


Visual Abstract

Abstract
[177Lu]Lu-PSMA is an effective class of therapy for patients with metastatic castration-resistant prostate cancer (mCRPC); however, progression is inevitable. The limited durability of response may be partially explained by the presence of micrometastatic deposits, which are energy-sheltered and receive low absorbed radiation with 177Lu due to the approximately 0.7-mm mean pathlength. 161Tb has abundant emission of Auger and conversion electrons that deposit a higher concentration of radiation over a shorter path, particularly to single tumor cells and micrometastases. 161Tb has shown in vitro and in vivo efficacy superior to that of 177Lu. We aim to demonstrate that [161Tb]Tb-PSMA-I&T will deliver effective radiation to sites of metastatic prostate cancer with an acceptable safety profile. Methods: This single-center, single-arm, phase I/II trial will recruit 30 patients with mCRPC. Key eligibility criteria include a diagnosis of mCRPC with progression after at least one line of taxane chemotherapy (unless medically unsuitable) and androgen receptor pathway inhibitor; prostate-specific membrane antigen–positive disease on [68Ga]Ga-PSMA-11 or [18F]DCFPyL PET/CT (SUVmax ≥ 20); no sites of discordance on [18F]FDG PET/CT; adequate bone marrow, hepatic, and renal function; an Eastern Cooperative Oncology Group performance status of no more than 2, and no prior treatment with another radioisotope. The dose escalation is a 3 + 3 design to establish the safety of 3 prespecified activities of [161Tb]Tb-PSMA-I&T (4.4, 5.5, and 7.4 GBq). The maximum tolerated dose will be defined as the highest activity level at which a dose-limiting toxicity occurs in fewer than 2 of 6 participants. The dose expansion will include 24 participants at the maximum tolerated dose. Up to 6 cycles of [161Tb]Tb-PSMA-I&T will be administered intravenously every 6 wk, with each subsequent activity reduced by 0.4 GBq. The coprimary objectives are to establish the maximum tolerated dose and safety profile (Common Terminology Criteria for Adverse Events version 5.0) of [161Tb]Tb-PSMA-I&T. Secondary objectives include measuring absorbed radiation dose (Gy), evaluating antitumor activity (prostate-specific antigen 50% response rate, radiographic and prostate-specific antigen progression-free survival, overall survival, objective response rate), and evaluating pain (Brief Pain Inventory–Short Form) and health-related quality of life (Functional Assessment of Cancer Therapy–Prostate and Functional Assessment of Cancer Therapy–Radionuclide Therapy). Conclusion: Enrollment was completed in February 2024. Patients are still receiving [161Tb]Tb-PSMA-I&T.
Although [177Lu]Lu-PSMA has been established as a safe and effective radiopharmaceutical therapy, enhancing overall survival and quality of life in patients with metastatic castration-resistant prostate cancer (mCRPC), disease progression remains inevitable (1). A likely contributing factor for the limited durability of response is the presence of micrometastatic deposits. Single tumor cells and micrometastases are considered energy-sheltered deposits. They receive vanishingly low absorbed radiation from 177Lu because of the approximately 0.7-mm mean pathlength (range, 0.04–1.8 mm). In comparison, the diameters of circulating tumor cells from prostate cancer patients and cultured prostate cancer cells measure on average 7.97 and 13.38 μm, respectively (2). Although a complete response of macroscopic disease is observed with [177Lu]Lu-PSMA, these small energy-sheltered deposits do not receive lethal radiation and eventually progress.
161Tb has physical properties that indicate it could be superior for medical use to the currently used 177Lu because of its abundant emission of Auger and conversion electrons. These ejected inner orbital electrons deposit their energy over a much shorter distance (nanometers to micrometers), with an order-of-magnitude higher linear energy transfer than β-electrons, resulting in greater cell death (3,4). 161Tb delivers higher radiation doses to single tumor cells and micrometastases than does 177Lu when calculating different scenarios of the radionuclide distribution (5). Several other studies have also supported theoretic dose calculations in favor of 161Tb (6–9). The physical characteristics of 161Tb and 177Lu are otherwise similar (Table 1).
Decay Characteristics of 177Lu and 161Tb (5)
161Tb has been shown superior to 177Lu in tumor cell killing and growth suppression in vitro and in vivo (10–12), supporting an additional contribution of Auger and conversion electrons. [161Tb]Tb-PSMA-617 was evaluated in tumor-bearing murine models, and the in vitro properties, pharmacokinetics, and biodistribution profile were similar to [177Lu]Lu-PSMA-617 (10). Theoretic dose calculations also confirmed an additive therapeutic effect of 161Tb when comparing both radiopharmaceutical therapies. Whether the promising theoretic and preclinical data will translate into clinical benefit has yet to be determined. We hypothesize that [161Tb]Tb-PSMA-I&T will deliver effective radiation to sites of metastatic prostate cancer with an acceptable safety profile.
MATERIALS AND METHODS
Design, Study Population, and Objectives
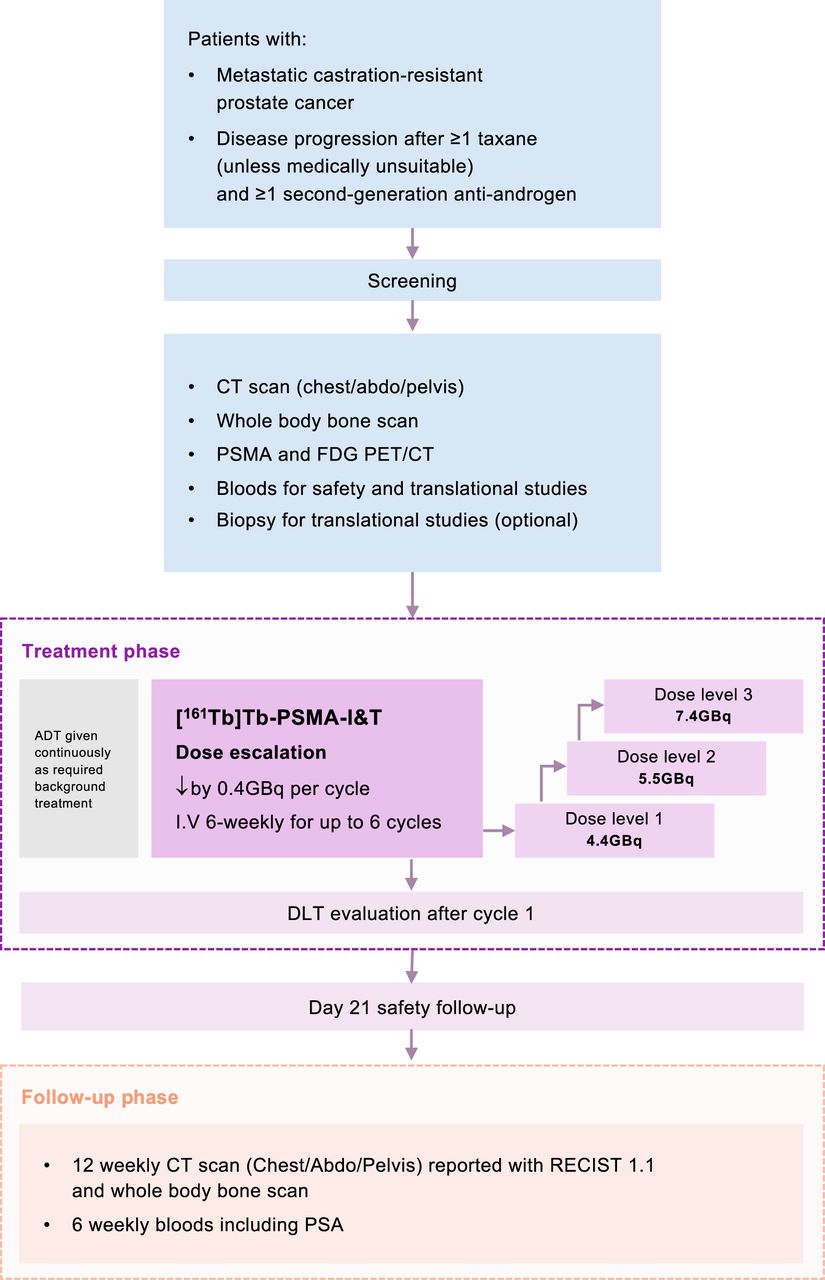
VIOLET is an investigator-initiated and -led, open-label, single-arm, single-center, phase I/II dose-escalation and -expansion study designed to evaluate the safety and efficacy of [161Tb]Tb-PSMA-I&T in patients with mCRPC. The dose-escalation phase is a traditional 3 + 3 design, with 3 prespecified activity levels. Recruitment will be halted after each cohort of 3 patients is enrolled until the dose-limiting toxicities (DLTs) have been assessed (Table 2) and the activity for the next cohort of 3 patients has been determined. The DLT assessment period is 6 wk from cycle 1 day 1. Once the maximum tolerated dose or maximum administered dose has been determined, the trial will proceed to the dose-expansion stage. The trial schema is illustrated in Figure 1.
Definitions of DLTs

Trial schema.
Patients with mCRPC who have progressed on at least one line of taxane chemotherapy (unless medically unsuitable) and an androgen receptor pathway inhibitor will be eligible for this study. Other key eligibility criteria include prostate-specific membrane antigen (PSMA)–positive disease on [68Ga]Ga-PSMA-11 or [18F]DCFPyL PET/CT (SUVmax ≥ 20); no sites of discordance on [18F]FDG PET/CT; adequate bone marrow, hepatic, and renal function; an Eastern Cooperative Oncology Group performance status of no more than 2; and no prior treatment with another radioisotope (Table 3).
Eligibility Criteria
The phase I primary objectives are to determine maximum tolerated dose, DLTs, and the recommended phase II dose of [161Tb]Tb-PSMA-I&T in patients with mCRPC. The phase II primary objective is to evaluate its safety. Secondary and exploratory objectives include estimating the absorbed radiation dose in normal organs and metastases, evaluating antitumor activity (prostate-specific antigen [PSA] response rate, radiographic and PSA progression-free survival, overall survival, objective response rate), evaluating dynamic changes in circulating tumor DNA, and evaluating pain (Brief Pain Inventory–Short Form) and health-related quality of life (Functional Assessment of Cancer Therapy–Prostate and Functional Assessment of Cancer Therapy–Radionuclide Therapy) (Table 4).
Study Objectives
The study protocol was approved by the Human Research Ethics Committee at Peter MacCallum Cancer Centre, and all participants will provide signed informed consent. The VIOLET trial is registered with ClinicalTrials.gov (NCT05521412) and sponsored by the Peter MacCallum Cancer Centre. The funders of the study had no role in study design or writing of this article.
[161Tb]Tb-PSMA-I&T Production, Administration, and Posttherapy Dosimetry
PSMA-I&T will be radiolabeled with no-carrier-added 161Tb using the same chelator (DOTAGA) as [177Lu]Lu-PSMA-I&T in the onsite hospital radiopharmacy. The production of [161Tb]Tb-PSMA-I&T will be automated on the iPHASE MultiSyn radiochemistry module using a kit formulation (Isotopia Molecular Imaging) encompassing labeling buffer and a precursor/stabilizer combination. [161Tb]Tb-PSMA-I&T production and quality control will undergo rigorous process validation leading to a product that meets all prerelease criteria matching or exceeding the Australasian Radiopharmaceutical Network production guidelines. This includes tests for radionuclidic purity, radiochemical purity, and radiochemical identity using high-pressure liquid chromatography and radio–thin-layer chromatography, as well as endotoxin and sterility testing.
[161Tb]Tb-PSMA-I&T will be administered by slow intravenous injection in an ambulatory treatment setting every 6 wk ± 1 wk, for a maximum of 6 cycles. A minimum of 500 mL of normal saline hydration over 1–2 h is recommended with radiopharmaceutical therapy, unless fluids are contraindicated. In the dose-escalation phase, there are 3 planned activity levels starting at 4.4, 5.5, and 7.4 GBq. The dose-escalation results will determine the activity of [161Tb]Tb-PSMA-I&T in the expansion phase. In both phases, the activity of [161Tb]Tb-PSMA-I&T will decrease by 0.4 GBq every cycle. All patients will receive androgen deprivation therapy continuously throughout the trial.
The subsequent activities of [161Tb]Tb-PSMA-I&T will be reduced by 20% for patients who have the following toxicities from the preceding cycle: dry mouth (grade 2), dry eyes (grade 2), nadir platelet count of less than 100 × 109/L, nadir neutrophil count of less than 1.0 × 109/L, or other significant dose-related toxicities (grade 3 or worse) that are considered both attributable to [161Tb]Tb-PSMA-I&T and activity-related. A maximum of 3 activity reductions per patient is permitted, and there are no reescalations.
Posttherapy SPECT/CT imaging will be performed after each cycle. In the dose-escalation phase, 3-time-point SPECT/CT from vertex to mid thigh will be acquired at 4 h (±2 h), 24 h (±4 h), and 96 h (±24 h) after cycle 1 and optionally thereafter. Acquisitions will be obtained on low-energy high-resolution collimators, using a double energy window peaked at 74 keV with lower scatter limits (13–15). Retention of [161Tb]Tb-PSMA-I&T will be estimated from voxel-based time–activity curves based on a multiphase exponential clearance model and convolved using a Geant4 Application for Emission Tomography–derived voxel dose kernel based on decay of 161Tb in International Commission on Radiological Protection soft tissue to yield 3-dimensional absorbed dose maps (16–18).
Assessment of Safety, Efficacy, and Patient-Reported Outcome Measures
During the treatment phase, patients will have weekly safety reviews and a full blood count performed during cycle 1. Adverse events will be graded and causality assigned according to Common Terminology Criteria for Adverse Events version 5.0 at each clinical review. For subsequent cycles, safety reviews and blood tests (full blood count, urea, creatinine, electrolytes, liver function tests, and PSA) will be repeated on day 22 and within 3 d before the next cycle. If the platelet count is less than 50 × 109/L, it will be rechecked 1 wk later and every week thereafter until it is rising. Administration of the next cycle can proceed if pretreatment bloods show a platelet level of at least 75 × 109/L and rising (≥100 × 109/L for cycle 1), a hemoglobin level of at least 80 g/L, and an absolute neutrophil count of at least 1.5 × 109/L. [161Tb]Tb-PSMA-I&T can be delayed for a maximum of 6 wk. Treatment will be withheld during grade 3 or 4 adverse events with the exception of fatigue or lymphocytopenia and not restarted until improvement to grade 0–2 or baseline.
Contrast-enhanced CT of the chest, abdomen, and pelvis, as well as whole-body bone scans, will be performed every 12 wk ± 1 wk from cycle 1 day 1, until radiologic progression. [68Ga]Ga-PSMA-11 or [18F]DCFPyL, and [18F]FDG PET/CT, will be repeated 12 wk ± 1 wk from cycle 1 day 1, and before cycle 3 day 1. Peripheral blood for translational research will be collected immediately before day 1 for cycles 1, 2, and 4 and within 10 d after unequivocal progression.
Three patient-reported outcome measure instruments will be used in this study. The Brief Pain Inventory–Short Form (19) will assess the location and severity of pain; the impact of pain on, or its interference with, daily functions; and the extent of pain relief. The Functional Assessment of Cancer Therapy–Prostate (20) will assess health-related quality of life in patients with prostate cancer, including physical, functional, emotional, and social well-being, as well as additional concerns specific to prostate cancer. The Functional Assessment of Cancer Therapy–Radionuclide Therapy is a 15-item questionnaire that has been developed for use with patients receiving radiopharmaceutical therapy and provides information about treatment-specific symptoms. Validation of the Functional Assessment of Cancer Therapy–Radionuclide Therapy and determination of the optimal subscales remain ongoing (21).
Statistical Considerations
This study will assess the toxicities associated with [161Tb]Tb-PSMA-I&T, recruiting 30–36 patients. Between 12 and 18 patients will be enrolled in the dose-escalation phase, with the number of enrolled patients dependent on the number of activity levels evaluated and the number of patients at each activity level. An additional 18 patients will be recruited in the dose-expansion phase, with a total of 24 patients treated at the maximum tolerated dose.
For the dose-escalation phase, analysis will focus primarily on adverse events, particularly DLTs. These will be tabulated descriptively for each activity level separately. The probability of observing toxicity in at least one patient is 90% for toxicities with true DLT incidence rates of 4.2% (95% CI, 0.3%–13.4%). The safety analysis will include all patients who received at least one cycle of [161Tb]Tb-PSMA-I&T. Safety will be assessed using Common Terminology Criteria for Adverse Events version 5.0, and the maximum toxicity grade per patient of each adverse event will be derived and presented in tabular form. All adverse events will be described overall and separately by activity level. The safety analysis will be reported once for all adverse events regardless of relatedness to treatment and once considering only adverse events related to treatment. The efficacy analysis will be reported for all patients who received at least one cycle of [161Tb]Tb-PSMA-I&T. PSA response rate and objective response rate will be described as percentages with 95% CIs using exact methods.
DISCUSSION
Targeting PSMA-expressing prostate cancer cells with 161Tb is a potent alternative to 177Lu. The additional clustered damage from Auger and conversion electrons within micrometastases may improve the durability of the response (Fig. 2). Preclinical data form the strong rationale for in-human investigation of [161Tb]Tb-PSMA-I&T. The VIOLET trial will establish the recommended activity and safety profile and provide the preliminary efficacy of [161Tb]Tb-PSMA-I&T in patients with mCRPC. The first patient was recruited on October 9, 2022 (Fig. 3). The VIOLET trial is named in honor of Dr. John Violet (22), who passed away unexpectedly in 2020 and had an enduring interest in Auger electron therapy.

High radiation delivery to micrometastases with [161Tb]Tb-PSMA-I&T Auger and conversion electrons from [161Tb]Tb-PSMA-I&T have high linear energy transfer, delivering more radiation to micrometastases as illustrated in enlarged prostate cancer cell (right). In contrast, β-particles deposit their energy over greater distance (left) and have lower probability of causing lethal damage to micrometastases.

Screening [68Ga]Ga-PSMA-11 PET and 3-time-point SPECT after cycle 1 of first patient recruited.
Limited use of terbium isotopes in humans has not reported any adverse events. Two studies using 152Tb, a positron emitter, provided important data because 161Tb has identical chemical characteristics and resultant pharmacokinetics. In these first-in-human studies, [152Tb]Tb-DOTATOC was administered to a patient with a metastatic neuroendocrine neoplasm (23), and [152Tb]Tb-PSMA-617 was administered to a patient with mCRPC (24). When terbium-labeled tracers were compared with their respective 68Ga-labeled tracers, all known metastases were visualized. The imaging procedures had no adverse events noted during or after the infusion. The first-in-human administration using 161Tb reported single-cycle administrations (596 and 1,300 MBq) of [161Tb]Tb-DOTATOC radiopharmaceutical therapy in 2 patients, without adverse events or significant laboratory abnormalities reported (25). After commencement of the VIOLET trial, 2 case reports of patients with mCRPC have described administration of single cycles (6.5 GBq and 5,550 MBq) of [161Tb]Tb-PSMA-617 (26,27). In both case reports, no immediate adverse events were reported. Posttherapy planar imaging and SPECT/CT were performed, without dosimetry calculations. To our knowledge, VIOLET is the first prospective clinical trial assessing [161Tb]Tb-PSMA in patients with mCRPC. A German prospective registry (REALITY; NCT04833517) is assessing various radionuclide therapies, including [161Tb]Tb-PSMA, in patients with advanced prostate cancer. The only other registered clinical trial investigating 161Tb is a Swiss randomized, crossover, prospective, single-center, open-label phase 0 study comparing the dosimetry of [177Lu]Lu-DOTATOC and [161Tb]Tb-DOTA-LM3 (Beta plus; NCT05359146) in 16 patients with neuroendocrine tumors.
We expect [161Tb]Tb-PSMA-I&T to be well tolerated, analogous to [177Lu]Lu-PSMA (28). The delivery of radiation is expected to mirror [177Lu]Lu-PSMA to off-target PSMA-expressing tissues including salivary glands, lacrimal glands, duodenum, and kidneys. The additional Auger emission from 161Tb is not expected to cause adverse events because of its subcellular (2–500 nm) pathlength range. Furthermore, 161Tb decays to the stable isotope 161Dy, which would not lead to subsequent radiation. Unbound 161Tb is not expected but would be tightly bound to diethylenetriaminepentaacetic acid with renal excretion. [161Tb]Tb-folate and [177Lu]Lu-folate have been compared to assess long-term renal damage in nude mice (29). The additional Auger and conversion electrons at similar activities did not worsen renal damage in this model. In the VIOLET trial population, delayed renal toxicity is not anticipated to be clinically relevant, as patients with mCRPC have a life expectancy in the 15- to 18-mo range after effective radiopharmaceutical therapy (30). The 6-wk DLT period chosen for VIOLET mirrors our definition in other clinical trials evaluating [177Lu]Lu-PSMA. The rationale is that hemotoxicity is expected in this window, as the nadir after 177Lu is less than 30 d. On the basis of the preclinical and clinical data available, we expect a similar nadir for 161Tb and all potential hematologic toxicities to be resolved or improving by the end of the 6-wk period.
The absorbed electron energy fraction per decay dose differs between 161Tb and 177Lu. On the basis of the biodistribution results and Monte Carlo simulations, the absorbed radiation dose from β-emission of [161Tb]Tb-PSMA-617 is approximately 35%–40% higher per decay than that of [177Lu]Lu-PSMA-617 (10,31). In the VIOLET trial, the starting activities of [161Tb]Tb-PSMA-I&T for each level are 4.4, 5.5, and 7.4 GBq, which are approximately equivalent to 5.9, 7.4, and 10.0 GBq of [177Lu]Lu-PSMA, respectively, and do not account for additional radiation from Auger and conversion electrons. Before the TheraP and VISION trials, few dose-escalation studies of [177Lu]Lu-PSMA were performed (32). The activities and cycle schedules were largely empiric based on experience using [177Lu]Lu-DOTATATE in neuroendocrine tumors and dosimetry safety data from [177Lu]Lu-PSMA. Nevertheless, we took a conservative approach by adopting a dose-escalation design to enable clinical evaluation for adverse effects and performing multiple-time-point posttherapy quantitative SPECT/CT enabling robust calculation of the actual radiation dose delivered to normal organs and tumors. This provides a significant layer of safety oversight that is not possible with conventional drugs. We chose PSMA-I&T as the small-molecule inhibitor to target PSMA, as [177Lu]Lu-PSMA-I&T is considered radioequivalent to [177Lu]Lu-PSMA-617. The European Association of Nuclear Medicine procedure guidelines for 177Lu-labeled PSMA ligands consider the current available data and do not indicate differences in efficacy between [177Lu]Lu-PSMA-617 and [177Lu]Lu-PSMA-I&T (33) because of equivalent clinical responses and toxicities (34).
The VIOLET trial will generate robust data for appropriate activities of [161Tb]Tb-PSMA in patients with mCRPC. High yields of no-carrier-added 161Tb can be produced for clinical application by neutron irradiation of 160Gd targets, analogous to the production of 177Lu (35,36). Upscaling the production is expected to be feasible, similar to 177Lu production (37). Since the physical properties of 161Tb and 177Lu are similar, no additional radioprotection measures would be required. The question of greatest interest is whether 161Tb will be superior to 177Lu in humans, particularly for killing micrometastases. In similar cohorts of patients with mCRPC treated with [177Lu]Lu-PSMA, median progression-free survival was 5.1 mo (95% CI, 3.4–5.7 mo) and overall survival was 16.4 mo (95% CI, 13.7–19.4 mo) in TheraP (38), and median radiographic PFS was 8.7 mo and median overall survival was 15.3 mo in VISION (30). This study will provide initial data on PFS and OS with [161Tb]Tb-PSMA but is not designed to enable direct comparison with [177Lu]Lu-PSMA.
CONCLUSION
VIOLET is a phase I/II clinical trial with enrollment completed in February 2024. Patients are still receiving [161Tb]Tb-PSMA-I&T.
DISCLOSURE
This study is funded by the Challenge Award from the Prostate Cancer Foundation through funds from CANICA AS and the Peter MacCallum Foundation. Isotopia is supplying [161Tb]Tb and PSMA-I&T kits as part of a commercialization agreement with the Peter MacCallum Cancer Centre. James Buteau receives support from a Prostate Cancer Foundation Young Investigator Award and PhD support through an Australian Government Research Training Program Scholarship. Louise Kostos received PhD support through an Australian Government Research Training Program Scholarship. Michael Hofman is supported through an NHMRC investigator grant. Shahneen Sandhu is supported through an NHMRC investigator grant. Brian Gonzalez received fees unrelated to this work from Sure Med Compliance and Elly Health. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Is [161Tb]Tb-PSMA-I&T a safe and effective radiopharmaceutical therapy for patients with mCRPC?
PERTINENT FINDINGS: We describe the protocol for the VIOLET trial, an investigator-initiated and -led, open-label, single-arm, single-center, phase I/II dose-escalation and expansion study designed to evaluate the safety and efficacy of [161Tb]Tb-PSMA-I&T. Recruitment was completed in February 2024.
IMPLICATIONS FOR PATIENT CARE: The VIOLET trial will generate robust data on the appropriate activity of [161Tb]Tb-PSMA in patients with mCRPC.
ACKNOWLEDGMENTS
The protocol of this study was presented as a poster at the ASCO Genitourinary Cancers Symposium, San Francisco, February 16–18, 2023. We thank the participating patients and their families; Annette Van Der Heyden (ProsTIC program manager); Claire Martin and Richelle Linklater (trial coordination with the Parkville Cancer Clinical Trials Unit); Petra Opar (clinical trial project management with the Centre of Biostatistics and Clinical Trials); the cancer imaging nuclear medicine and radiology consultants, technologists, and nurses; and Nicole Ng, Maria Docanto, and Patricia Bukczynska (Cancer Research Division).
Footnotes
↵* Contributed equally to this work.
Published online Jul. 11, 2024.
- © 2024 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication February 21, 2024.
- Accepted for publication May 28, 2024.
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.