


Abstract
Imaging of mild traumatic brain injury (TBI) using conventional techniques such as CT or MRI often results in no specific imaging correlation that would explain cognitive and clinical symptoms. Molecular imaging of mild TBI suggests that secondary events after injury can be detected using PET. However, no single specific pattern emerges that can aid in diagnosing the injury or determining the prognosis of the long-term behavioral profiles, indicating the heterogeneous and diffuse nature of TBI. Chronic traumatic encephalopathy, a primary tauopathy, has been shown to be strongly associated with repetitive TBI. In vivo data on the available tau PET tracers, however, have produced mixed results and overall low retention profiles in athletes with a history of repetitive mild TBI. Here, we emphasize that the lack of a mechanistic understanding of chronic TBI has posed a challenge when interpreting the results of molecular imaging biomarkers. We advocate for better target identification, improved analysis techniques such as machine learning or artificial intelligence, and novel tracer development.
Traumatic brain injury (TBI) is a significant public health problem worldwide. It is the leading cause of death and disability in children and young adults in the United States, with variable long-term outcomes in survivors (1–3). There is increasing evidence that for some individuals, TBI might be not only an acute and self-limiting event but also a trigger, which can initiate a chronic, sometimes lifelong degenerative process (4).
Acute brain injuries are traditionally classified clinically by the Glasgow coma scale (GCS) as mild, moderate, or severe (5). A detailed summary of TBI levels with the corresponding clinical definitions and reported symptoms is listed in Table 1.
Terminology for Head Trauma in TBI
Incidence rates vary because there is no consensus on a common or standardized classification system. Evidence from European epidemiologic studies calculate a TBI severity ratio of 22:1.5:1 for mild, moderate, and severe injuries, respectively (6), whereas in the United States, recent reports suggest that 82.5% of TBIs are mild, 8.3% moderate, and 1.0% severe (7).
The GCS score is a useful method to assess the level of consciousness among trauma victims but can be inaccurate because of levels of sedation and the need for intubation. More importantly, most TBIs are mild, including so-called concussions, and the GCS score is not useful to assess such cases. In addition, GCS scores do not provide pathophysiologic or mechanical information on the injury.
Imaging can provide valuable in vivo information on the extent and severity of an acute brain injury. In particular, CT of the head or MRI is used in the clinical work-up of injuries for GCS scores of below 13. Unenhanced CT is the imaging modality of choice for the evaluation of acute moderate to severe TBI. It has many benefits, which include low cost, wide availability, and high accuracy for triaging TBI patients in need of surgical intervention. However, for mild TBI (mTBI), CT may fail to detect more subtle anatomic findings of acute injury.
For mild injuries with GCS scores of 13–15, specific criteria to warrant the use of CT or MRI should be present. These include a GCS of less than 15 at 2 h after trauma, a suspected open or depressed skull fracture, any sign of skull base fracture, an age of 65 y or older, amnesia for more than 30 min, 2 or more episodes of vomiting, and a dangerous mechanism of injury (8). However, there is no consensus on the clinical indications to perform MRI for acute trauma. MRI may be considered when there are discrepancies between CT and clinical findings, such as negative CT results and poor neurologic status, or when there are persistent symptoms.
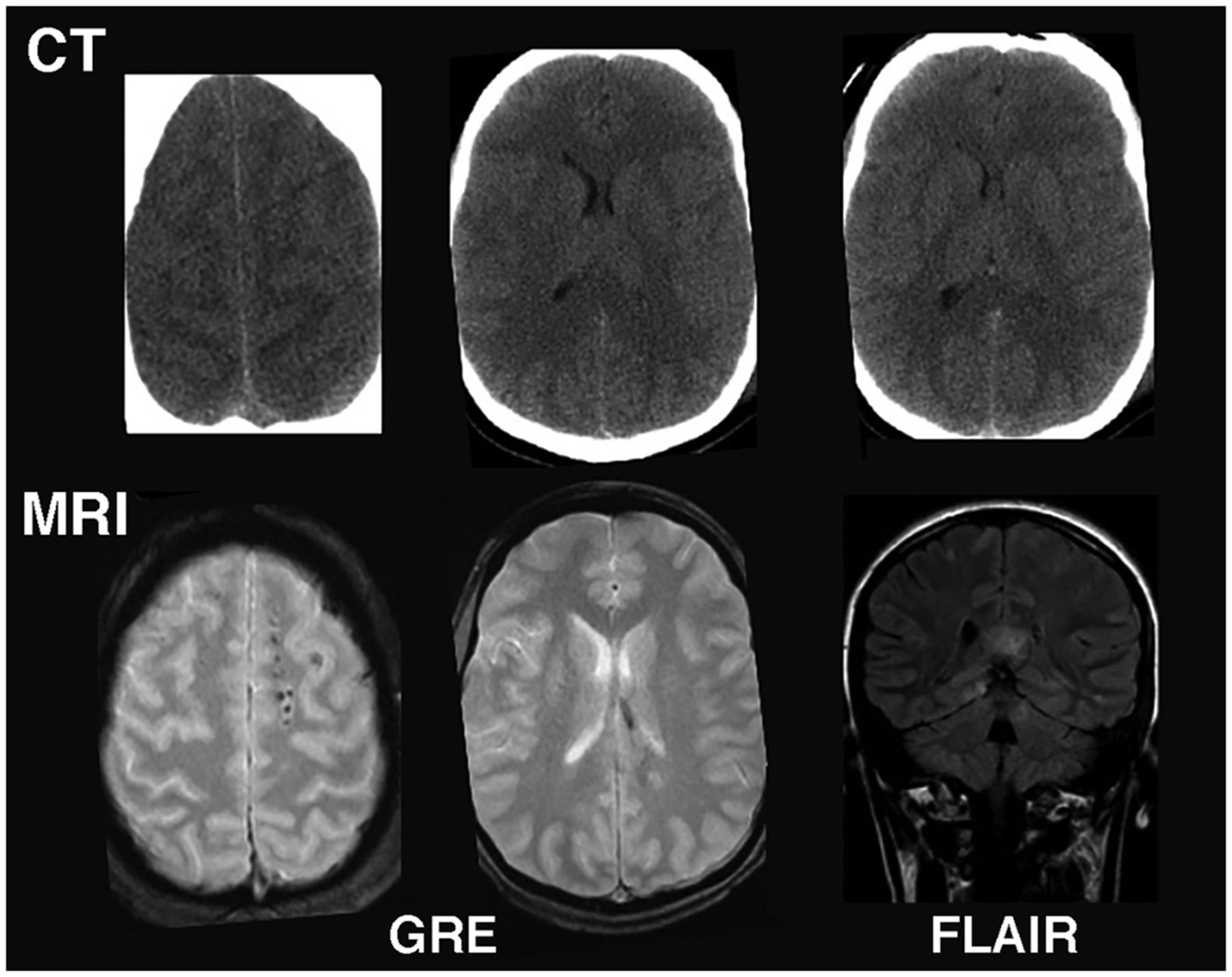
A typical brain MRI protocol in TBI might include standard T1-weighted imaging, T2-weighted fast spin-echo imaging, axial gradient-echo imaging, and fluid-attenuated inversion recovery imaging. MRI is more sensitive in detecting subtle indications of diffuse axonal injury than is CT (Fig. 1). However, one study indicated that the use of MRI for acute mTBI did not result in changes in patient management (9), and better guidelines are needed.
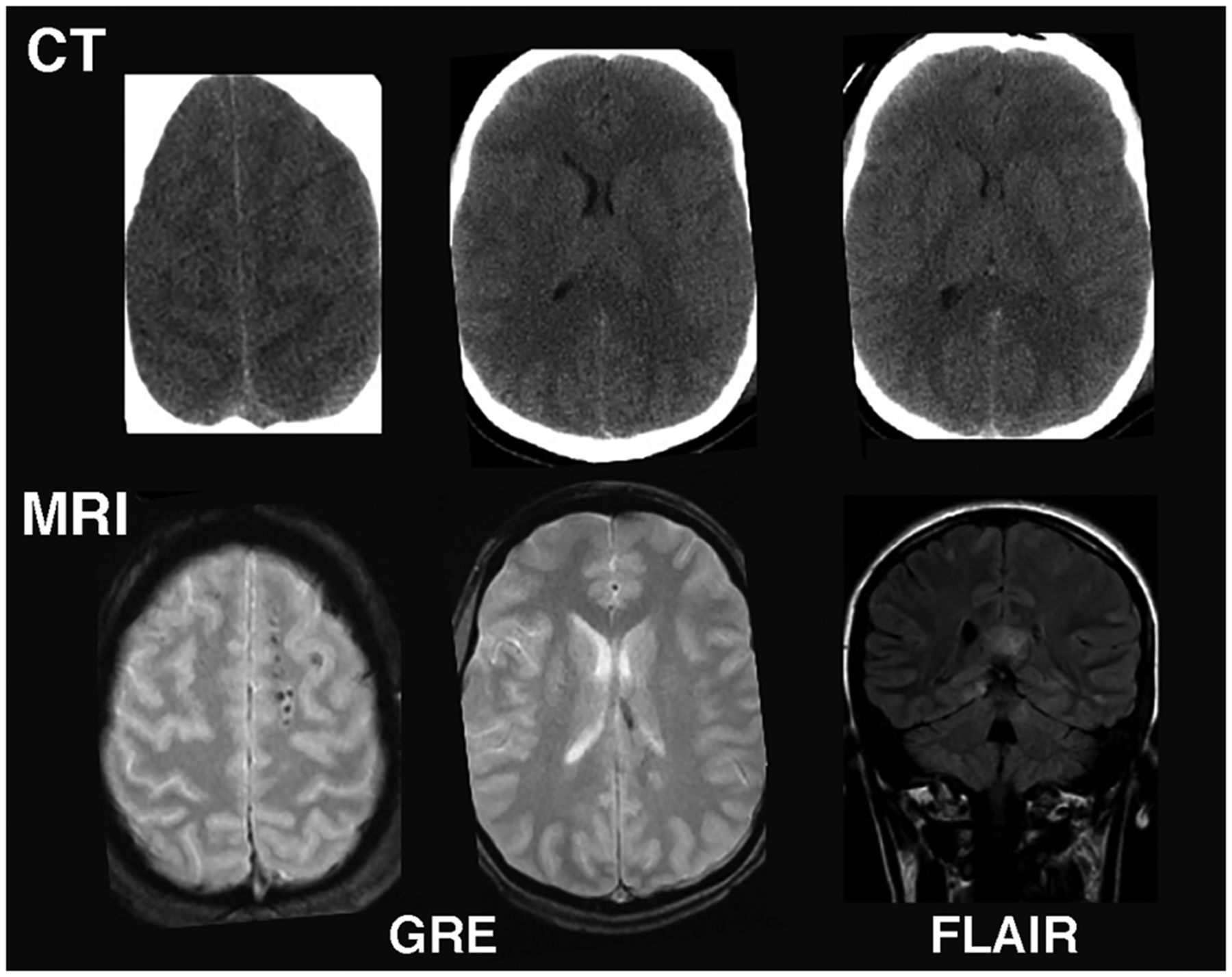
MRI vs. CT for imaging acute mTBI: Example from mTBI patient in which CT was read as normal. However, subsequent MRI showed multiple small, elliptic lesions in white matter on gradient-echo image and callosal hyperintensity on fluid-attenuated inversion recovery image, indicating diffuse axonal injury. FLAIR = fluid-attenuated inversion recovery; GRE = gradient echo.
Over the past decade, the potential long-term consequences of repeated mTBIs have been recognized. Multiple pathologic processes link single TBIs with dementia (3,10), and repeated concussions and subconcussive injuries may trigger pathogenic processes that later manifest as neurodegenerative disorders (11). In particular, chronic traumatic encephalopathy (CTE) is the most common neurodegenerative disorder linked to repeated mTBIs. CTE can present symptoms and pathologic findings comparable with other neurologic diseases, including Alzheimer disease (AD), Parkinson disease, frontotemporal dementia, and amyotrophic lateral sclerosis (12). Pathologically, CTE is characterized on postmortem examination by widespread brain atrophy and microscopic accumulation of neurofibrillary tangles in the superficial neocortical layers and depths of the sulci. In addition, diffuse gliosis may be apparent (13). Efforts to establish antemortem clinical diagnostic criteria for traumatic encephalopathy syndrome (TES), the clinical disorder associated with pathologically defined CTE, are ongoing. In the first National Institute of Neurologic Disorders and Stroke consensus workshop for TES in 2019, panelists agreed that biomarker evidence, including imaging, was not sufficiently advanced to be included as diagnostic criteria at that time (14).
At present, molecular imaging using PET combined with specific radiotracers such as 18F-FDG for glucose metabolism and proteomic specific imaging for amyloid and tau deposition have been applied, mainly for research of TBI and CTE. This review will present the current status and challenges of these various imaging modalities for TBI and suggest future directions that may significantly aid diagnosis and treatment.
ACUTE AND SECONDARY FEATURES OF TRAUMATIC BRAIN INJURY
Microscopically, acute moderate to severe TBIs involve mechanical disruption of ion channels and cellular processes such as axonal transport, which if not reestablished can result in axonal swelling and, ultimately, Wallerian degeneration (13). On a global scale, these injuries result mainly in hematomas, contusions, and what is termed diffuse axonal or diffuse traumatic injury. Downstream macroscopic features of severe to moderate TBI might include cerebral edema, ischemia, and herniation, as well as blood–brain barrier impairment and neuroinflammation (15).
Mild TBIs may also involve acute dysregulation of ion channels and axonal injury, including loss of cytoskeletal integrity. White matter shearing by mechanical impact occurs primarily in transition regions between different tissue densities or structural rigidities, such as white matter/gray matter boundaries (13). In addition, neurons with longer axons, such as corticocortical projection neurons, are more susceptible. Even mild impacts can interrupt axonal transport and other homeostatic processes such as blood–brain barrier permeability and can result in a prolonged inflammatory response (16). Postconcussive symptoms can range from none to one or more, including headache, dizziness, loss of concentration, blurry vision, anxiety, fatigue, and insomnia, which may persist over time (15).
In addition to blunt force trauma, TBIs from exposure to explosive blasts can occur. Blast-related hazards are not restricted to collisions with objects or a violent impact with something such as the wall of a vehicle. By itself, primary blast overpressure can damage the brain. In war, soldiers and civilians can be exposed to repeated subconcussive blast overpressure events (17). Many features of impact TBI overlap with blast-related TBI; however, there are additional aspects such as a vascular surge, which add to the pathophysiology of the injury (18). How these differences may alter the eventual risk for later-life neurodegenerative disorders, including CTE, is not known but remains an active area of research for military institutions.
In both acute and chronic phases, it can be challenging to detect subtle, diffuse white matter injuries and microhemorrhages in individual patients by clinical imaging modalities. Research is ongoing to develop new methods, including imaging, to serve as biomarkers of mild injuries. The subsequent sections of this article present such investigations with a goal of characterizing the pathophysiology of chronic mTBIs and of identifying biomarkers that may represent incipient neurodegenerative processes.
IMAGING IN TBI RESEARCH
MRI of Brain Injury
MRI may be used clinically for acute mTBI to detect diffuse axonal injury and other subtle indications of tissue injury. However, in research, various MRI modalities have been used to investigate alterations in a chronic TBI, which may be associated with poor long-term outcomes, including neurodegenerative diseases (Fig. 1). Since the primary focus of this review is molecular imaging, we will briefly summarize such MRI modalities, which have been described in more detail elsewhere (19).
Trifan et al. retrospectively investigated MRI correlates of persistent symptoms in young adults (<45 y old, n = 180) with a variety of single injury mechanisms and severity, but most were diagnosed as mild (20). MRI included T1-weighted, fluid-attenuated inversion recovery, and susceptibility-weighted imaging, which is sensitive to iron deposition. Fluid-attenuated inversion recovery imaging was most sensitive in detecting mild abnormalities such as white matter hyperintensities, and susceptibility-weighted imaging became more sensitive as the injury severity and likelihood of microhemorrhages increased. However, the authors concluded that although abnormalities on MRI can explain persistent symptoms in TBI, in many of the mild cases (38%) no such abnormalities could be detected.
Diffusion tensor imaging is a quantitative measure of structural integrity, which assesses the direction of water diffusion within white matter tracts. The approximate ratio of axial to radial diffusivity within a tract is calculated as the fractional anisotropy, in which values approaching 1 represent more intact white matter. This is because myelin and other axonal components restrict water diffusion primarily in the radial direction, whereas diffusion in the axial direction is relatively unrestricted. Diffusion tensor imaging has been widely applied to TBI research (21). In chronic mTBI, reductions in fractional anisotropy have been reported (22) that may indicate the presence of neurodegeneration in the brain. However, the findings are somewhat contradictory because the fractional anisotropy value may be affected by transient alterations in cellularity, such as gliosis, which can also restrict diffusion in the radial direction, thereby altering the overall ratio (23). In addition, fractional anisotropy values in white matter can change in chronic TBI and are fairly nonspecific to a particular pathologic process. Because of these limitations, diffusion tensor imaging may not be suitable as a clinical biomarker of mTBI or a diagnostic indication of incipient neurodegenerative disease.
Other MR modalities are more sensitive to specific pathologic processes and have been used to investigate chronic TBI. MR spectroscopy can measure small alterations in cortical metabolites such as choline for axonal injury and inflammation, N-acetyl-aspartate for neuronal viability, lactate for hypoxia, glutamate and glutamine for excitatory neurotransmission, creatine for cerebral energetics, and myoinositol for glia activation and inflammation (24). Numerous studies have indicated alterations in these metabolites in acute and chronic TBI and have been summarized elsewhere (24). However, MRS acquisition and analyses can be challenging to perform and interpret (25). More research is needed to determine its potential value as a biomarker of injury and neurodegeneration in TBI.
Resting-state functional MRI is a measure of the functional connectivity in the brain and has been used to evaluate consciousness in acutely brain-injured patients (26). However, a systematic review of more than 45 peer-reviewed articles found no consistent changes or convergent findings to reflect a relationship between postconcussive symptoms and resting-state functional MRI. The authors suggest this may be due to heterogeneity in both injury patterns and long-term behavioral profiles in patients.
Other advanced MRI modalities such as MR elastography, which assesses stiffness and elastic properties of tissue, and magnetization transfer imaging, which correlates with myelin integrity, have been used to investigate particular features of TBI that may be relevant to long-term outcomes. Changes in MR elastography are thought to reflect neuroinflammation, demyelination, and neurodegeneration as markers of a chronic degenerative state in the brain. Limited studies, primarily in preclinical models, have indicated promising results to sensitively detect injured brain tissue (27). Although this lack of specificity for a particular mechanism related to post-trauma degeneration does not provide information on the injured brain, there may be diagnostic utility as a sensitive biomarker of incipient irreversible brain damage to guide patient management decisions. More research is needed to determine whether MR elastography can detect the onset of a chronic degenerative condition. Magnetization transfer imaging techniques such as macromolecular proton fraction imaging have been shown to be associated with myelin integrity (28). Macromolecular proton fraction was used to investigate chronic changes associated with repeated mTBI from blasts in veterans (22). Voxelwise analyses demonstrated that macromolecular proton fraction values were 6.8%–24.7% lower in blast mTBI than in non–blast-exposed veterans in multiple white matter tracts, indicating a sensitive measure of chronic white matter injury. However, more research is needed to determine the clinical utility of these more esoteric modalities.
PET Imaging
18F-FDG
Chronic mTBI can be difficult to characterize using conventional MRI or CT. Imaging of glucose metabolism using 18F-FDG PET may be better suited to examining functional alterations related not only to the metabolic activity of any given brain region but also to additional neurovascular changes, which may be linked to symptomatology.
Previous studies of metabolic patterns in patients with mTBIs have found much heterogeneity, including no change in glucose metabolism (29), and hypometabolism in the parietal regions (22) or the amygdala. Many of these studies, however, were characterized by a low sample size and a mixed patient population with or without clinical or cognitive symptoms, which may in part contribute to the heterogeneity in the observed findings. A recent study with a larger sample size (n = 89, with a single blunt mTBI from a vehicle accident and without visible lesions on MRI/CT, vs. 93 age-matched controls) found both hypermetabolism (parahippocampal gyrus, middle temporal gyrus, cingulate, precuneus, and brain stem) and concurrent hypometabolism (angular gyrus, calcarine cortex, and middle/superior frontal brain regions) (30). In addition, the study found that the hypometabolism in frontal brain regions was associated with decreased cognitive scores. Such a finding provides evidence for the sensitivity of 18F-FDG PET to relate changes in glucose metabolism with reduced cognitive function. Further evaluation is needed to determine whether 18F-FDG PET can be used as a diagnostic marker of specific long-term effects in mTBI, as the underlying cause for alterations in the 18F-FDG PET signal can be difficult to determine. Formal testing of the sensitivity of 18FDG PET to track longitudinal changes in glucose patterns indicative of long-term cognitive and clinical impairments are needed to establish a role for glucose metabolism in diagnosing and treating chronic mTBI symptoms. For example, 18F-FDG PET in AD aids the diagnosis and is suitable to evaluate disease progression (31). In contrast, there is no clear pattern of 18F-FDG specific to TBI compared with other neurodegenerative diseases such as AD or dementia with Lewy bodies (Figs. 2A and 2B) (32). In addition to heterogeneity in the hypometabolic patterns, research on individuals with multiple blast exposures indicated that metabolic patterns may change over time in the same individuals. This finding suggests a degree of functional reorganization in the chronic rTBI patients and hampers efforts to use 18F-FDG PET to assess whether the brain injury is improving or declining longitudinally (Fig. 3).
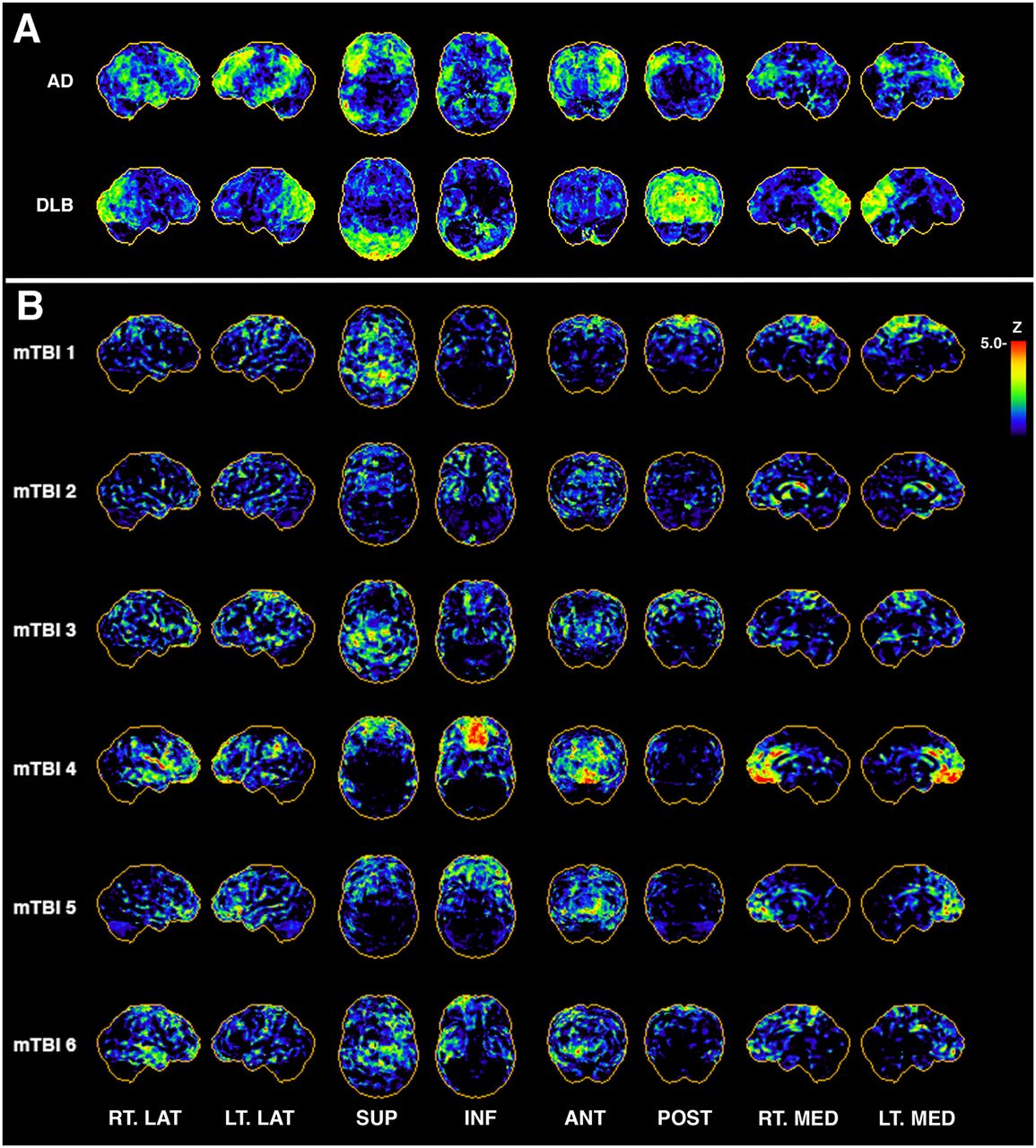
In patients with AD and dementia with Lewy bodies, typical hypometabolic patterns vs. randomly selected cases of repeated mTBI from exposure to blasts. (A) Representative 3-dimensional stereotactic surface projection (3D-SSP) z score map from patient clinically diagnosed with AD shows typical AD pattern of hypometabolism in posterior cingulate cortex and in parietal, temporal, and frontal lobes, with relative sparing of sensorimotor cortex. Diffuse Lewy body dementia is generally characterized by hypometabolism in occipital regions on 18F-FDG PET imaging. (B) Selected examples of 3D-SSP maps from veterans with multiple blast TBI exposures indicate pattern heterogeneity across subjects. Rows are single-subject 3D-SSP z score maps of hypometabolic voxels compared with reference database of age-matched community controls (n = 9). ANT = anterior; DLB = dementia with Lewy bodies; INF = inferior; LT. LAT = left lateral; LT. MED = left medial; POST = posterior; RT. LAT = right lateral; RT. MED = right medial; SUP = superior.
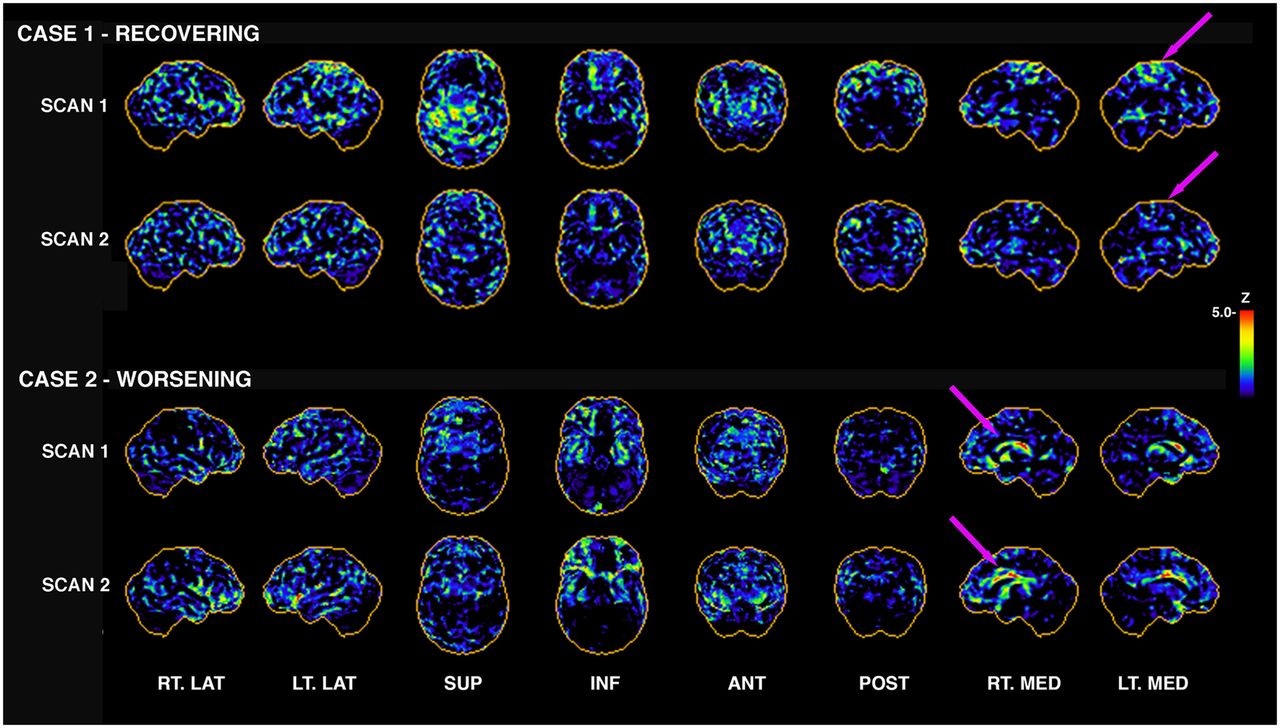
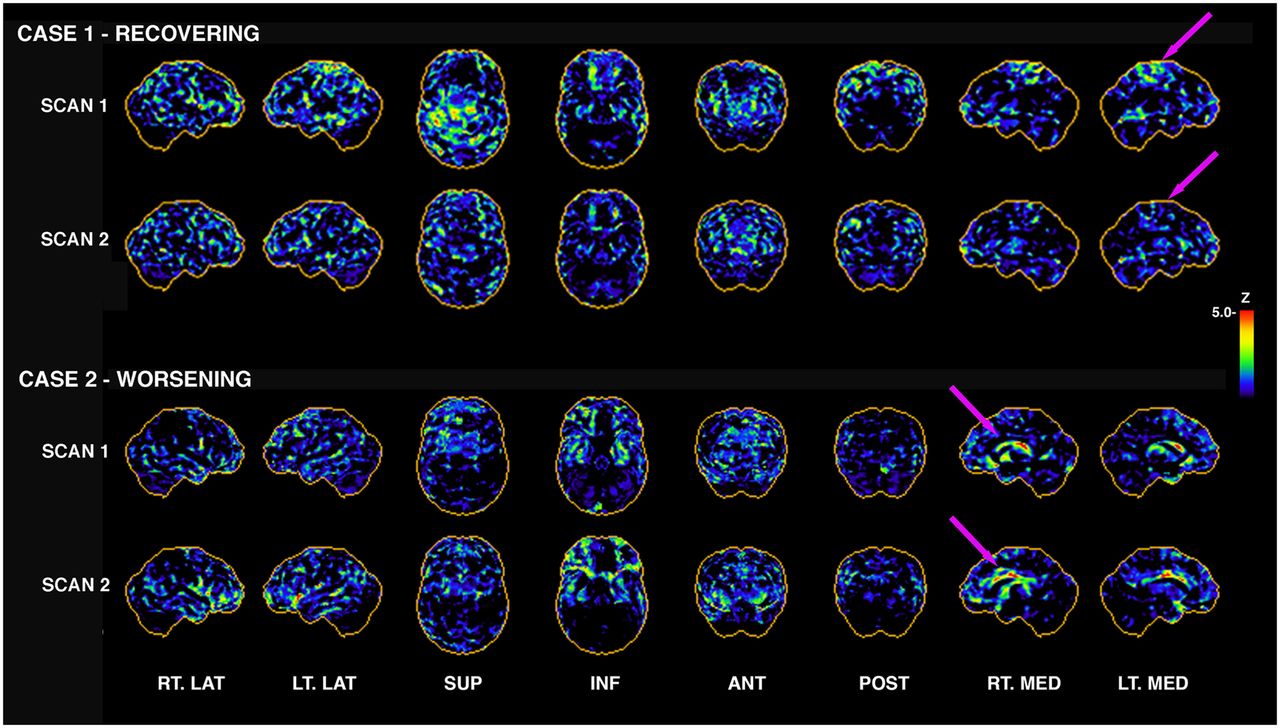
Longitudinal 18F-FDG PET in individuals with repeated mTBI from blast. Subjectively, one can appreciate that 18F-FDG PET from case 1 appears to be recovering; that is, there are fewer apparent hypometabolic pixels in scan 2 than in scan 1 (arrows indicate some regional differences). This subject had 44% decrease in number of hypometabolic voxels globally from scan 1 to scan 2. However case 2 indicates the opposite, with 46% increase in number of hypometabolic voxels globally. Not all cases are this clear-cut; some showed clear evidence of reorganization, with some regions increasing and others decreasing in total number of hypometabolic pixels (Fig. 4). Initial scans were 3–5 y after last blast, with 1–2 y interval between scans. ANT = anterior; INF = inferior; LT. LAT = left lateral; LT. MED = left medial; POST = posterior; RT. LAT = right lateral; RT. MED = right medial; SUP = superior.
These limitations and overall lack of specificity for 18F-FDG PET to discriminate the long-term sequelae of mTBI may diminish its utility as a diagnostic biomarker of trauma-associated brain injury and progression to a neurodegenerative condition in individual patients. Therefore, PET imaging using radionuclides with protein-specific binding are also under investigation.
Tau PET
Pathologically, after a single TBI only one third of subjects showed abundant neurofibrillary tangles at autopsy years after injury (33). Although the mechanistic pathway by which a single impact triggers tangle formation is not well understood, it is thought that axonal injury leads to tau hyperphosphorylation and subsequent aggregation (34). In recent years, extensive development of tau-selective radiotracers of the first generation (i.e., 18F-T807 [18F-AV-1451], 18F-T808, 18F-THK-5351, 18F-THK-5105, and 18F-THK-5117) and second generation (i.e., 18F-RO-948, 18F-MK-6240, 18F-PI-2620, 18F-JNJ-311, and 18F-GTP1) has offered a unique opportunity to image tau pathology in patients with TBI (35). The most widely used tau PET tracer, 18F-AV-1451 (18F-flortaucipir), was recently evaluated in a cohort of 21 patients with a history of a single moderate to severe TBI and a median follow-up of 32 y after injury (36). The spatial extent of the 18F-flortaucipir signal in gray and white matter was greater in the TBI group than in age-matched controls and was most pronounced in the right lateral occipital area, whereas the medial temporal lobes did not show significant differences (36). A multimodal imaging study using both an inflammatory PET radioligand (i.e., 11C-PK11195) and 18F-THK5317 as a tau selective tracer in 6 moderate-to severe TBI patients observed tau deposition in the thalami, temporal white matter, and midbrain regions in conjunction with evidence of widespread neuroinflammation (37). A larger cohort of 27 moderate to severe TBI patients was imaged using 11C-PBB3 as the tau-targeting tracer, and uptake was most pronounced in the temporal and occipital gray and white matter, in addition to frontal white matter (38). Importantly, the authors separately analyzed patients with repeated TBIs (i.e., potential TES) versus those with a single TBI and found that the white matter signal retention was enhanced in the repeated-TBI patients. Interestingly, the binding of 11C-PBB3 correlated with psychosis severity in the TBI cohort. The authors suggest that TBI presents a unique pattern of tau pathology distinguishable from the spatial distribution of other tauopathies such as AD or progressive supranuclear palsy. The development of tau pathology in chronic TBI and how it relates to the severity of persistent clinical symptoms remains a critically important area of research.
Among the current tau tracers used for TBI research, some heterogeneity in the binding patterns has emerged. In some cases, subcortical but not cortical gray matter was involved, whereas in others, white matter but not gray matter was most significantly associated with clinical symptoms. The origin of such disparate findings could be tracer- or cohort-dependent. For example, the amplitude of the signal retention for tau PET tracers for a single moderate to severe TBI is relatively low compared with AD. It is possible that the accumulation of tau pathology increases in a chronic phase and is therefore not as evident in the acute phase of a single TBI. Alternatively, the affinity of the tau tracers may be less sensitive to isoforms of tau pathology from TBI (39), resulting in lower tracer retention. Therefore, the target sensitivity of the currently available tau PET tracers for TBI needs further investigation.
Amyloid Imaging
Several radiotracers are used to visualize β-amyloid in the brain, including the Food and Drug Administration/European Medicines Agency–approved 18F-florbetapir, 18F-florbetaben, 18F-flutemetamol, 18F-florbetapir, and others such as 18F-NAV4694 and the first β-amyloid tracer, 11C-Pittsburgh compound B. Case reports using 18F-florbetapir at 1, 12, and 24 mo after TBI found an increase in β-amyloid deposition initially, which was cleared by 1 and 12 mo in the left hippocampus and caudate nucleus, whereas regions such as the right hippocampus and left anterior putamen showed a steady increase to the 24-mo time point (40). In one study, 15 patients with moderate to severe TBI were scanned between 1 and 365 d after injury using 11C-Pittsburgh compound B and showed increased binding in cortical gray matter and the striatum (41). The elevated binding in gray matter regions (i.e., frontal, parietal, occipital, and lateral temporal) was ubiquitous except in the hippocampus, where no significant differences from controls were observed. Interestingly, the effect of a history of mTBI on β-amyloid burden in clinically normal older adults was investigated (42) in a cross-sectional and longitudinal study design. The authors observed trend-level increases in β-amyloid aggregation over time in individuals with (n = 11) or without (n = 19) a history of TBI, and no differences in β-amyloid burden between groups or interactions with cognitive performance were observed (42). Research comparing older adults with (n = 81) and without (n = 240) prior TBI showed significantly increased orbitofrontal, prefrontal, superior frontal, and posterior cingulate SUV ratios using 18F-florbetapir (43). Although β-amyloid deposition can be initially elevated after brain injury, in chronic phases the β-amyloid deposition may be cleared. Different factors may impact clearance mechanisms in patients after a single TBI, which may alter the aggregation of β-amyloid in a chronic phase. For example, it was shown that amyloid-precursor protein secretase has a detrimental role in the initiation of secondary injury after TBI in mice (44). Decreasing secretase activity was shown to block secondary injury, suggesting that the level of amyloid-precursor protein secretase after injury influences differential outcomes after mTBI. Other known risk factors and comorbidities for β-amyloid deposition such as age, genetic risk, or vascular factors may contribute to observed differences and need to be carefully controlled for in future studies.
Imaging of Neuroinflammation in Brain Trauma
Inflammation in the central nervous system is associated with many psychiatric and neurodegenerative diseases. In TBI, the mechanical impact initiates a host of secondary events, including free radical generation and an inflammatory response via infiltrating microglia, activated astrocytes, and the endothelial cells of the blood–brain barrier. PET imaging using radiotracers such as 11C-PK11195, which binds to the translocator protein, have been used to evaluate the inflammatory response after mTBI. Translocator protein is a protein expressed in high levels in activated microglia, as well as in peripheral monocytes and astrocytes. At various times after TBI (i.e., 11 mo to 17 y), in a small number of patients (n = 10) with moderate to severe injury, significantly higher 11C-PK11195 binding was observed in the thalamus, putamen, occipital cortices, and posterior region of the internal capsule (45). Even shorter follow-up scans at only 6 mo after injury found enhanced 11C-PK11195 signal in 8 patients with moderate to severe mTBI. Another translocator protein marker, 11C-DPA-713, was imaged in active and retired football players (7–42 y after the last self-reported concussion), and increased signal retention was found in many brain regions (46). These results suggest that activated microglia remain present in patients with a remote history of TBI. One major limitation with 11C-DPA-713 is different binding potentials based on the rs6971 genotypes, in which a polymorphism results in reduced binding affinity. PET analysis of DPA-713 needs to be corrected for this polymorphism for accurate interpretation of findings.
An experimental study on patients after moderate to severe TBI targeted chronic microglia activation after 12 wks of treatment with minocycline, an tetracycline antibiotic that inhibits microglia activation (47). Plasma neurofilament light levels, white matter injury by MRI, and 11C-PBR28 PET were assessed before and after treatment. The authors reported a significant 25% reduction in 11C-PBR28 PET signal in both white matter and gray matter, as well as the thalamus, in the treated versus nontreated subjects. Interestingly, plasma neurofilament light levels increased after treatment but returned to baseline at 6 mo after drug discontinuation. The authors interpreted their results as evidence for therapeutic benefit of inhibition of microglia activation after mTBI (47).
These studies indicate that persistent neuroinflammation is a characteristic feature of chronic TBI. Importantly, translocator protein radiotracers detect not only activated microglia but also astrocytic activation and peripheral monocytes, suggesting that multiple mechanisms contribute to signal retention. Although current evidence suggests targeting inflammation as a potential therapeutic strategy, additional well-powered studies are needed. Moreover, the long-term consequences of chronic inflammation on neurodegenerative processes are not well understood.
Other Molecular Imaging Tracers for TBI Research
Because γ-aminobutyric acid is the principal inhibitory neurotransmitter in the brain, efforts to develop a sensitive tracer to measure γ-aminobutyric acid concentration has applications to many neurologic (e.g., epilepsy) and psychiatric (e.g., posttraumatic stress disorder) clinical syndromes. The tracer, 11C-flumazenil, is sensitive to γ-aminobutyric acid A-type benzodiazepine receptor binding and has been investigated in small cohorts of mTBI. In 1 study, 8 patients with diffuse axonal injury showed regionally low 11C-flumazenil uptake bilaterally in the medial frontal gyri, anterior cingulate gyri, and thalamus compared with controls (48). In another study, decreased uptake of 11C-flumazenil was observed in 60% of a cohort compromising 10 TBI patients and partially coincided with hypometabolism assessed by the cerebral metabolic rate of oxygen (49). Additionally, some regions did not show reduced binding of 11C-flumazenil, despite significant hypometabolism. The authors suggested that 11C-flumazenil binding may indicate regions where loss of neuronal integrity may be the underlying molecular correlate of the hypometabolic regions in mTBI (49). These studies indicate that abnormalities in γ-aminobutyric acid may play a role in chronic TBI, but neurotransmission is dependent on several factors affecting excitatory and inhibitory processes (50). Therefore, multimodal imaging, including imaging of neurotransmitter integrity, may unravel the contribution of neurotransmitter alterations in chronic TBI.
Synaptic vesicles store neurotransmitters within neurons and mediate release into the synaptic cleft via voltage-dependent calcium channels. The synaptic vesicle protein 2A is an integral 12-transmembrane domain glycoprotein present in neurons and is widely expressed in the brain. Currently used radiotracers for synaptic density are 11C-UCB-J and the fluorine-labeled analog 18F-SynVesT-1, and the pharmacokinetic characteristics have been described elsewhere (51). To date, no research on synaptic density in mTBI patients has been published; however, this tracer may prove valuable to link alterations in synaptic density with long-term clinical and cognitive consequences in mTBI.
Molecular Imaging of CTE and TES
CTE is a neurodegenerative condition most commonly associated with a history of repetitive TBI (rTBI), which often occurs in athletes or military personnel. CTE is a neuropathologic diagnosis characterized by phosphorylated tau protein aggregates in the depths of the cortical sulci, in neurons, and around blood vessels (52). It has been debated whether additional features such as brain stem pathology, β-amyloid deposition, α-synuclein Lewy bodies, TDP-43 pathology, vascular degeneration, or gliosis should be considered core features of CTE (53) or whether they are not distinctive enough to uniquely identify CTE at autopsy (53). To classify the clinical syndrome associated with rTBI, new criteria were proposed for the associated cognitive, clinical, neurologic, and psychiatric symptoms representing TES (54). The potential detrimental effects of rTBIs have been actively discussed in the research community as more evidence accumulates to link rTBIs to CTE and other neurodegenerative diseases, including AD (55), Parkinson disease (56), and amyotrophic lateral sclerosis (57). Therefore, it is important to stress that histopathologically defined CTE is causally related to rTBI but that not all rTBI leads to a histopathologic diagnosis of CTE (58).
Because phosphorylated tau is a key pathophysiologic feature of CTE, the advent of tau PET tracers was initially thought to present an opportunity to image the spatial distribution of tau in living rTBI patients. A study comparing 18F-flortaucipir signal in retired amyloid-negative professional football athletes (n = 26) with controls (n = 31) found elevated binding in the medial temporal, parietal, and superior frontal brain regions, which correlated with years of active play but not with cognitive performance (59). A study using the TES research criteria to classify individuals who had a history of contact sports (n = 11; mostly American football players) and were exhibiting objective and subjective cognitive impairments compared 18F-flortaucipir uptake with amyloid-negative controls and AD patients and validated the proposed TES criteria (60). The authors observed only minute differences in increased uptake of 18F-flortaucipir in the medial frontal regions compared with controls but no significant differences in uptake compared with the AD cohort (60). Somewhat dampening the enthusiasm for 18F-flortaucipir to image tau pathology in CTE were autoradiographic studies showing only limited binding affinity for autopsy-confirmed cases of CTE with abundant tau inclusions (61,62). Potential differences in the binding affinity of 18F-flortaucipir could be due to fibril-folding differences recently revealed to be distinct between AD and CTE brains (39). Other tracers selective to tau pathology, such as 11C-PBB3, have indicated increased white-matter signal congruent with CTE criteria (38). However in a case report (63), 18F-MK6240 binding bilaterally in the superior frontal and medial temporal lobe regions did not correspond to autoradiography, suggesting low binding affinity of this tracer to CTE pathology (64). Together, these studies using currently available PET tracers for the in vivo depiction of phosphorylated tau have provided limited and inconsistent results. To move tau PET into the clinical realm for TES, more sensitive ligands need to be developed—ligands that incorporate specific binding characteristics for the fibril-folding structures of CTE tau pathology.
Other molecular targets have been investigated in cohorts of rTBI and probable CTE. In small cohorts of boxers (n = 5 (65) and n = 19 (66)), hypometabolism using 18F-FDG PET in the frontal, parietooccipital, cerebellar, and cingulate regions was observed, whereas in former football players, hypometabolism in the frontotemporal regions was most pronounced (60). The presence of β-amyloid burden has been suggested as an exclusion criterion for underlying CTE pathology with cognitive deficits (52), and most research evidence is confirmatory and indicates a lack of association with β-amyloid burden and previous history of rTBI (67,68).
Despite evidence of a relationship between postmortem CTE pathology and rTBI, current research studies using tau PET tracers in rTBI cohorts have not identified a universal pattern of tau deposition that is characteristic of rTBI. Newly outlined research criteria for TES will hopefully assist in identifying individuals at high risk for the pathophysiologic cascade of CTE. The advancement of novel tau and other protein- or mechanism-specific PET tracers may help unravel any distinct patterns of protein deposition. In addition, other tracers, such as 18F-FDG or neuroinflammation ligands, may help characterize brain alterations indicative of incipient neurodegenerative processes after rTBI.
IMAGE ANALYSIS AND CHALLENGES
A key challenge in imaging of acute and chronic TBI, rTBI, and TES/probable CTE is that research across all modalities explored to date has not revealed a cohesive pattern of structural or functional deficits—or of protein- or mechanism-specific uptake—that is common to most patients within a particular study cohort (Fig. 2). This lack of a cohesive pattern is due primarily to the unique profile of the underlying substrate for the condition—that is, brain “injuries”—as well as individual differences in the longitudinal response and recovery after injury. Figure 3 shows examples of 2 veterans who experienced multiple mild blast exposures, with the initial 18F-FDG PET acquired 3–5 y after the last exposure and follow-up scans acquired approximately 2 y later. Characteristic patterns of both recovery of glucose metabolism and reduction of glucose metabolism are visible. Such regional reorganization in combination with regions of prolonged hypometabolism (Fig. 4) need to be further evaluated in the context of clinical decline for rTBI patients.
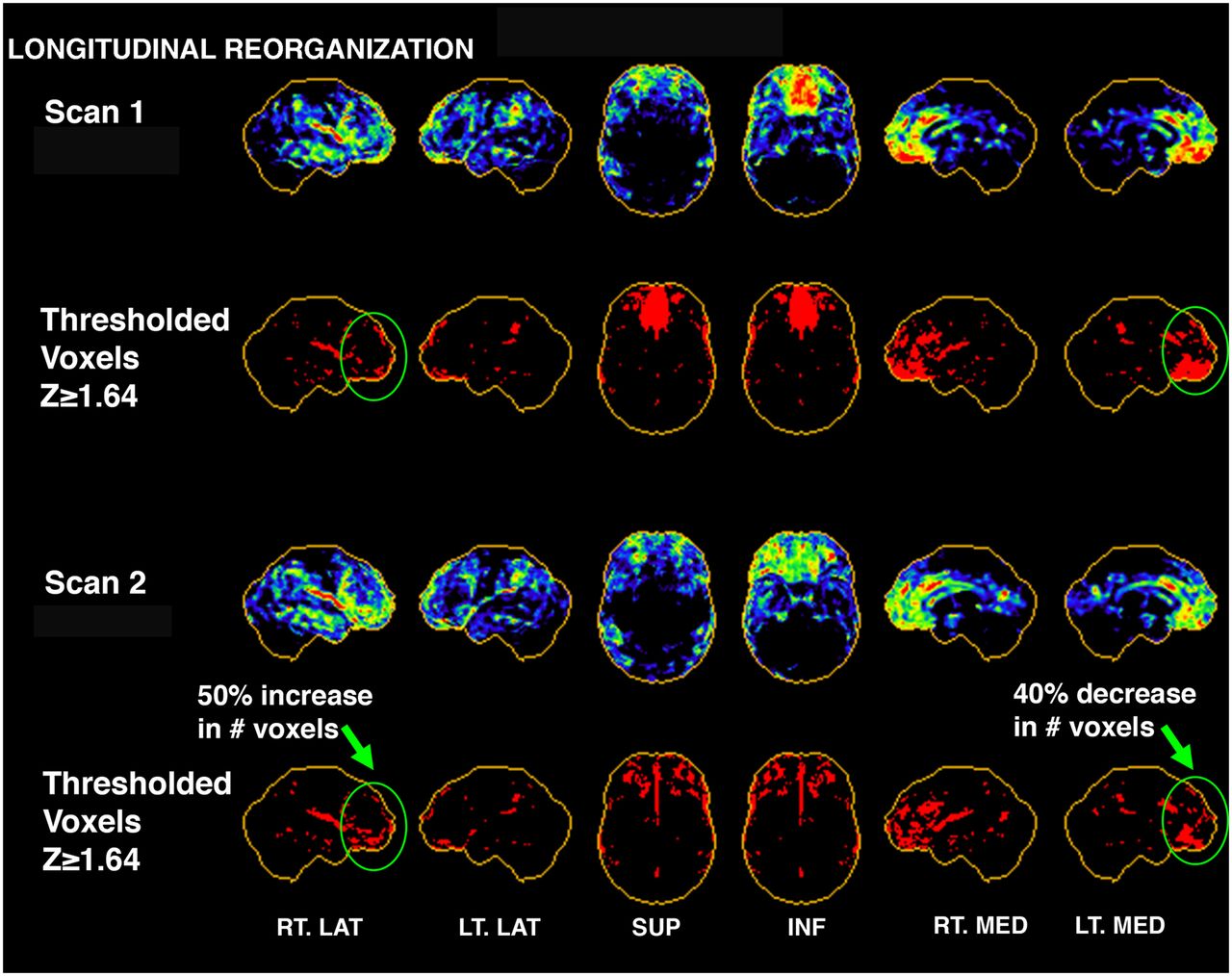
Three-dimensional stereotactic surface projection maps showing example of reorganization in single mTBI subject from scan 1 to scan 2. Casual visual inspection may conclude that subject’s scans have improved because severely hypometabolic region of inferior medial frontal lobe on scan 1 has improved subjectively by scan 2. This was supported in thresholding analysis (second and bottom rows), which indicated 40% decrease in number of voxels with z score ≥ 1.64 (hypometabolic voxels) for that region. However, analysis of 26 regions covering entire brain indicated that other regions, such as right lateral frontal region, actually became more hypometabolic (50% increase in hypometabolic voxels)—likely a result of reorganization and compensation over time. This subject also showed an 8% increase in hypometabolic voxels globally from scan 1 to scan 2. ANT = anterior; INF = inferior; LT. LAT = left lateral; LT. MED = left medial; POST = posterior; RT. LAT = right lateral; RT. MED = right medial; SUP = superior.
Regardless of modality, most imaging research and clinical studies to date have used groupwise analytic techniques to investigate the common features of acute and chronic TBI, rTBI, and probable TES/CTE. However, these studies, taken collectively, have failed to converge on such common features, which could have diagnostic or discriminatory utility in a clinical setting for individual patients. There is a distinct and critical need for new quantitative biomarkers of either protein or mechanism-specific tracers combined with novel image analysis methods, which may not depend on a specific regional pattern to sensitively detect and classify TBI and related neurodegenerative processes.
Supervised learning techniques such as scaled subprofile model principal-component analysis may improve the information inferred from PET imaging by capitalizing both on abnormal patterns of tracer retention and on the covariance across regional uptake (69). Such methods have identified distinct and reliable patterns in tau PET imaging between primary and secondary tauopathies (70) and metabolic brain patterns selective for Parkinson disease, progressive supranuclear palsy, and multiple-system atrophy (69). This approach in TBI may reveal some important regional covariance patterns from which quantitative thresholds can be derived.
Other approaches have focused on improving the measurement of PET imaging itself. For example, the estimation of glucose metabolism in 18F-FDG PET can be improved by assessing plasma tracer concentrations during the imaging time window. There was 25%–33% increased hypometabolism in TBI patients when plasma tracer concentration was included in the quantification (71). Importantly, this improved quantification of glucose metabolism correlated with level of consciousness.
These studies emphasize that novel quantitative and qualitative approaches may overcome current challenges in imaging of brain trauma and may enhance the utility of PET for the heterogeneous disease phenotypes commonly observed in TBI.
FUTURE DIRECTIONS
A lack of a mechanistic understanding of chronic TBI has posed challenges with interpreting the results of any single molecular imaging biomarker. We believe that future research should address the following issues: better target identification, improved analysis techniques using machine learning and artificial intelligence algorithms, and novel tracer development.
Better Target Identification
Multiple neural and molecular events follow TBI, such as axonal injury, energy crises, vascular impairments, blood–brain barrier disruptions, and inflammation via activation of microglia and astrocytes. It is not well understood which of these changes persist in a chronic phase and are subsequently associated with, or causal to, clinical symptoms. First, more consistent samples of individuals with and without clinical symptoms are needed, along with a detailed definition of the TBI itself. Then, a detailed program of imaging and nonimaging biomarkers that assess the differential processes triggered by TBI should be investigated to identify the biomarker most imperative for relating long-term consequences to brain injury. We believe that a better understanding of such substrates that underlie the cognitive and clinical phenotypes is necessary to advance disease understanding, infer potential long-term sequalae, and identify new and appropriate targets for molecular imaging.
Improved Analysis Techniques Using Machine Learning and Artificial Intelligence Algorithms
Heterogeneous variations in observed patterns have challenged the distinct identification of a one-fits-all approach in TBI. Data-driven approaches such as machine-learning and artificial intelligence algorithms may improve quantification and determine which aspects most contribute to the distinction of TBI patients with and without long-term cognitive and clinical consequences. We speculate that such outcome measures may be best expressed in a threshold or ratio combining different biomarker information that gives rise to persistent neural, vascular, or molecular injury. Such methods require the development of large, well-characterized imaging databases, such as the TRACK-TBI (Transforming Research and Clinical Knowledge in Traumatic Brain Injury) study (72). However, that particular study has limited the imaging database to CT/MRI and, thus, does not include PET imaging modalities at this time.
Novel Tracer Development
It may be necessary to shift research efforts from established imaging biomarkers toward the development of novel tracers specific to TBI, TES, and CTE. One promising avenue that could potentially inform tracer development is the application of cryogenic electron microscopy. The imaging of the macromolecular structure of biomolecules has identified the common structure of tau pathology in different disease populations (39,73). Such techniques could potentially improve the definition of tau protein strains after TBI or in CTE and, subsequently, the development of tracers more specific to such targets. Another direction would be to develop tracers that can specifically and sensitively image cytoskeletal alterations. More subtle mechanisms such as deficits in axonal transport or microtubule stabilization, alterations in neuroplasticity, or disruption of cerebrovascular reactivity may underlie the observed heterogeneity in post-mTBI recovery. Such cytoskeletal characteristics might be important targets for future tracer development.
CONCLUSION
Brain trauma imaging is a diverse field that ranges from the clinical criteria for imaging in the assessment of acute TBI to research of biomarkers underlying the presence of symptoms in chronic mTBI, as well as the diagnosis of TES/CTE. The role of molecular imaging is not well established within these areas. The multitude of recent studies has indicated some common findings but also contradictory and disparate results with regard to imaging of TBI. In fact, no specific biomarker has emerged to aid in diagnosing injury or determining the prognosis of long-term behavioral profiles. Lastly, with respect to the development of future treatments for TBI, it is critical to establish such validated biomarkers, which can be implemented to evaluate treatment response and improvement in brain health.
ACKNOWLEDGMENTS
We acknowledge contributions to this article from Satoshi Minoshima, MD, PhD, and Yoshimi Anzai, MD, of the University of Utah Department of Radiology and Imaging Sciences and from Elaine R. Peskind, MD, and Garth E. Terry, MD, of the Mental Illness Research, Education, and Clinical Center (MIRECC), VA Puget Sound Health Care System.
- © 2023 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication March 30, 2022.
- Revision received September 2, 2022.





{kind=link}
{kind=link}
{kind=link}
{kind=link}