


Abstract
Comorbid Lewy body (LB) pathology is common in Alzheimer disease (AD). The effect of LB copathology on 18F-FDG PET patterns in AD is yet to be studied. We analyzed associations of neuropathologically assessed tau pathology, LB pathology, and substantia nigra neuronal loss (SNnl) with antemortem 18F-FDG PET hypometabolism in patients with a clinical AD presentation. Methods: Twenty-one patients with autopsy-confirmed AD without LB neuropathologic changes (LBNC) (pure-AD), 24 with AD and LBNC copathology (AD-LB), and 7 with LBNC without fulfilling neuropathologic criteria for AD (pure-LB) were studied. Pathologic groups were compared regarding regional and voxelwise 18F-FDG PET patterns, the cingulate island sign ratio (CISr), and neuropathologic ratings of SNnl. Additional analyses assessed continuous associations of Braak tangle stage and SNnl with 18F-FDG PET patterns. Results: Pure-AD and AD-LB showed highly similar patterns of AD-typical temporoparietal hypometabolism and did not differ in CISr, regional 18F-FDG SUVR, or SNnl. By contrast, pure-LB showed the expected pattern of pronounced posterior-occipital hypometabolism typical for dementia with LB (DLB), and both CISr and SNnl were significantly higher compared with the AD groups. In continuous analyses, Braak tangle stage correlated significantly with more AD-like, and SNnl with more DLB-like, 18F-FDG PET patterns. Conclusion: In autopsy-confirmed AD dementia patients, comorbid LB pathology did not have a notable effect on the regional 18F-FDG PET pattern. A more DLB-like 18F-FDG PET pattern was observed in relation to SNnl, but advanced SNnl was mostly limited to relatively pure LB cases. AD pathology may have a dominant effect over LB pathology in determining the regional neurodegeneration phenotype.
Alzheimer disease (AD) and dementia with Lewy bodies (DLB) are 2 distinct neurodegenerative conditions defined by the cerebral accumulation of amyloid-β plaques and tau neurofibrillary tangles (NFTs) and of α-synuclein containing Lewy bodies (LB), respectively (1,2). In contrast to AD, DLB typically presents with denervation of the nigrostriatal dopaminergic pathway caused by degeneration of dopaminergic substantia nigra neurons (3), as well as more predominant executive and visuospatial deficits accompanied by visual hallucinations, cognitive fluctuations, parkinsonism, and rapid-eye-movement sleep behavioral disorder (4). Although AD and DLB have unique neuropathologic profiles, up to 60% of clinical AD and DLB patients present with neuropathologic findings of both diseases (5,6). Concomitant LB pathology in clinical AD has been associated with faster cognitive decline (7–9), younger age at death (8), and usually more DLB-like clinical features (9–12), although this could not be confirmed by others (7,13,14). In the era of disease-modifying therapies, these patients may benefit less from amyloid-lowering therapies (15) and may potentially show a better response to cholinesterase inhibitors (16). Biomarkers identifying these patients may thus allow for a more targeted treatment of AD (11,17).
PET with the glucose analog 18F-FDG is a well-established modality for imaging neurodegeneration, and differentiated hypometabolism patterns have been established for different conditions (18). Particularly, in contrast to the characteristic temporoparietal pattern of hypometabolism in AD, patients with DLB are characterized by a more pronounced posterior–occipital pattern of hypometabolism with relatively preserved metabolism in the medial temporal lobe (MTL) and in the posterior cingulate, with the latter being known as the cingulate island sign (19,20). The cingulate island sign is a well-established biomarker for distinguishing patients with DLB and AD (19,21), even at prodromal stages (22).
Previous imaging–pathologic association studies have demonstrated that AD copathology in DLB associates with a less DLB-typical hypometabolic pattern (20,23), but the potential contributions of 18F-FDG PET to the identification of mixed pathology in AD-like presentations are yet to be explored. Here, we assessed antemortem 18F-FDG PET patterns of clinically diagnosed AD patients in relation to AD and LB neuropathology at autopsy.
MATERIALS AND METHODS
Study Participants
Our cohort included 59 participants enrolled in the Alzheimer’s Disease Neuroimaging Initiative (ADNI, https://adni.loni.usc.edu/) who had neuropathologic examinations at autopsy, a clinical diagnosis of AD dementia or amnestic mild cognitive impairment at the last clinical evaluation, and available antemortem 18F-FDG PET scans. The average interval between the last available 18F-FDG PET acquisition and death was 3.0 ± 2.6 y.
Neuropathologic Assessments
Neuropathologic assessments were performed by the ADNI Neuropathology Core following the National Institute on Aging–Alzheimer’s Association guidelines (24–26). Standard rating scales for AD pathology (amyloid, tau, neuritic plaques) were further merged into the AD neuropathologic change (ADNC) composite, whereas LB pathology assessment followed the criteria of McKeith et al. (4). Patients were considered to have autopsy-confirmed AD when presenting with intermediate or high ADNC (24), and the presence of LB neuropathologic changes (LBNC) was denoted when LBs were present in limbic or neocortical regions or the amygdala (4). Amygdala-predominant LBs, which have been suggested to be characteristic of advanced AD and less likely related to DLB (27), were considered as positive for LBNC. Patients with LBNC restricted to the brain stem were excluded. Patients were stratified as having autopsy-confirmed AD without LBNC (pure-AD), autopsy-confirmed AD with comorbid LBNC (AD-LB), LBNC without fulfilling neuropathologic criteria for AD (pure-LB), or none of these (negative). We also studied semiquantitative ratings (assessed on a scale from 0 to 3) of substantia nigra neuronal loss (SNnl) as a marker of DLB-specific neurodegeneration (4,28). For a subset of patients (n = 45/59), semiquantitative ratings of the regional loads of tau NFTs and LBs were available (Supplemental Table 1; supplemental materials are available at http://jnm.snmjournals.org).
Genetics
APOE genotype was determined by Cogenics using standard methods to genotype the 2 APOE-ɛ4–defining single-nucleotide polymorphisms (rs429358 and rs7412). Patients were labeled as having 0, 1 or 2 ɛ4 copies.
Neuropsychologic Evaluation
The Mini-Mental State Examination was used to characterize global cognitive performance (29). Domain-specific composite scores were used to assess memory (MEM) (30) and executive function (EXEC) (31). In addition, we calculated a cognitive profile variable, Δ (MEM − EXEC), to characterize relative impairments between these 2 domains (32). The average interval between neuropsychologic evaluation and death was 1.9 ± 2.0 y.
18F-FDG PET Acquisition and Processing
We used 18F-FDG PET images in fully preprocessed format (level 4) as provided by ADNI. The acquisition and preprocessing are detailed elsewhere (33). Blood glucose levels, previously associated with changes in posterior–occipital hypometabolism (34), are reported. 18F-FDG PET images were spatially normalized using SPM, version 12 (https://www.fil.ion.ucl.ac.uk/spm/), and intensity normalized using a previously validated data-driven method (35) and 18F-FDG PET data from 179 cognitively normal ADNI subjects (the control group). Region-of-interest (ROI) analysis was performed to calculate the average 18F-FDG uptake in the occipital cortex and the MTL (21), as well as the cingulate island sign ratio (CISr) (20,22) between the posterior cingulate cortex and the precuneus and cuneus uptake. To this end, we used the corresponding ROIs from the Harvard–Oxford neuroanatomic atlas.
Statistical Analysis
Two-sample t tests and Mann–Whitney U tests were used for comparing normally distributed continuous variables and nonnormally distributed and ordinal variables, respectively. Effect sizes were reported as Cohen’s d.
Hypometabolism patterns were determined by voxel-wise 2-sample t tests between each pathologic group and the control group using SPM. Age, sex, and blood glucose levels were used as confounding nuisance covariates (34,36). T-score maps were transformed to Cohen’s d maps, and a threshold was applied using a P value of less than 0.05 (corrected using the false discovery rate [FDR]) and a cluster size of more than 250 voxels. For secondary analysis, the AD-LB group was separated into AD with amygdala-predominant LBs and AD with limbic or neocortical LBs. The different pathologic groups were also directly compared. Spatial similarities between hypometabolism patterns were assessed using spatial Spearman correlation analysis across the 52 ROIs defined in the Harvard–Oxford atlas (37).
In addition, we performed regional and voxel-wise Spearman correlation analyses of the association of AD-specific (Braak tau stage) and DLB-specific (SNnl) neuropathologic markers with 18F-FDG PET patterns. In complementary analyses, we also assessed associations between 18F-FDG PET and semiquantitative ratings of regional LB and NFT load (supplemental materials).
RESULTS
Demographics and Neuropathology
Seven subjects (11.9%) did not fulfil the criteria for either ADNC or LBNC, including 2 cases that had LBs restricted to the brain stem (and ADNC ≤ 1). Of the remaining 52 subjects, 21 (35.6%) had autopsy-confirmed AD without LBNC (pure-AD), 24 (40.7%) had autopsy-confirmed AD with LBNC copathology (AD-LB), and 7 (11.9%) had LBNC without fulfilling pathologic criteria for AD (ADNC ≤ 1) (pure-LB). Among AD-LB, 16 patients presented limbic/transitional or neocortical LBNC (67%), whereas 8 patients presented amygdala-predominant LBNC (33%).
Patients in the pure-AD and AD-LB groups did not differ with respect to age, sex, APOE ɛ4 positivity, or blood glucose levels, but pure-LB patients were significantly older (P = 0.039) and less often carriers of the APOE ɛ4 allele (P = 0.018) than the other groups (Table 1). Regarding neuropathology, the pure-AD and the AD-LB groups did not differ in severity of Braak stages (P = 0.695) or regional NFT burden (Supplemental Fig. 1). Amygdala-predominant LBs were significantly more frequent in the AD-LB group than in the pure-LB group (P = 0.047), but semiquantitative ratings of regional LB burden did not differ between these groups (Supplemental Fig. 1). Finally, SNnl was significantly higher for pure-LB than for pure-AD (P = 0.005) and AD-LB (P = 0.020) but similar between pure-AD and AD-LB (P = 0.210).
Demographic Characteristics of the Different Pathologic Subgroups
In terms of cognition, there were no significant differences in Mini-Mental State Examination between groups, but patients in the AD-LB group showed significantly worse memory performance than the pure-AD (P = 0.012) and pure-LB (P = 0.004) groups, whereas executive function was similar among groups. Accordingly, AD-LB subjects showed a memory-predominant cognitive profile in Δ (MEM − EXEC) (1-sample t test, P = 0.045), whereas pure-LB subjects showed a disproportionate executive impairment (P = 0.057), and pure-AD cases showed balanced deficits in both domains (P = 0.161). Limbic LB load correlated negatively with MEM (after correcting for the effect of tau NFT burden) across the whole cohort but not in the AD-LB group alone (Supplemental Table 2).
18F-FDG Patterns of Pathologically Defined Groups
Compared with healthy controls, the pure-AD group showed the classic AD hypometabolism pattern, with pronounced medial and lateral temporal effects extending to the lateral parietal cortex, posterior cingulate, and precuneus; mild frontal hypometabolism; and well-preserved occipital metabolism (Fig. 1A). The mixed-pathology AD-LB group was characterized by a spatial pattern remarkably similar to the pure-AD group (spatial correlation, ρ = 0.82). Interestingly, the same pattern was also observed when analyzing AD-LB cases with limbic/neocortical or amygdala-predominant LB separately (Fig. 1B). By contrast, the pure-LB group showed the typical DLB pattern of pronounced posterior–occipital hypometabolism with relative sparing of the MTL and the posterior cingulate, which as expected did not spatially correlate with the pure-AD pattern (ρ = 0.09). In direct comparisons, only non-significant differences were observed between the pure-AD and AD-LB groups, whereas the pure-LB group showed significant posterior–occipital hypometabolism and a relative sparing of frontal and temporal regions in comparison to both the pure-AD and the AD-LB groups (Fig. 1C).

(A) Hypometabolism patterns of pathologic groups compared with controls. (B) Patterns of limbic/neocortical and amygdala-predominant LB subgroups in AD-LB. (C) Direct comparisons between pathologic groups. Color represents effect size. White bars in color bars: P < 0.05 (FDR-corrected).
ROI-based analyses fully reproduced and quantified the voxel-wise observations, revealing significant MTL hypometabolism in the pure-AD and AD-LB groups, and occipital hypometabolism in the pure-LB group (Fig. 2).

Comparison of MTL and occipital cortex 18F-FDG SUV ratios (SUVR) between different neuropathologic groups and the control group (CN).
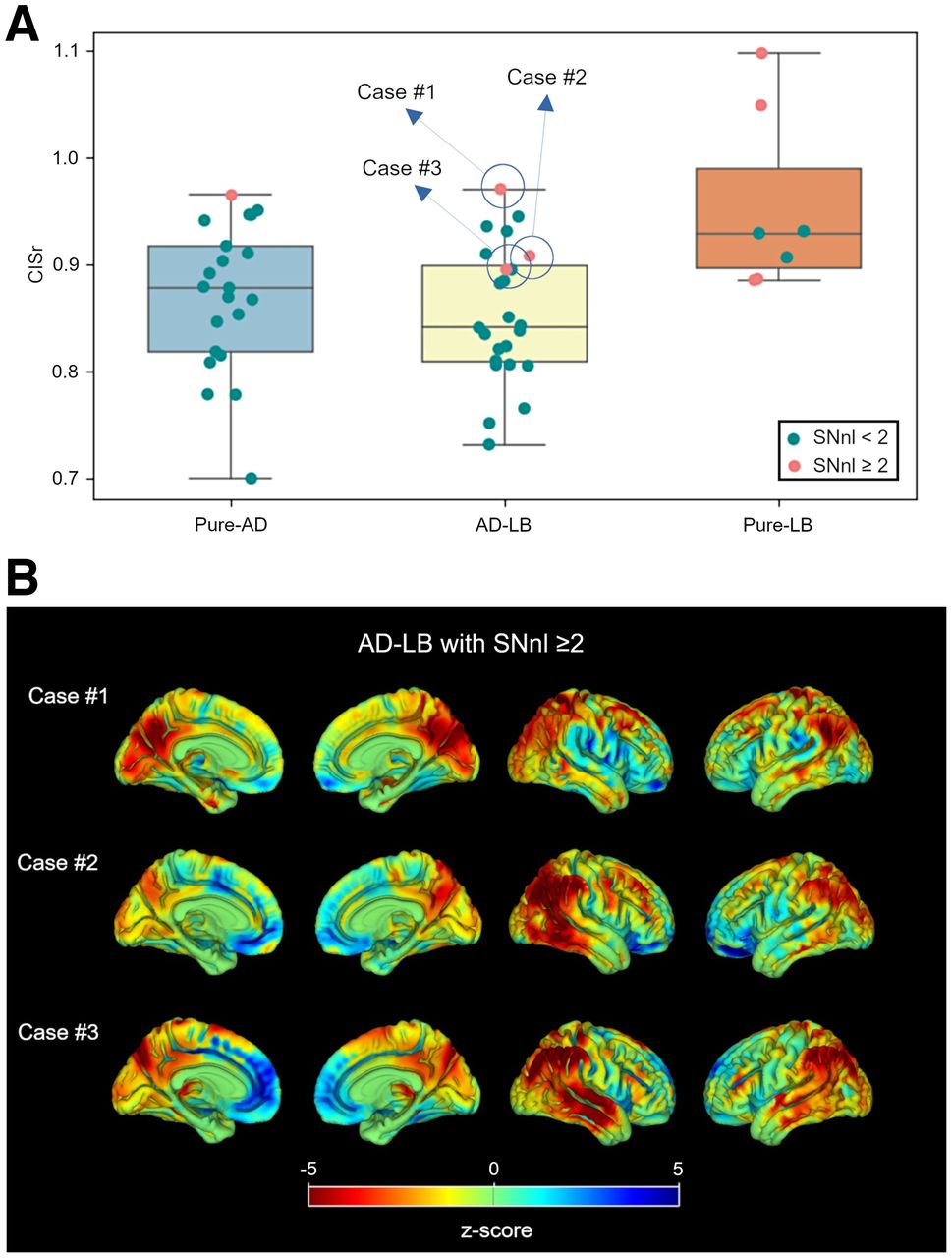
Additionally, patients in the pure-LB group exhibited significantly higher CISr than those in the pure-AD (d = 0.78, P = 0.010) and the AD-LB (d = 0.95, P = 0.002) groups, but the CISr did not differ between the pure-AD and AD-LB groups (d = 0.15, P = 0.375) (Fig. 3A). CISr was also similar for limbic/neocortical and amygdala-predominant AD-LB patients (d = 0.18, P = 0.516). By contrast, patients with an elevated SNnl (≥2) showed a significantly higher CISr (d = 1.49, P < 0.001), even when considering only the pure-AD and AD-LB groups (d = 1.31, P = 0.016). Moreover, individual z score maps of the 3 AD-LB patients with elevated SNnl revealed a more prominent DLB-like or mixed hypometabolism pattern (Fig. 3B; spatial correlations: case 1, ρ = 0.82 and 0.26; case 2, ρ = 0.57 and 0.17; case 3, ρ = 0.57 and 0.41, for the pure-LB and pure-AD patterns, respectively).

(A) CISr comparisons between pathologic groups. Cases with high SNnl (≥ 2) are highlighted in orange. (B) Individual z score maps of 3 AD-LB cases with high SNnl.
Continuous Associations of Braak Tau Stage and SNnl with 18F-FDG PET Patterns
To better understand the role of AD- and DLB-specific neuropathologic markers in shaping the observed 18F-FDG PET patterns, we studied the continuous associations of Braak tau stage and SNnl with 18F-FDG PET ROI values across the full sample (Fig. 4). Braak stage correlated negatively with CISr and MTL metabolism, whereas SNnl correlated positively with CISr and negatively with occipital metabolism. In complementary analyses, similar associations with regional 18F-FDG PET markers were observed when using regional tau NFT loads instead of Braak tau stage (Supplemental Fig. 2), but regional LB burden did not significantly correlate with 18F-FDG PET features (Supplemental Fig. 3).

Correlations of Braak tau stage and SNnl with regional 18F-FDG PET markers.
In additional voxel-wise analyses, higher Braak tau stages correlated with more hypometabolism in the posterior cingulate, MTL, and temporoparietal cortex, as well as with less hypometabolism in the occipital and the paracentral cortex (Fig. 5, top). SNnl was correlated with more hypometabolism in occipital and parietotemporal regions and less hypometabolism in the orbitofrontal cortex and the posterior cingulate (Fig. 5, bottom).

Voxel-wise correlations of Braak tau stage and SNnl with 18F-FDG uptake.
DISCUSSION
In the present work, we analyzed antemortem 18F-FDG PET patterns in relation to AD and LB pathology in a cohort of clinical AD patients. Concomitant AD-LB patients did not show more DLB-like 18F-FDG PET features but, rather, a pattern regionally very similar to the pure-AD pattern (Figs. 1 and 2). Accordingly, the CISr (20–22) did not differ between the pure-AD and AD-LB groups (Fig. 3). In contrast to our results, in a previous work comparing pathologically verified DLB (n = 3) and AD-LB (n = 3) patients (38), the authors reported similar occipital hypometabolism in both groups. These differences may be explained by different definitions of the AD-LB group, since AD-LB patients in this previous work presented DLB symptomatology whereas those in our study exhibited a relatively pure AD phenotype. Interestingly, the amygdala-predominant LB type, which has been previously linked to AD (27), was indeed higher in the AD-LB group than in the pure-LB group (from which it was completely absent), but this did not seem to affect the neurodegeneration phenotype (Fig. 1B). Although the little effect of comorbid LB pathology on the regional 18F-FDG PET pattern may come as a surprise, it is in line with the lack of elevated SNnl, a pathologic hallmark of LB-typical neurodegeneration, in these comorbid AD-LB cases (28,39).
Although AD-LB patients showed an even more amnestic-predominant cognitive profile than the pure-AD patients, rather than a more dysexecutive phenotype typical of DLB (4), this difference is unlikely to result from a more advanced AD pathology in the AD-LB group, as severity of both Braak tau stage (Table 1) and regional NFT burden (Supplemental Fig. 1) were comparable between pure-AD and AD-LB. Previous studies have similarly suggested that comorbid LB pathology exacerbates AD-typical cognitive deficits but does not necessarily produce a mixed clinical phenotype (7,13,14), whereas others did observe more DLB symptomatology in AD-LB cases (8–12,40). These differences may be explained by different clinicopathologic definitions of the AD-LB groups, as some autopsy studies define the different pathology groups based solely on neuropathologic criteria (8,40), whereas others also restrict their samples to a particular clinical phenotype as in our study (6,13,20,23).
Interestingly, a smaller group of patients (12%) who had relatively pure LBNC with no or low ADNC did indeed show the expected DLB-like posterior-occipital hypometabolism pattern (20), which was accompanied by significantly elevated SNnl. Moreover, quantitative neuropsychologic analysis showed these patients to have a more dysexecutive rather than amnestic-predominant profile. Thus, it is likely that these cases may reflect misdiagnosed DLB patients with no DLB-specific symptomatology and a clinical profile more similar to AD (4,41). Our findings indicate that 18F-FDG PET may serve as a useful imaging marker to identify this nonnegligible and clinically highly relevant portion of misdiagnosed AD patients in vivo. Interestingly, despite having similar regional loads of LB pathology (Supplemental Fig. 1), the comorbid AD-LB group did not exhibit a DLB-like 18F-FDG PET pattern or elevated SNnl, suggesting that AD pathology may have a dominant effect over LB pathology in determining the regional neurodegeneration phenotype in these patients. Altogether, these results suggest that the role of LBs in AD-LB may be different from that of pure-LBs. Although recent studies have provided evidence of in vivo interactions between tau and α-synuclein (42), more work is needed to better understand how these interactions may modify the effect of LB pathology on the neurodegeneration phenotype in AD-LB.
In continuous association analyses, we observed that Braak tau stage (Figs. 4 and 5) and tau NFT load (Supplemental Fig. 2) correlated significantly with more AD-like 18F-FDG PET features, confirming and expanding recent findings obtained in a smaller subsample of the ADNI autopsy cohort (43) as well as similar observations previously reported in clinical DLB (20,23). Interestingly, SNnl showed the opposite pattern of associations, being associated with a higher CISr, lower occipital SUVR, and a more DLB-like hypometabolism pattern in voxelwise analysis (Figs. 4 and 5). Most interestingly, this association was even observed on an individual basis in a small subset of AD-LB patients who did have advanced SNnl (Fig. 3B). Nevertheless, this applied only to 3 AD-LB cases (12.5%), suggesting that comorbid LB rarely affects the neurodegeneration phenotype in cases with fully developed AD pathology. More research is necessary to better understand the neurobiologic factors that determine why comorbid LB pathology leads to SNnl and a DLB-typical neurodegeneration pattern in some patients but not in others (44).
Altogether, our results suggest that it may not be the presence of LB pathology by itself, but rather the associated SNnl, that links with a more DLB-like hypometabolic pattern in these clinical AD patients. This notion was further corroborated by the fact that semiquantitative ratings of regional LB burden were not significantly associated with 18F-FDG PET markers (Supplemental Fig. 3). To the best of our knowledge, our study is the first report demonstrating these associations. A very recent multimodal neuroimaging study (n = 55) has pointed to an association between nigrostriatal degeneration (as assessed by dopamine transporter SPECT) and cortical hypometabolism in clinical DLB (45). However, such associations had not yet been assessed using neuropathologic evaluations or in the context of clinical AD. Additional studies combining 18F-FDG PET with imaging modalities aimed to evaluate SN degeneration in vivo (45–48) would be of great interest for replicating and studying these associations in larger observational cohorts.
Regarding the clinical implications of our work, our novel finding of comparable 18F-FDG PET patterns in AD-LB and pure-AD suggests that 18F-FDG PET may not be able to readily detect comorbid LB pathology in AD patients, which may be a disappointing finding that is nevertheless of the utmost clinical relevance. Although larger studies might be useful to corroborate these findings, the comparably large sample used here (n = 21 for pure-AD vs. n = 23 for AD-LB) and the low effect size estimates indicate that this finding would be unlikely to change with higher sample sizes. However, according to our findings, 18F-FDG PET may be useful for identifying a subset of clinically diagnosed AD patients who have relatively pure LB pathology, as well as those pathologic AD patients for whom the comorbid LB pathology is accompanied by substantia nigra neurodegeneration. Identifying these patients has important clinical implications because these will most likely also show different clinical trajectories (7), including development of more DLB-typical symptomatology (9), and may possibly also exhibit the typical susceptibility to antagonistic dopaminergic neuroleptics known for DLB patients (49).
Our work also presents a series of limitations. First, the restriction to patients with typical AD-like clinical presentations limits the reach of our conclusions to this particular clinical setting, and different effects of comorbid AD-LB pathology may be observed in clinically more diverse dementia cohorts (8). Nevertheless, identifying (comorbid) LB pathology in clinical AD patients poses a distinct diagnostic challenge—which has not been addressed so far using 18F-FDG PET—that has potentially high relevance for individual patient management and recruitment into AD clinical trials (6). In close relation, neuropsychologic data collected within the ADNI study allow for the assessment of a specific dysexecutive or amnestic-predominant neuropsychologic profile, but DLB core features are not assessed in enough detail (or are not assessed at all). Finally, quantitative assessments of regional pathologic load may represent a closer pathologic correlate of phenotypic differences than the standardized semiquantitative rating scales used here (44).
CONCLUSION
18F-FDG PET may not be able to readily detect comorbid LB pathology in clinical AD, but it may be useful for identifying a subset of patients with prominent LB-related neurodegeneration—a capability that may have important implications for patient management, individualized disease prognostication, and selection for treatment trials.
DISCLOSURE
This study has been funded by Instituto de Salud Carlos III through the projects “PI20/00613 and PI19/01576” (co-funded by European Regional Development Fund; “A way to make Europe”) and the Junta de Andalucía (CVI-02526, CTS-7685, PE-0186–2019, and PI-0046-2021). Jesús Silva-Rodríguez is a “Sara Borrell” fellow (CD21/00067), and Michel Grothe is a “Miguel Servet” fellow (CP19/00031). Miguel Labrador-Espinosa is supported by VI-PPIT-US (University of Seville, USE-19094-G). Alexis Moscoso is supported by Gamla Tjänarinnor. Michael Schöll is supported by the Knut and Alice Wallenberg Foundation (KAW 2014.0363), the Swedish Research Council (2017-02869), the Swedish state under the ALF-agreement (ALFGBG-813971), and the Swedish Alzheimer Foundation (AF-740191). Data collection and sharing for this project were funded by the ADNI, a public–private partnership program supported by national funding agencies, as well as contributions from several companies (https://adni-dup.loni.usc.edu/about/funding/). No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Is the presence of LB pathology, or related SNnl, associated with a differential 18F-FDG PET pattern in clinical AD?
PERTINENT FINDINGS: LB copathology did not affect the 18F-FDG PET pattern in autopsy-confirmed AD, but a distinct posterior–occipital 18F-FDG PET pattern was observed in relation to SNnl, most commonly in clinical AD cases with relatively pure LB pathology at autopsy.
IMPLICATIONS FOR PATIENT CARE: 18F-FDG PET can identify clinically diagnosed AD patients who have relatively pure LB pathology and substantia nigra neurodegeneration at autopsy. In vivo identification of these patients has important implications for clinical patient management, individualized disease prognostication, and selection for treatment trials.
ACKNOWLEDGMENT
Data used in preparation of this article were obtained from the ADNI database. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Footnotes
Published online Aug. 25, 2022.
- © 2023 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication April 5, 2022.
- Revision received August 3, 2022.
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.