in the deoxyribonucleoside salvage pathway (1). dCK is unique among salvage enzymes because of its ability to provide cells with all four deoxyribonucleoside triphosphates via direct (deoxycytidine triphosphate, deoxyadenosine triphosphate, and deoxyguanosine triphosphate) and indirect (thymidine triphosphate) mechanisms (Supplemental Fig. 1; supplemental materials are available online only at http://jnm.snmjournals.org) (1). The highest levels of dCK are found in lymphocytes (2,3), particularly during lymphopoiesis (1). dCK expression is upregulated in activated T cells (4). dCK is also expressed in most lymphoid and myeloid malignancies and in some solid tumors (1).
We have recently demonstrated a critical requirement for dCK in normal lymphocyte development in vivo (5). This requirement suggests that the rate-limiting function of dCK in deoxyribonucleoside phosphorylation is biologically significant. Moreover, dCK is therapeutically important. dCK phosphorylates and activates cytarabine, fludarabine, gemcitabine, cladribine, decitabine, and clofarabine (6). These nucleoside analogs are mainstay cytotoxic prodrugs in the treatment of hematologic and solid tumors. Cladribine has completed phase III clinical trials in multiple sclerosis (7). Lamivudine and emtricitabine, two other dCK-dependent nucleoside analogs, are approved by the Food and Drug Administration for treatment of HIV infections (8).
A common thread among dCK-dependent prodrugs is the high degree of variability in their therapeutic responses. This heterogeneity may reflect genetic variations in deoxyribonucleoside metabolism (9), which confer resistance to therapy. Because decreased dCK activity is a common drug-resistance mechanism (10), dCK-specific PET probes may enable patient stratification into likely responders and nonresponders to dCK-activated prodrugs.
We have recently described the development of 1-(2′-deoxy-2′-18F-fluoro-β-d-arabinofuranosyl)cytosine (18F-FAC) (Fig. 1A) (4,11), a new probe for PET of the deoxyribonucleoside salvage pathway. In mice, 18F-FAC small-animal PET enables imaging of adaptive immunity and several types of cancer (4). 18F-FAC accumulation in tumor tissues is predictive of responses to gemcitabine (12). Here, we analyzed the metabolic stability of 18F-FAC. In mice, 18F-FAC was found to rapidly undergo deamination to 1-(2′-deoxy-2′-18F-fluoro-β-d-arabinofuranosyl) uracil (18F-FAU). Deamination confounds the specificity of 18F-FAC for dCK, and it may decrease its sensitivity in species with high deaminase activity such as humans (13). To bypass tracer deamination, a series of 18F-FAC analogs was synthesized and evaluated. These studies indicated that 18F-FAC analogs with the unnatural l-enantiomeric sugar configuration resist deamination and allow PET imaging of dCK activity in vivo.

In vivo metabolite analysis of 18F-FAC. (A) Chemical structures of 18F-FAC and 18F-FAU and schematic showing extracellular and intracellular routes by which 18F-FAC can be deaminated to 18F-FAU. (B) Percentage of total detected radioactivity in plasma that is attributable to 18F-FAC over time as determined by HLPC analysis. Analysis is performed after intravenous injection of 18F-FAC in mice. 5′-NT = 5′nucleotidase; DCTD = deoxycytidylate deaminase (catabolic enzymes are shown in red fonts); MP = monophosphate.
MATERIALS AND METHODS
Radiochemical Synthesis of 18F-Labeled PET Probes
18F-FAC was synthesized as previously described (4,11). The radiochemical syntheses of the new probes are described in the “Supporting Methods” section of the supplemental material.
PET Probe Metabolite Analyses
Mice were injected intravenously with 18F-labeled probes and allowed up to 45 min of uptake. Tissues were removed and homogenized. Whole-blood sampling was performed via retroorbital eye bleeding, followed by centrifugation to obtain plasma. Tissues and plasma were treated with a mixture of methanol:acetonitrile (1:9) to extract nucleosides. After centrifugation, supernatants were evaporated at 50°C under nitrogen. The residue was dissolved in 100 mL of mobile phase (5 mM pentane-1-sulfonic acid, pH 3.1, and methanol [96:4]), filtered, and injected into a μBondapak C18 column (Waters) with a flow rate of 1.5 mL/min. Peak radioactivity was measured using a Bioscan coincidence detector.
In Vitro Enzymatic Assays
The production of recombinant human cytidine deaminase (CDA) and dCK is described in the “Supporting Methods” section of the supplemental material. The deamination assay solution contained purified CDA (0.05 μg/μL) and 140 μM nucleoside analog in 0.125 M phosphate buffer, pH 7.6. The assay was performed at 37°C for 1 h; products were resolved via high-performance liquid chromatography (HPLC) using a μBondapak C18 column (3.9 × 300 mm), as previously reported (14). Elution was isocratic, using 3% methanol in 0.l M phosphate buffer (pH 5.5) at a flow rate of 1 mL/min. The rate of dCK-catalyzed phosphorylation was measured continuously at 340 nm using an iEMS Reader MF (LabSystems) system and a modification of a previously described spectrophotometric assay consisting of a coupled lactate dehydrogenase (LDH)–pyruvate kinase (PK) reaction (15). Reactions were performed in triplicate at 37°C with 50 mM Tris-HCl, pH 7.6; 50 mM KCl; 10 mM MgCl2; 5 mM adenosine triphosphate; 0.2 mM NADH; 1 mM phosphoenolpyruvate; 1 mM DTT; 1.4 μM dCK; and 10 units of PK (Sigma) and 15 units of LDH per milliliter (Sigma). Kinetic constants were calculated using GraphPad Prism 5 (GraphPad Software).
Cell-Based Phosphorylation and Uptake Competition Assays Using 3H-Labeled Deoxycytidine (3H-dC)
L1210 cells (12) were lysed by three cycles of freeze-thawing in 50 mM Tris-HCl, pH 7.6; 2 mM DTT; 20% glycerol; and 0.5% Nonident P40 (Roche). The lysate was clarified by centrifugation (15,000g for 10 min), and the protein concentration in the supernatants was determined using the Bradford assay. The kinase reaction mixture contained 4.2 μg of L1210 lysate; 3 μM 3H-dC (Moravek Biochemicals; specific activity, 1.295 TBq [35 Ci]/mmol); 50 mM Tris-HCl, pH 7.6; 5 mM MgCl2; 1 mM UTP (Sigma); 2 mM DTT; 10 mM NaF; 1 mM thymidine (to inhibit thymidine kinase 2); and 30 μM nucleoside analog. The reaction was incubated at 37°C for 20 min and was stopped by the addition of 30 μL of ice-cold water, followed by heating at 90°C for 3 min. The reaction was then spotted on Whatman DE81 filter disks; these were washed 3 times with 4 mM ammonium formate and 2 times with ethanol. The disks were placed in scintillation vials and counted using a Beckman Liquid Scintillation counter. For uptake assays, 38.85 kBq (1.05 μCi) of 3H-dC were added to L1210 cells (105 cells per well in 96-well plates), followed by 3 μL of 100 μM stocks of each nucleoside analog (at a final volume of 100 μL/well). After 1 h at 37°C, wells were washed 3 times with ice-cold phosphate-buffered saline using a vacuum filtration system (Millipore). Plates were oven-dried at 45°C−50°C for 10 min, and 200 μL of scintillation fluid were added per well. Radioactivity was measured using a Trilux MicroBeta scintillation counter (Perkin Elmer).
Small-Animal PET/CT Studies in Mice
In vivo studies followed the guidelines of the Department of Laboratory Animal Medicine at UCLA and were performed as previously described (4,12) (Supplemental “Supporting Methods” section). Autoimmune B6.MRL-Faslpr/J mice were obtained from the Jackson Laboratory (stock no. 000482).
Statistical Analysis
Data are presented as mean ± SD. All P values are 2-tailed, and P values of less than 0.05 were considered to be statistically significant. Graphs were generated using GraphPad Prism 5 software.
RESULTS
18F-FAC Is Rapidly Deaminated In Vivo
With the exception of the fluorine substitution of the ara-2′ hydrogen, FAC is identical to deoxycytidine. Therefore, it is likely that 18F-FAC and deoxycytidine are competitive substrates and share the same metabolic pathway. According to the model shown in Figure 1A, 18F-FAC is either phosphorylated by cytosolic dCK to 18F-FAC-monophosphate or converted to 18F-FAU by extracellular or cytosolic CDA. 18F-FAU, a weak substrate for thymidine kinase 1 (TK1) (16), cannot be phosphorylated by dCK. 18F-FAU can also be produced from 18F-FAC by a CDA-independent mechanism involving deoxycytidylate deaminase (which converts 18F-FAC-monophosphate to 18F-FAU-monophosphate) and 5′nucleotidase (which dephosphorylates 18F-FAU-monophosphate, producing 18F-FAU; Fig. 1A). To validate this model, mice were intravenously injected with 18F-FAC and plasma samples were analyzed by HPLC. 18F-FAU was the only 18F-FAC metabolite detected in plasma by this HPLC method (Supplemental Fig. 2A). As early as 1 min after injection, 18F-FAU contributed to approximately 14% of the total radioactivity in the plasma (Fig. 1B). Ten minutes later, the contribution of 18F-FAU increased to approximately 60% and reached approximately 90% at 45 min. The plasma clearance rate of 18F-FAU was significantly slower than that of 18F-FAC (Supplemental Fig. 2B). Similar to plasma, thymus and liver samples from 18F-FAC–injected mice also contained high concentrations of 18F-FAU (Supplemental Fig. 2C) at 45 min after injection.
2-Chloro-2′-Deoxy-2′-18F-Fluoro-9-β-d-Arabinofuranosyl-Adenine (18F-Clofarabine), a Potential dCK-Specific PET Probe That Resists Deamination
The fast exponential decline of 18F-FAC in plasma and the accumulation of the 18F-FAU metabolite in plasma and tissues reduce the specificity of 18F-FAC. They may also affect the sensitivity of 18F-FAC PET in humans, who express twice as much deaminase activity as do mice in most tissues (13). This problem can be solved by identifying dCK substrates that are amenable to 18F labeling and have low affinity for deaminase enzymes. Clofarabine (Supplemental Fig. 3) is a dCK-dependent prodrug with excellent metabolic stability (17). The 2-halogenated aglycone of clofarabine confers resistance to deamination by adenosine deaminase. The 2′ fluorine atom blocks cleavage of the glycosidic bond by bacterial purine nucleoside phosphorylase (17). Clofarabine is efficiently phosphorylated by dCK (18) and is amenable to 18F labeling (Supplemental Fig. 3). Surprisingly, 18F-FAC and 18F-labeled clofarabine showed distinct biodistribution patterns in immunocompetent (C57BL/6J) mice (Fig. 2). In contrast to 18F-FAC (4), 18F-clofarabine accumulation in expected target organs such as the thymus, bone marrow, and spleen was undetectable. Whether the differences in the biodistribution of 18F-FAC and 18F-clofarabine are unique to mice or can also be found in other species, including humans, will be addressed in future studies.

18F-FAC and 18F-clofarabine (18F-CA) small-animal PET/CT scans of C57BL/6J mice (images are representative of pattern observed in 3 mice scanned with each probe). %ID/g = percentage injected dose per gram of tissue; Bl = urinary bladder; BF = brown fat; BM = bone marrow; GI = gastrointestinal tract; K = kidney; L = liver; S = spleen; Thy = thymus.
FAC Analogs as Potential dCK-Specific Probes
The disappointing performance of 18F-clofarabine in mice indicated that a different approach was required to solve the tracer deamination problem. Toward this end, the FAC pharmacophore was explored to seek pyrimidine analogs matching three criteria: amenability to routine 18F radiolabeling with high specific activity, resistance to deamination, and affinity for dCK comparable to that of 18F-FAC. Nine potential dCK-specific probes (Fig. 3) were designed and synthesized. The rationale for selecting these compounds was two-fold. First, modifications of deoxycytidine at position 5 of the nucleobase have been shown to reduce susceptibility to deamination (19). Second, dCK has the unusual property of phosphorylating both d-enantiomers (the natural configuration of nucleosides) and unnatural l-enantiomers (20,21). In contrast, CDA has a strong preference for d- over l-deoxycytidine analogs (20). The susceptibility of FAC analogs to deamination was determined by incubating them with purified recombinant human CDA, followed by HPLC analysis to determine the formation of deaminated products. Figure 3 shows that the nucleosides with the d-chirality were completely deaminated, whereas the unnatural l-nucleosides were resistant to deamination. Although these modifications solve the deamination problem, it is possible that they also reduce the affinity of the FAC analogs for dCK. This possibility was addressed using several assays.
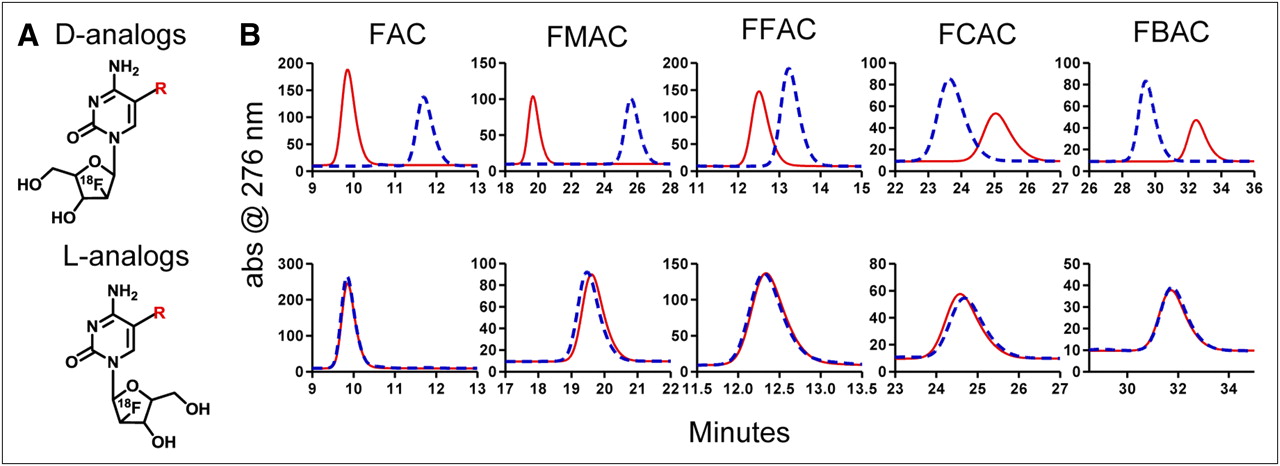
Candidate dCK-specific PET probes that resist deamination. (A) Chemical structures of deoxycytidine analogs amenable to 18F labeling. (B) In vitro deamination assay. After incubation at 37°C for 1 h in presence (blue dashed traces) or absence (red solid traces) of recombinant purified CDA, candidate probes were analyzed on HPLC. d-analogs are shown in top row, and l-analogs are shown in bottom row. abs = absorbance. R = H (FAC); R = CH3 (FMAC); R = F (FFAC); R = Cl (FCAC); R = Br (FBAC).
First, an LDH and PK–coupled reaction (15) was used to determine the kinetic parameters of human recombinant dCK for l-analog candidate probes. Kinetic parameters for 3 of the 5 l-analogs were obtained (Table 1). l-18F-FMAC and 1-(2′-deoxy-2′-fluoro-β-l-arabinofuranosyl)-5-chloro-cytosine (l-FCAC) had similar kinetics, with KM values of 1.0 and 0.6 μM, respectively. These KM values were similar to those of FAC (0.8 μM). l-FMAC and l-FCAC also had specificity constants (kcat/KM) for dCK that were similar to those of FAC (0.96–1.2 times that of FAC). However, this was not the case for 1-(2′-deoxy-2′-fluoro-β-l-arabinofuranosyl)-5-bromo-cytosine (l-FBAC), which had a higher KM and a lower specificity constant than FAC.
Kinetic Parameters for FAC and Its Analogs with l-Chirality
The LDH–PK assay determines the kinetics only for phosphorylation reactions that are faster than the ultraviolet-induced decomposition of NADH (22). The inability to obtain the kinetic parameters for l-FAC and 1-(2′-deoxy-2′-fluoro-β-l-arabinofuranosyl)-5-fluoro-cytosine (l-FBAC) using the LDH–PK assay indicated that these nucleoside analogs either are high-affinity substrates for dCK, with low enzymatic turnover, or that they lack affinity for dCK. To determine whether l-FAC and l-FFAC were phosphorylated by dCK, kinase assays (23) were performed using 18F-labeled probes (synthesized according to the scheme shown in Supplemental Fig. 4), purified recombinant human dCK, and lysates from L1210 dCK–positive and –negative cell lines (12). The kinase assays showed conclusively that l-FAC and l-FFAC are indeed dCK substrates (Supplemental Fig. 5).
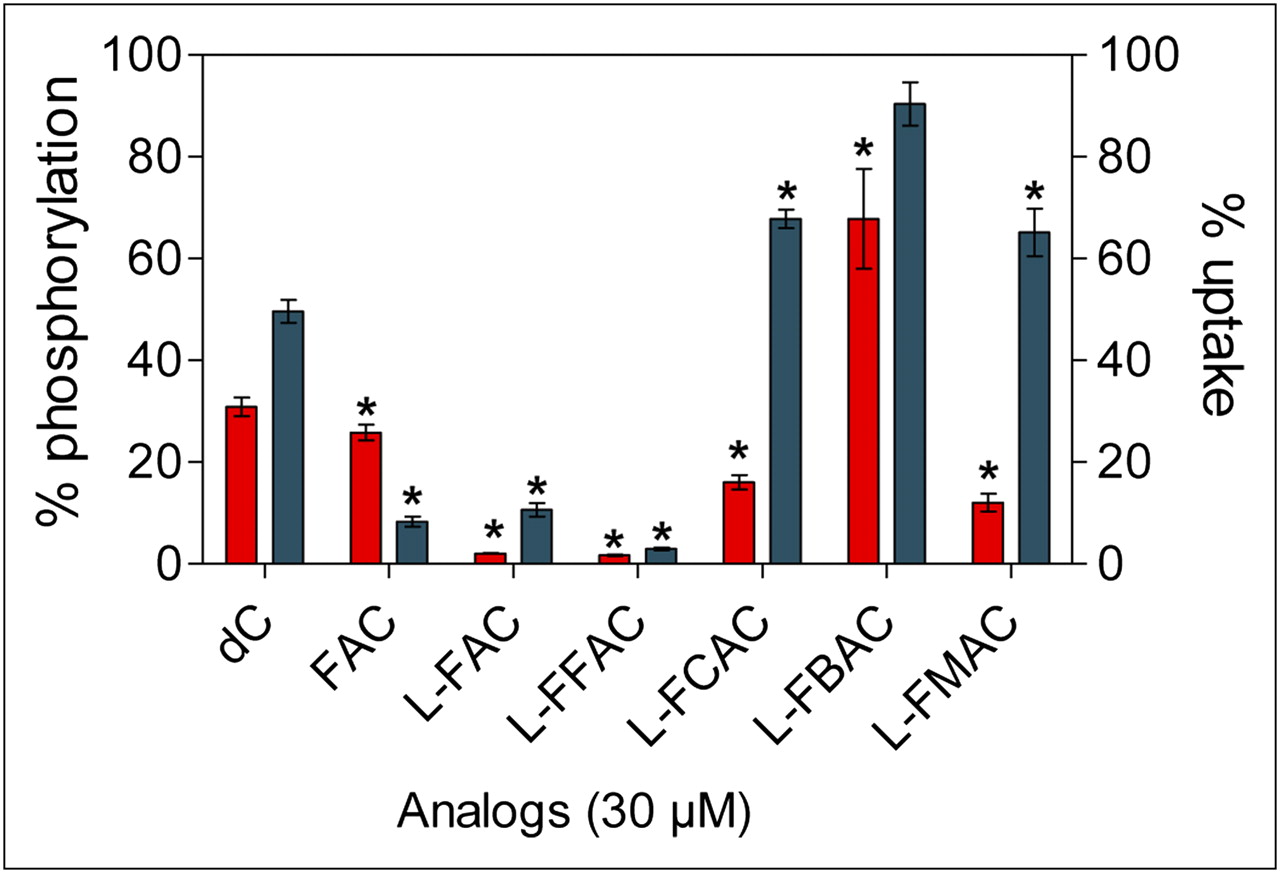
Two cell-based assays were then used to determine whether the l-analogs can compete with the endogenous dCK substrate deoxycytidine. In a kinase assay using whole-cell lysates of the dCK-positive murine leukemia cell line L1210 (12), l-FAC and l-FFAC outperformed the other nucleoside analogs by inhibiting 3H-dC phosphorylation by over 95% (Fig. 4). The enhanced activity of dCK toward l-FAC and l-FFAC, compared with FAC, is not entirely surprising because the enzyme has been previously shown to favor the l-conformation of other pyrimidine analogs (21,24). Confirming the results from the LDH–PK assay, l-FBAC inhibited deoxycytidine phosphorylation by only 32%. The ability of l-nucleosides to competitively inhibit 3H-dC uptake by L1210 cells was also determined (Fig. 4). In addition to dCK-mediated phosphorylation, the uptake assay took into account the efficiency of transport across the cell membrane. l-FAC and l-FFAC reduced the uptake of 3H-dC by at least 90%. The inhibition by l-FCAC and l-FMAC was 32%−35%.
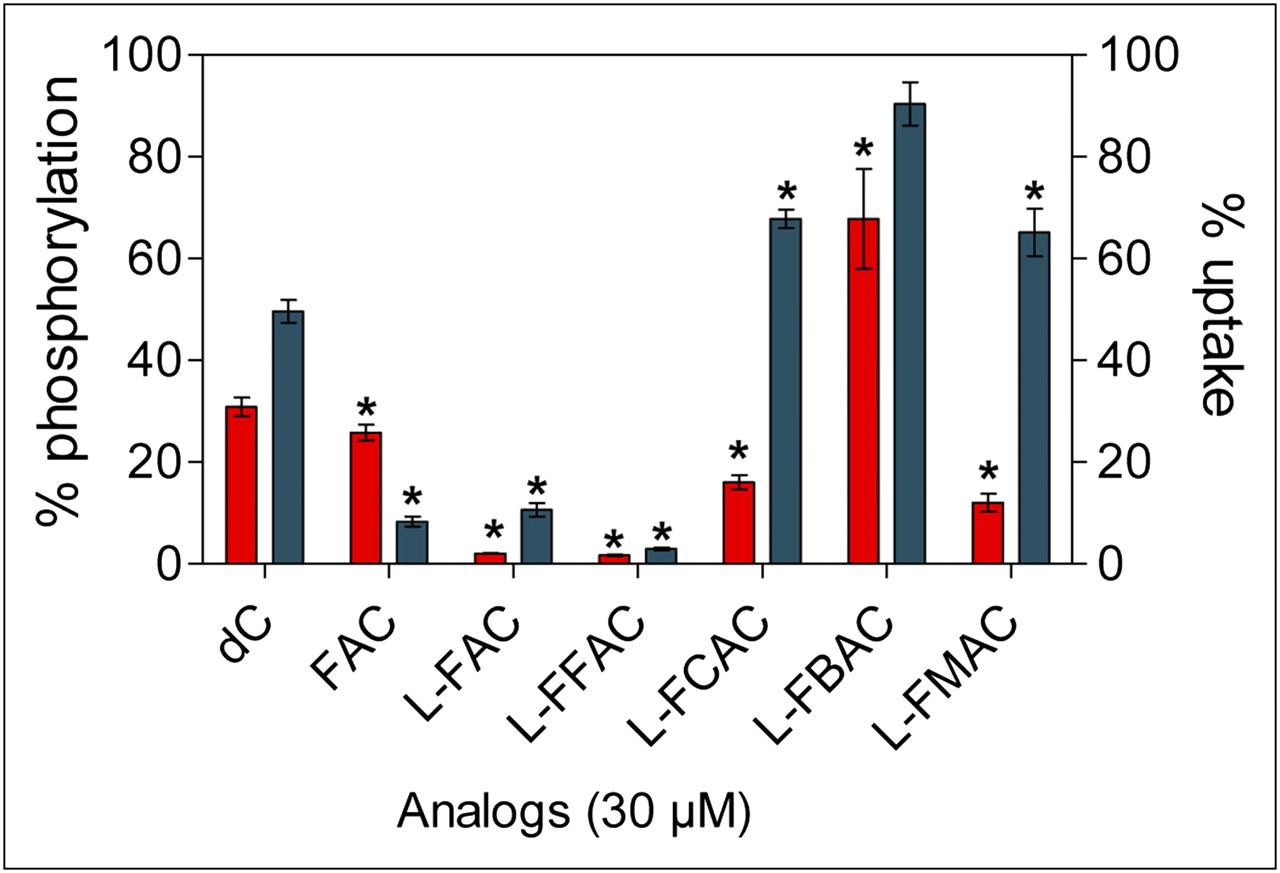
Analyses of affinity of l-analogs for dCK. l-analogs were tested for their ability to competitively inhibit phosphorylation (red bars; left axis) and uptake (blue bars; right axis) of tritium-labeled deoxycytidine (3H-dC) using dCK-expressing L1210 cells. Results represent 2 independent experiments. P values are calculated relative to water (n = 3). *P < 0.05. dC = deoxycytidine.
Biodistribution of 18F-Labeled l-Nucleosides in Immunocompetent C57BL/6J Mice
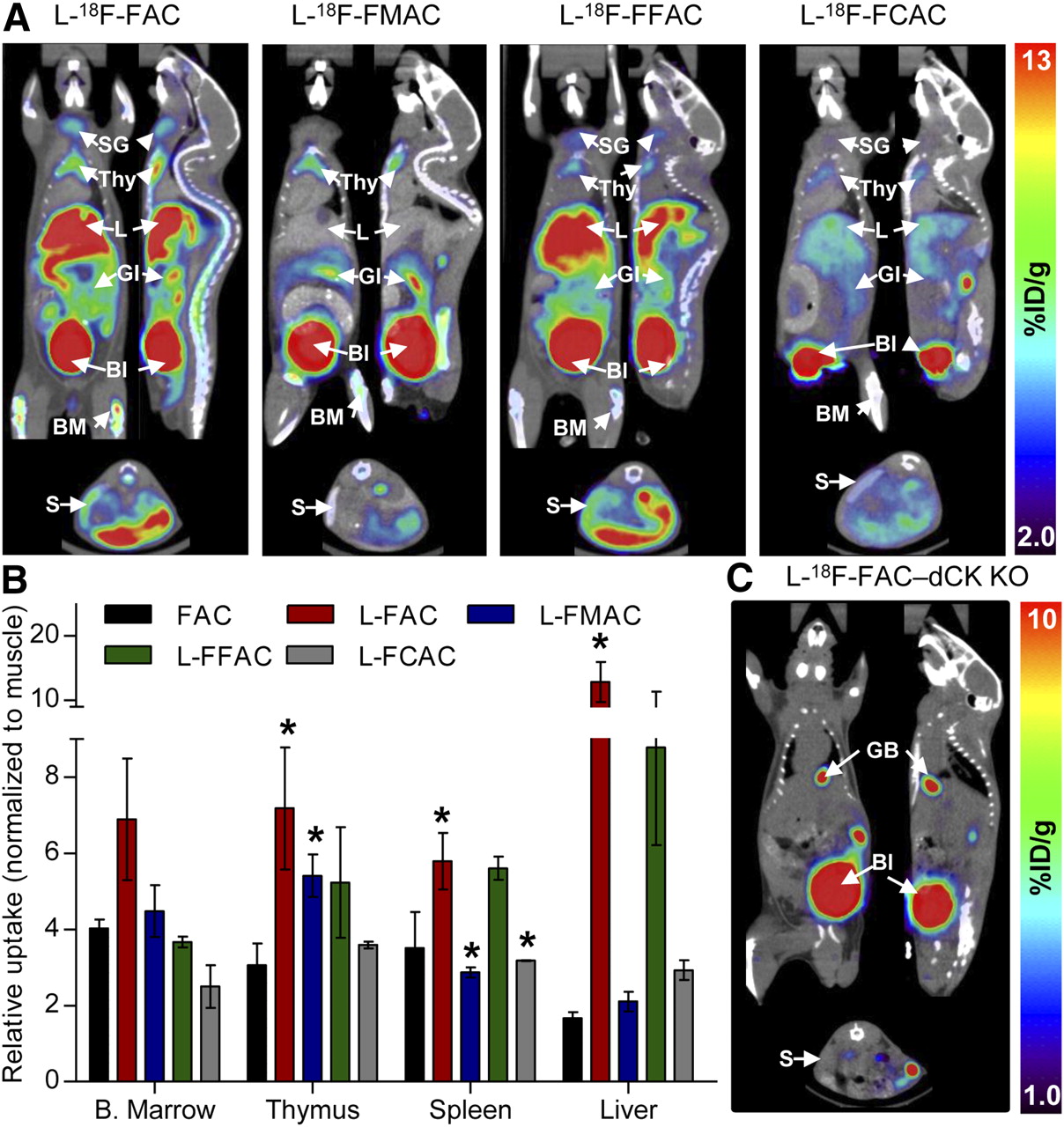
Results from the in vitro phosphorylation and uptake assays narrowed down the selection of candidate l-enantiomer probes to l-18F-FAC, l-18F-FMAC, l-18F-FFAC, and l-18F-FCAC. Similar to 18F-FAC and unlike 18F-clofarabine, all four candidate probes accumulated in the thymus and to varying degrees in the bone marrow and spleen (Fig. 5A). Figure 5B shows the relative probe uptake values in dCK-positive tissues normalized to uptake in the muscle (a dCK-negative tissue); absolute probe uptake values are shown in Supplemental Figure 6. Among the tested l-nucleosides, l-18F-FCAC had the lowest uptake in lymphoid tissues and l-18F-FAC had the highest uptake in thymus and spleen. An intermediate profile was observed for l-18F-FMAC, which had higher uptake in the thymus and lower uptake in the spleen than did 18F-FAC.
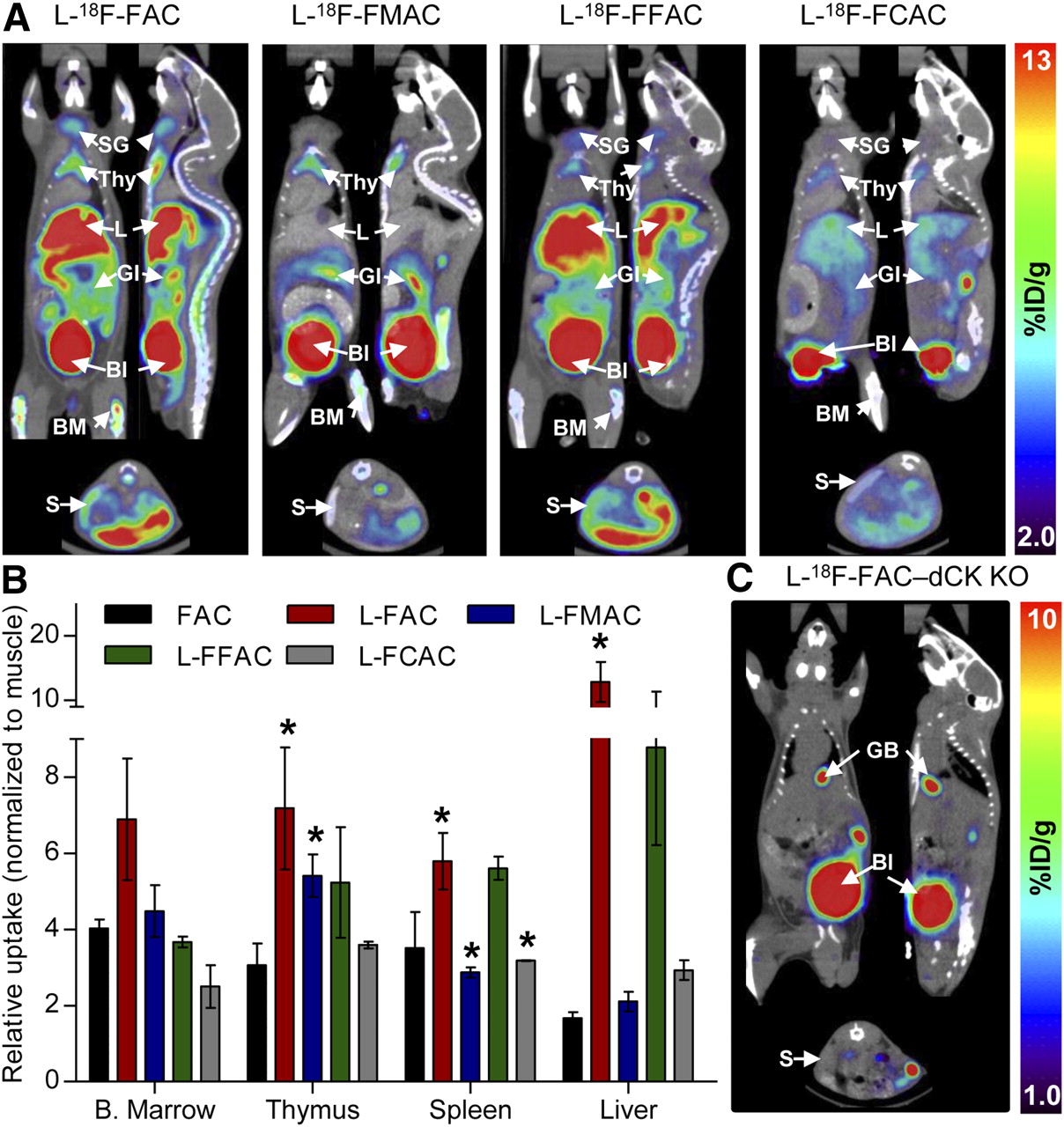
Biodistribution of 18F-labeled unnatural nucleosides in mice. (A) Small-animal PET/CT images of C57BL/6J mice. (B) Quantification of PET data. Probe uptake was normalized to muscle background (absolute uptake values are shown in Supplemental Fig. 6). (C) l-18F-FAC small-animal PET/CT scan of dCK knockout mouse. P values were calculated relative to FAC for each specific tissue. *P < 0.05; n = 5 (18F-FAC), n = 3 (l-18F-FAC), n = 3 (l-18F-FMAC), n = 2 (l-18F-FFAC), and n = 2 (l-18F-FCAC). %ID/g = percentage injected dose per gram of tissue; Bl = urinary bladder; B. Marrow = bone marrow; BM = bone marrow; GI = gastrointestinal tract; KO = knockout; L = liver; S = spleen; SG = salivary gland; Thy = thymus.
In addition to bone marrow and thymus retention, l-18F-FAC and l-18F-FFAC are also retained in the liver. Similar to 18F-3′-deoxy-3′-fluorothymidine (18F-FLT) (25), these probes accumulate in the liver by a nonspecific mechanism (26). Alternatively, the liver uptake may be dCK-specific. A novel dCK knockout mouse (5) can be used to evaluate these possibilities. l-18F-FAC small-animal PET/CT scans (Fig. 5C and Supplemental Fig. 7) indicate that, with the exception of excretory organs such as gallbladder and urinary bladder, the l-18F-FAC organ distribution typically observed in wild-type mice (Fig. 5A) was absent in the dCK knockout mouse. Although these data demonstrate that the liver trapping of l-18F-FAC requires dCK expression, other mechanisms such as uptake via liver-specific transporters for unnatural l-nucleosides may also play a role.
l-18F-FAC and l-18F-FMAC Small-Animal PET/CT of Malignant and Autoimmune Lymphoproliferative Disorders in Mice
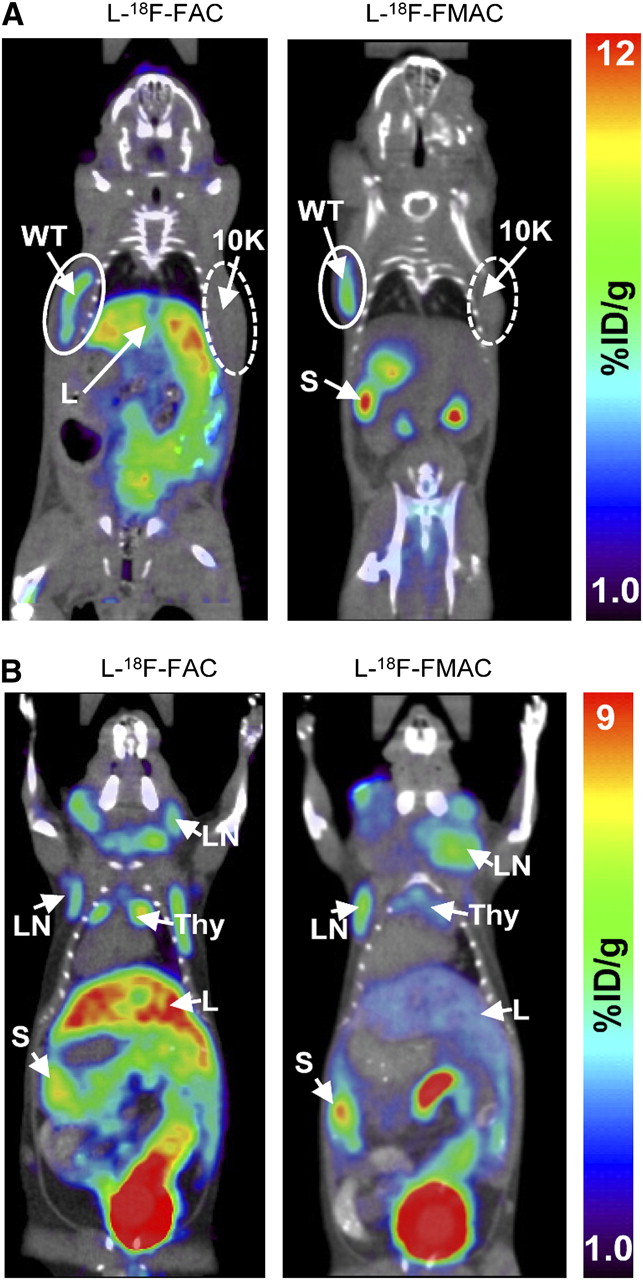
Biodistribution studies in healthy mice indicate that the deamination-resistant probes l-18F-FAC, l-18F-FFAC, and l-18F-FMAC compare favorably with 18F-FAC. Although l-18F-FMAC had a slightly lower sensitivity than l-18F-FAC and l-18F-FFAC, its low liver uptake may be advantageous in certain applications. Because the biodistribution of l-18F-FFAC was similar to that of l-18F-FAC, this probe was not selected for further evaluations. l-18F-FMAC and l-18F-FAC, the two remaining candidate probes, were compared in two mouse models of cancer and autoimmunity, previously used to evaluate 18F-FAC (4,12). The first test was to determine whether l-18F-FAC and l-18F-FMAC could distinguish between isogenic murine L1210 leukemia cells that either express dCK (wild-type cells) or lack this enzyme (L1210-10K cells) (12). As shown in Figure 6A, dCK-positive tumors were detected with both l-18F-FAC and l-18F-FMAC. Neither of these probes accumulated in the dCK-negative L1210-10K tumors. l-18F-FAC and l-18F-FMAC were then evaluated in the B6.MRL-Faslpr/J autoimmune mice (27,28). Figure 6B shows that both probes detected the cervical, axillary, and brachial lymphadenopathies characteristic of the Faslpr model.

l-18F-FAC and l-18F-FMAC small-animal PET/CT images of malignant and autoimmune lymphoproliferative disorders. (A) l-18F-FAC and l-18F-FMAC small-animal PET/CT of L1210 lymphoma tumors. L1210 parental cell line (WT, solid-lined circle) and dCK-deficient variant L1210-10K (10K, dash-lined circle) were injected subcutaneously under the left and right shoulders of the mouse, respectively. Only the parental cell line accumulated both probes. (B) l-18F-FAC and l-18F-FMAC small-animal PET/CT of autoimmune B6.MRL-Faslpr/J mice. Both probes detected cervical, axillary, and brachial lymphadenopathy in these mice. %ID/g = percentage injected dose per gram of tissue; L = liver; LN = lymph nodes; S = spleen; Thy = thymus; WT = wild-type. Number of mice per probe ≥ 3.
DISCUSSION
Factors That May Affect Performance of 18F-FAC, l-18F-FAC, and l-18F-FMAC in Preclinical and Clinical Applications
Despite its rapid deamination in vivo (Fig. 1B), 18F-FAC performs well in mice (4,12). A potential limitation of 18F-FAC concerns its muscle background, which is the highest among tested compounds (Supplemental Fig. 6). In applications in which muscle background may interfere with specific signals, 18F-FAC could be replaced by l-18F-FMAC, a probe with a similar biodistribution pattern and sensitivity but with a lower nonspecific uptake in the muscle. Regarding l-18F-FAC, its utility in mice may be limited by the high liver uptake. Furthermore, it is conceivable that, because of their unnatural l-chirality, l-18F-FAC and l-18F-FMAC may be transported less efficiently than 18F-FAC across the plasma membrane.
In the context of clinical applications, the deamination-resistant probes l-18F-FAC and l-18F-FMAC may have an advantage over 18F-FAC given the high deamination activity in certain human tissues (13). Concerning the utility of the FAC probes for treatment stratification, 18F-FAC could be the best probe to use for predicting responses to deamination-susceptible prodrugs such as cytarabine, gemcitabine, and decitabine. In contrast, l-18F-FAC and l-18F-FMAC may be more suitable for predicting responses to deamination-resistant drugs such as cladribine and clofarabine.
Comparison Between TK1 and dCK-Specific PET Probes
PET imaging of TK1 and dCK—the two rate-limiting enzymes in the deoxyribonucleoside salvage pathway—is now possible given the development of 18F-FLT by Shields et al. in 1998 (26), followed by the identification of 18F-FAC in 2008 (4) and optimized 18F-FAC analogs described herein. Direct comparisons between 18F-FLT and the 18F-FAC series of probes should take into account the fact that high levels of endogenous thymidine in rodent serum compete with 18F-FLT and reduce its sensitivity in mice (29). Nonetheless, it is likely that PET measurements of TK1 and dCK provide nonoverlapping information. Therefore, although 18F-FLT provides measurements of DNA-synthesizing activity and cell proliferation (30), dCK-specific probes may enable PET-guided identification of cancer patients who are more likely to respond to cytotoxic chemotherapy nucleoside analog prodrugs. Furthermore, recently described mouse genetic models of TK1 (31) and dCK deficiency (5) provide invaluable tools to enable the identification of biologic processes that are critically dependent on the activity of these two cytosolic deoxyribonucleoside kinases.
CONCLUSION
We describe the development of a novel set of PET probes that can be used to measure the metabolic flux through the deoxyribonucleoside salvage pathway. A better understanding of the salvage pathway in terms of its physiologic function and potential role in cancer development and progression may expand the utility of TK1 and dCK-specific PET probes beyond their current status of surrogate markers for cell proliferation, immune activation, treatment stratification, and monitoring.
Acknowledgments
We thank David Stout, Waldemar Ladno, and Judy Edwards for performing small-animal PET/CT; Rachel Laing for helping with experiments; and the cyclotron group for producing PET probes. This work was supported by the In Vivo Cellular and Molecular Imaging Centers Developmental Project Award NIH P50 CA86306; by R24CA92865, U.S. Department of Energy contract DE-FG02-06ER64249; by California Institute for Regenerative Medicine grant RT1-01126-1; and by funding from the Dana Foundation. Caius Radu, Owen Witte, and Johannes Czernin are among the inventors of the national and Patent Cooperation Treaty (PCT) patent applications for the FAC technology referred to in the article. That patent application was filed on September 19, 2008. A group of UCLA faculty members including Caius Radu, Johannes Czernin, Michael Phelps, and Owen Witte are involved in Sofie Biosciences, a startup company that has licensed this intellectual property.
Footnotes
-
↵* Contributed equally to this work.
-
Guest Editor: Leonard Wiebe, University of Alberta
-
COPYRIGHT © 2010 by the Society of Nuclear Medicine, Inc.
References
- Received for publication November 30, 2009.
- Accepted for publication March 12, 2010.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- STING-driven interferon signaling triggers metabolic alterations in pancreas cancer cells visualized by [18F]FLT PET imaging
- Predicting Gemcitabine Delivery by 18F-FAC PET in Murine Models of Pancreatic Cancer
- 18F-FAC PET Visualizes Brain-Infiltrating Leukocytes in a Mouse Model of Multiple Sclerosis
- Imaging of Activated T Cells as an Early Predictor of Immune Response to Anti-PD-1 Therapy
- 18F-FAC PET Selectively Images Liver-Infiltrating CD4 and CD8 T Cells in a Mouse Model of Autoimmune Hepatitis
- Production of diverse PET probes with limited resources: 24 18F-labeled compounds prepared with a single radiosynthesizer
- Detection of immune responses after immunotherapy in glioblastoma using PET and MRI
- A PET Imaging Strategy to Visualize Activated T Cells in Acute Graft-versus-Host Disease Elicited by Allogenic Hematopoietic Cell Transplant
- Human Biodistribution and Radiation Dosimetry of 18F-Clofarabine, a PET Probe Targeting the Deoxyribonucleoside Salvage Pathway
- [18F]CFA as a clinically translatable probe for PET imaging of deoxycytidine kinase activity
- Co-targeting of convergent nucleotide biosynthetic pathways for leukemia eradication
- Stratification of Nucleoside Analog Chemotherapy Using 1-(2'-Deoxy-2'-18F-Fluoro-{beta}-D-Arabinofuranosyl)Cytosine and 1-(2'-Deoxy-2'-18F-Fluoro-{beta}-L-Arabinofuranosyl)-5-Methylcytosine PET
- Structure-guided Engineering of Human Thymidine Kinase 2 as a Positron Emission Tomography Reporter Gene for Enhanced Phosphorylation of Non-natural Thymidine Analog Reporter Probe