


Abstract
Targeted α-therapy (TAT) is an emerging powerful tool treating late-stage cancers for which therapeutic options are limited. At the core of TAT are targeted radiopharmaceuticals, where isotopes are paired with targeting vectors to enable tissue- or cell-specific delivery of α-emitters. DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) and DTPA (diethylenetriamine pentaacetic acid) are commonly used to chelate metallic radionuclides but have limitations. Significant efforts are underway to develop effective stable chelators for α-emitters and are at various stages of development and community adoption. Isotopes such as 149Tb, 212/213Bi, 212Pb (for 212Bi), 225Ac, and 226/227Th have found suitable chelators, although further studies, especially in vivo studies, are required. For others, including 223Ra, 230U, and, arguably 211At, the ideal chemistry remains elusive. This review summarizes the methods reported to date for the incorporation of 149Tb, 211At, 212/213Bi, 212Pb (for 212Bi), 223Ra, 225Ac, 226/227Th, and 230U into radiopharmaceuticals, with a focus on new discoveries and remaining challenges.
Targeted radionuclide therapy has demonstrated significant therapeutic efficacy and survival benefit for some conditions, especially for late-stage disease with limited therapeutic alternatives (1,2). Several isotopes, such as 177Lu, 90Y, 89Sr, 153Sm, and 131I, are in clinical use for treatment of neuroendocrine tumors (177Lu-DOTATATE [Lutathera; Advanced Accelerator Applications]), hepatobiliary tumors (90Y-glass microspheres [TheraSphere; Boston Scientific]), bone metastases (89Sr-chloride [Metastron; GE Healthcare] and 153Sm-lexidronam [Quadramet; Lantheus Medical Imaging]), and non-Hodgkin lymphoma (tositumomab and 131I-tosutumomab [Bexxar; GlaxoSmithKline]), respectively. Most isotopes used for targeted radionuclide therapy are β-emitters, with 223Ra ([223Ra]RaCl2, Xofigo; Bayer) being the only Food and Drug Administration–approved α-emitter to date for the treatment of patients with metastatic castration-resistant prostate cancer with symptomatic bone metastases and no known visceral metastatic disease. α-emitters have much higher linear energy transfer (energy deposition per unit pathlength) than do β-emitters (∼100 keV/μm vs. 1–2 keV/μm) and generate substantially more free radicals and lethal DNA double-strand breaks (3,4).
When delivered via tumor-specific targeting vectors, the short range of α-particles (40–100 μm) enables highly selective targeting of cancers, including micrometastases, while potentially sparing surrounding healthy tissues. The cytotoxicity of α-emitters is also independent of cell cycle or oxygen concentration (5,6), providing an advantage for treating hypoxic, often radiation-resistant tumors (7).
Targeted α-therapy (TAT) has been fast-tracked by the Food and Drug Administration approval of [223Ra]RaCl2 for the treatment of bone metastases of metastatic castration-resistant prostate cancer, and 225Ac-PSMA617, which is in pilot trials to treat metastatic castration-resistant prostate cancer, has shown remarkable clinical effectiveness (8,9).
Few α-emitting radionuclides suitable for clinical use are available, considering half-life, decay mode, and availability. Isotopes with potential medical applications and their properties are listed in Table 1. With the exception of [223Ra]RaCl2, TAT typically comprises an isotope, a targeting vector (e.g., small molecule, peptide, antibody, or engineered antibody), and a chelator that can form a stable complex and carry the isotope to deliver high–linear-energy-transfer radiation directly to cancer cells and the tumor microenvironment. An ideal chelator exhibits fast metal-complexation kinetics, selectivity for the radionuclide (because of inevitable metal impurities), high thermodynamic stability, high in vivo stability, and the ability to bind an imaging isotope for theranostics applications. Many of the isotopes in this review decay through multiple daughter products, each with its own unique coordination chemistry. The issue of daughter recoil and coordination presents a unique set of physics and chemistry challenges that are beyond the scope of this review (10).
Isotopes for TAT
DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) has been a standard chelator for radiometals and is still useful for coordinating Ac, Bi, Tb, Th, and Pb; however, issues remain with this chelate, including decreased thermodynamic stability toward large metal ions and slow chelation kinetics. High ligand concentration, which reduces the molar activity, and heating are required, which may compromise some targeting vectors. Sensitivity to metal impurities is another shortcoming. Thus, much work is required to develop suitable chelators for α-emitters, particularly Ra, U, and arguably At. The need for novel, suitable labeling methods remains unmet for the rapidly expanding field of TAT. In this review, we will discuss the current state of the art of labeling method development for medically relevant α-emitters and their applications in TAT.
CHEMISTRY TO LABEL α-EMITTERS
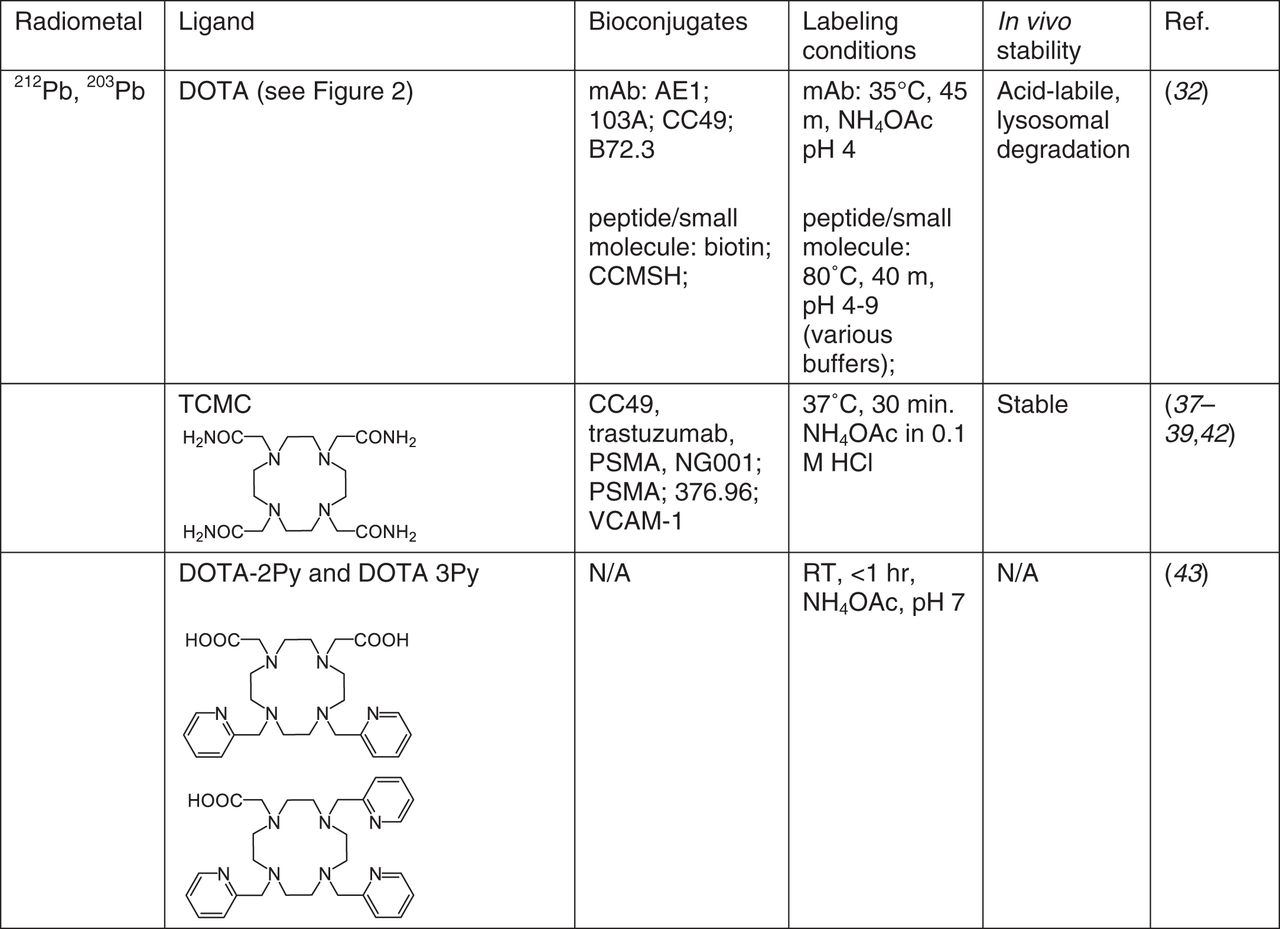
149Tb
Cyclen-based chelators, diethylenetriamine pentaacetic acid (DTPA), and dipicolinic acids are among the many chelators reported to complex Tb3+ (11,12). Chelators that form stable complexes with Lu3+ may also do the same with Tb3+, although binding may be weaker, as Tb3+ has a larger atomic radius.
Medical applications of Tb isotopes are at an early stage; they bind DOTA effectively, and also (S,S)-cyclohexane-1,2-diamine-pentaacetic acid (CHX-A″-DTPA), preferentially for antibody labeling because of the fast kinetics and ambient temperature metal incorporation. PET imaging with 149Tb-DOTANOC has shown excellent tumor visualization (13), 149Tb-DOTA-folate (cm09) was found to delay tumor growth in a dose-dependent manner (14), and 149Tb-CHX-A″-DTPA-rituximab (anti–CD-20) demonstrated tumor-free survival in 89% of the mice treated 2 d after tumor inoculation (15).
To date, only DOTA and CHX-A″-DTPA have been studied with radio-Tb isotopes, and reports on their preclinical applications are limited (Fig. 1); additional Tb chelates would yield more ideal candidates with improved kinetics or stability. The intense luminescent properties of various Tb complexes may be leveraged for use in optical/radio multimodality imaging.
NOTEWORTHY
▪ α-emitting radionuclides may require new methods to be efficiently incorporated and stably delivered in vivo.
▪ New chemistry (chelators or astination methods) is quickly emerging, although more in-depth investigation of the applications and limitations are required.
▪ For 223Ra and 230U, stable chelators are yet to be discovered.

Chelates for 149Tb. RT = room temperature.
211At
Comprehensive reviews summarizing organic and inorganic chemical methods for producing radioastatinated compounds have been published (16–18). Briefly here, isotopes of At have relatively short half-lives (the longest is 8.1 h, none are stable), and radioiodine is commonly used as a surrogate when studying At chemistry, despite a divergence in reactivity with different labeling reagents and in vivo stability of the products formed. Although At has metallic character, to date no chelating ligand evaluated provides an 211At complex with enough in vivo stability for TAT applications (16–18). An exception may be a rhodium(III) astatide complex stabilized by a macrocyclic crown thioether (16aneS4-diol) (19). Differences in the biodistribution of Rh[16aneS4-diol]131I and Rh[16aneS4-diol]211At suggest some deastatination of the latter, but the in vivo stability may be potentially useful (19). How stable the 211At label must be before it can be useful for patient treatment has not been established.
A few At labeling methods use a nonactivated aryl-At bonding approach (16–18), with the most widely used method involving organometallic compounds such as N-succinimidyl 3-(tri-alkylstannyl)benzoate, compound 2, in electrophilic substitution reactions (Fig. 2). This approach was used in many preclinical studies and more recently in two phase I clinical trials for treating brain or ovarian cancer (20,21). In these trials, the 211At-labeled radiopharmaceutical was prepared using a 2-step labeling approach. A 1-step labeling approach has been demonstrated by the Gothenburg team wherein the tri-alkylstannyl benzoate is conjugated before radiolabeling (22).
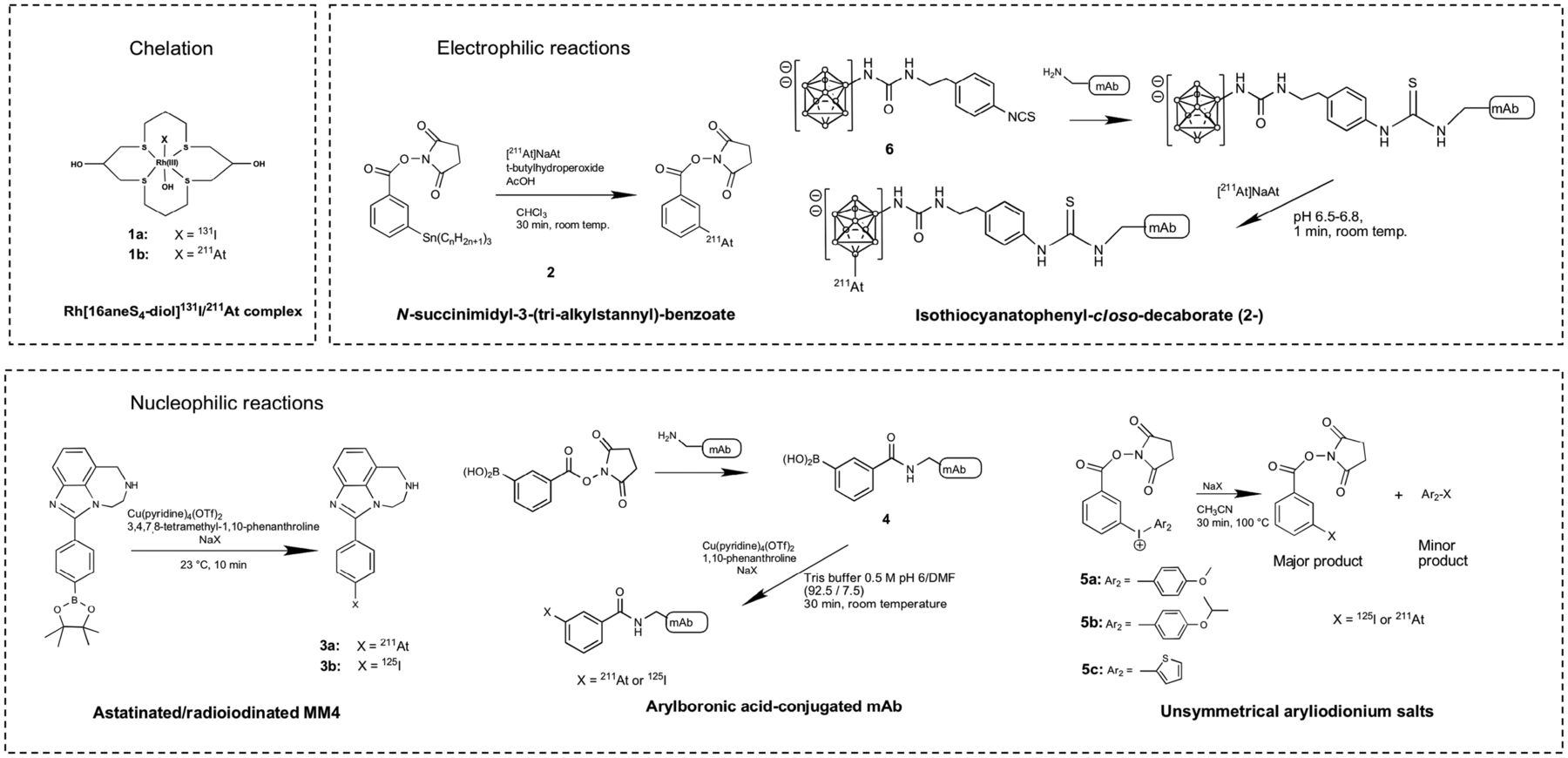
211At labeling methods. DMF = dimethylformamide; NaX = [125I]NaI or [211At]NaAt.
Nonactivated arylboronic acid derivatives were shown to react rapidly with electrophilic At species. More recently, nucleophilic substitution reactions using aryl boronic acids/esters have been shown to provide highly efficient 211At-labeling. A series of 211At-labeled compounds was prepared using boronic ester precursors and a Cu catalyst, including the 211At-labeled poly(adenosine diphosphate-ribose)polymerase-1 inhibitor [211At]MM4 (compound 3a, Fig. 2), which exhibited a biodistribution profile similar to that of its 18F and 125I analogs and could potentially be useful for TAT applications (23,24). Cu-catalyzed astatination and radioiodination reactions using arylboronic acids can be conducted in aqueous solution at ambient temperature and thus are suitable for monoclonal antibody (mAb) labeling. Cu-catalyzed astatination and radioiodination of arylboronic acid–conjugated mAbs (compound 4, Fig. 2), such as anti-CD138 antibody, 9E7.4 (25), have been reported.
Aryliodonium salts can also be used for mAb conjugate labeling via nucleophilic substitution. For example, asymmetric aryliodonium salts with one of the aryl rings bearing an N-hydroxysuccinimidyl ester (compounds 5a–5c, Fig. 2) were conjugated to anti-CD138 mAb for astatination and radioiodination studies (25). Striking differences were observed between radioiodination and astatination reactions in regioselectivity and in reaction conditions required to obtain optimal radiochemical yield; the astatination reaction using aryliodonium salts is more efficient than the radioiodination.
Another 211At-labeling reagent that has been used in preclinical and clinical studies is the isothiocyanatophenyl-closo-decaborate(2−) (B10) boron cage molecule (compound 6, Fig. 2) (25). The 211At-labeled B10-conjugated anti-CD45 mAb BC8 is currently being evaluated in 2 phase I/II trials for hematopoietic cell transplantation in patients with leukemia, myelodysplastic syndrome, and nonmalignant diseases. The aromatic B10 moiety provides high 211At labeling efficiency (75%–90% radiochemical yield, 1 min) with high in vivo stability. New boron cage 211At-labeling reagents to potentially stabilize 211At in a higher oxidation state, +3 or +5, are being evaluated for improved tissue distribution and more favorable pharmacokinetic properties (25).
Overall, 211At is a highly attractive radionuclide for TAT applications. To date, electrophilic substitution on unactivated aromatic rings is the most widely used astatination method for TAT preclinical and clinical radiopharmaceutical synthesis. Other recent studies demonstrate the potential of nucleophilic substitution reactions for astatination of small molecules and mAbs (23,24). In addition to the aryl-At bonding approach, boron-At bonding provides an alternative astatination strategy. Other astatination strategies that involve the use of chelation chemistry, nanoparticles, and others have been explored but have yet to show potential for use in humans (26).
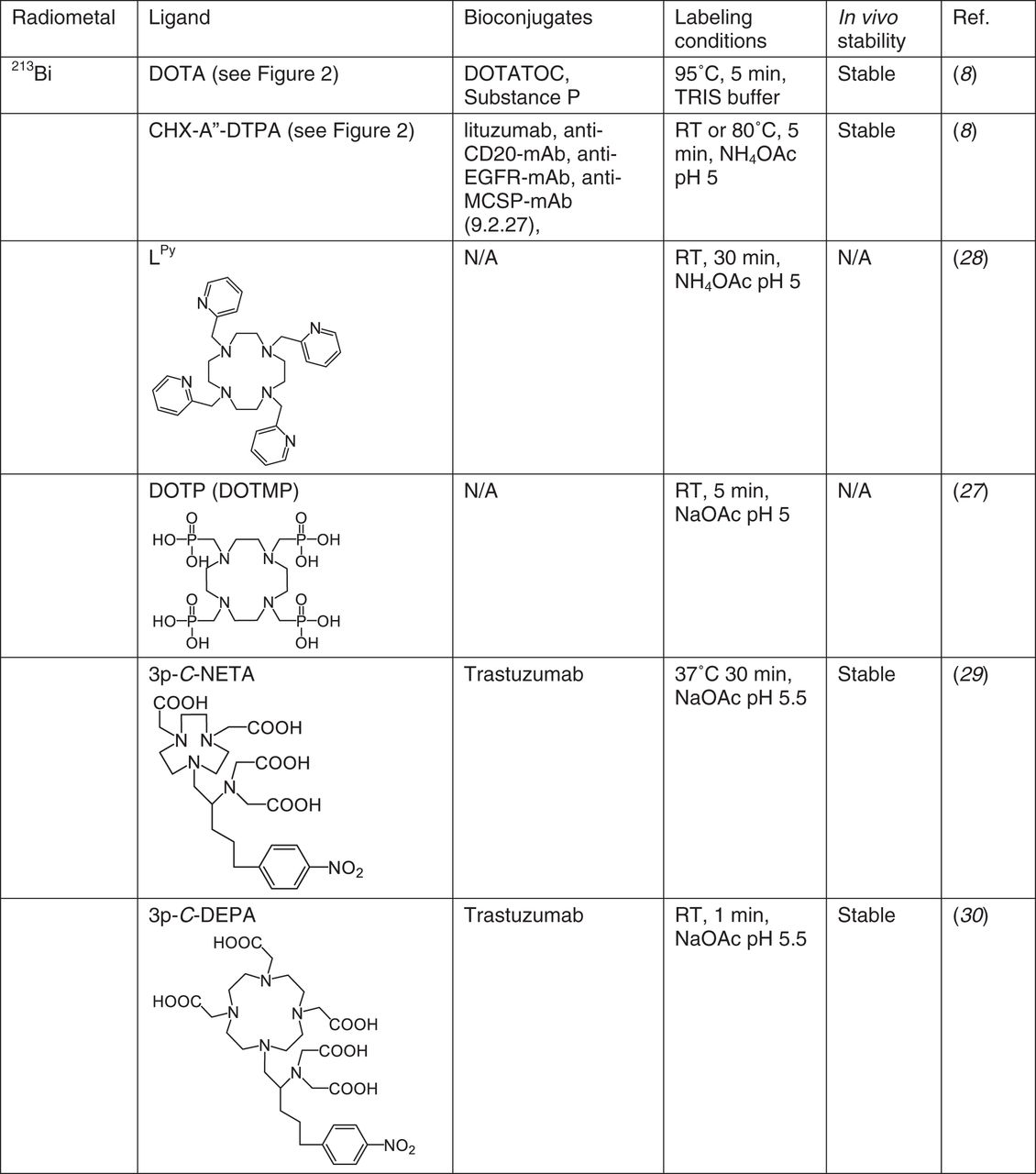
212/213Bi
DOTA and CHX-A″-DTPA are frequently used for Bi labeling, but neither chelate is a good match for Bi3+. Bi-DOTA or Bi-CHX-A″-DTPA complexes are not sufficiently stable in human plasma (85% DOTA, 76% CHX-A″-DTPA, 2 h) (27). Nonetheless, their efficacy was tested successfully in vivo for antibodies and peptides, with CHX-A″-DTPA used primarily for antibody conjugates and DOTA for peptides (Fig. 3). Radiopharmaceuticals in clinical studies include 213Bi-HuM195, 213Bi-anti-CD20-mAb, 213Bi-anti-EGFR-mAb, 213Bi-anti-MCSP-mAb (9.2.27), 213Bi-DOTA-substance P, and 213Bi-DOTATOC. The reader is directed toward another recent review for additional information on 213Bi radiopharmaceuticals (8).

212/213Bi chelators. N/A = not applicable; RT = room temperature.
The nitrogen-rich cyclen-pyridine ligand Lpy and analogs are reported to form highly stable complexes with 213/207Bi at room temperature (28). Lpy showed selectivity toward Bi3+ over Ac3+ and showed resistance to transmetalation when challenged with Cu2+, Zn2+, Fe2+, or Bi3+, but in vivo studies are not yet reported. The phosphorus-containing cyclen chelates DOTP (also named DOTMP, Fig. 3) and analogs form stable complexes with 213Bi at room temperature as well, and 213Bi-DOTP showed higher stability in human serum at 120 min than did 213Bi-DOTA or 213Bi-CHX-A″-DTPA (27). 213Bi-DOTP accumulates primarily in bone, unlike free Bi3+, which accumulates in the kidneys.
205/206Bi has been used to evaluate novel chelates such as {4-[2-(bis-carboxymethyl-amino)-ethyl]-7-carboxymethyl-[1,4,7]triazonan-1-yl}-acetic acid (NETA, a NOTA derivative) and 7-[2-(bis-carboxymethyl-amino)-ethyl]-4,10-bis-carboxymethyl-1,4,7,10-tetraaza-cyclododec-1-yl-acetic acid (DEPA, a DOTA derivative), synthesized specifically to provide a larger cavity to coordinate with Bi3+. Both coordinate 205/206Bi at low or ambient temperature with high efficiency (29,30). Using a 3-carbon linker in the bifunctional chelates, 205/206Bi-3p-C-NETA- and 205/206Bi-3p-C-DEPA-trastuzumab both showed effective tumor accumulation and low kidney uptake (29,30).
Overall, because of the short half-life of the Bi isotopes, development of novel chelators such as Lpy, DOTP, NETA, and DEPA, capable of rapid, ambient temperature metal incorporation, may help to improve the radiochemical yields and molar activity of potential radiopharmaceuticals. Additional in vivo studies are required to evaluate stability and demonstrate suitability for TAT. There also remains potential for further development of S-donor chelates, informed by meso-2,3-dimercaptosuccinic and 2,3-dimercaptopropane-1-sulfonic acid, which were used for chelation therapy to prevent Bi overdose in humans (31).
212Pb
Although not an α-emitter itself, 212Pb is used as an internal generator for 212Bi. DOTA was one of the first chelators studied with 212Pb (Fig. 4). Although data suggest favorable kinetic stability of Pb-DOTA complexes (32), release of Pb in vivo in the acidic tumor environment was reported in some cases and may prove to be a source of toxicity upon internalization and metabolic processing (33).
Chelators for 212Pb. N/A = not applicable; RT = room temperature.
In a pretargeting approach where 212Pb-DOTA-biotin was injected into mice preinoculated with streptavidin-NR-LU-10 mAb, despite the elevated dose to kidneys from free 212Bi, the biodistribution relative to the 203Pb counterpart was identical for all other organs, indicating little migration of 212Bi once in tissue (34). 212Pb-DOTA-Re(Arg11)CCMSH, a melanoma-targeting peptide, was evaluated in B16/F1 xenografts (35) and provided measurable therapeutic outcomes. 212Pb-DOTA-103A mAb was studied for treating Rauscher leukemia virus (RVB3) (36). Despite success in eradicating the target, all animals succumbed to bone marrow toxicity, in contrast to the lack of marrow toxicity in a similar study using 212Bi-103A-mAb. Given the observed susceptibility for some DOTA derivatives to be acid-labile at lower pH, it has been postulated that 212Pb dissociation, followed by clearance from the target tissue and localization of the free Pb/Bi in bone marrow, leads to the observed toxicity.
A more efficient and stable chelate for Pb, TCMC (1,4,7,10-tetraaza-1,4,7,10-tetra-(2-carbamoyl methyl)-cyclododecane), has since become the standard for Pb labeling (37). Bifunctional 4-nitrobenzylisothiocyanate derivatives were used to generate 203Pb-TCMC-CC49 (37) and 212Pb-TCMC-trastuzumab (38). TCMC-trastuzumab conjugates were determined to be more stable and more efficient at metal conjugation than their DOTA counterparts. Pb-TCMC-trastuzumab radioconjugate stability was analyzed in vitro, coupled with the demonstrated therapeutic effect and minimal toxicity of 212Pb-trastuzumab in an orthotopic model of human prostate cancer cells (38). 212Pb-TCMC-trastuzumab was used in a first-in-humans dose escalation clinical trial and was well tolerated (39).
In 2020, a biodistribution study of several 203Pb- and 212Pb-labeled TCMC-PSMA derivatives was also reported (40). One derivative demonstrated tumor growth delay, with the kidney as the dose-limiting organ. Another PSMA ligand, [212Pb]Pb-NG001, showed that 212Pb and 212Bi colocalized during the 24-h study period, leading the authors to suggest that the rapid target internalization and nontarget clearance of [212Pb]Pb-NG001 prevented measurable amounts of 212Bi from being released from the target tissue (41). Recently, 212Pb-TCMC mAb376.96 was shown to bind the B7-H3 epitope found on ovarian cancer cells, with a 2- to 3-fold increased survival in tumor-bearing mice over the control group (42).
Recently, a series of cyclen-based chelators (described as DOTA-1Py, -2Py, and -3Py) was compared with the established chelators DOTA and TCMC. All chelates incorporated 212Pb (and 203Pb) efficiently, with radiochemical yields higher than for DOTA and lower than for TCMC (43). A separate report examined the complexation between Pb and calix[4]arene-1,3-crown-6, where the formation of a 1:1-Pb(II)-calix complex was confirmed by 207Pb NMR (44). The phosphonic acid chelates DOTP (DOTMP) and tetra-methylenephosphonic acid (EDTMP) were labeled with Pb but found to have low stability in vivo (32).
Overall, 212Pb coordination chemistry appears to have emerged from a multidecade preclinical development phase and has advanced into a limited number of human clinical trials. The availability of the isotope and an imageable element-equivalent companion (203Pb), coupled with extensive development of key chelators (i.e., TCMC), suggests an exciting future for this isotope.
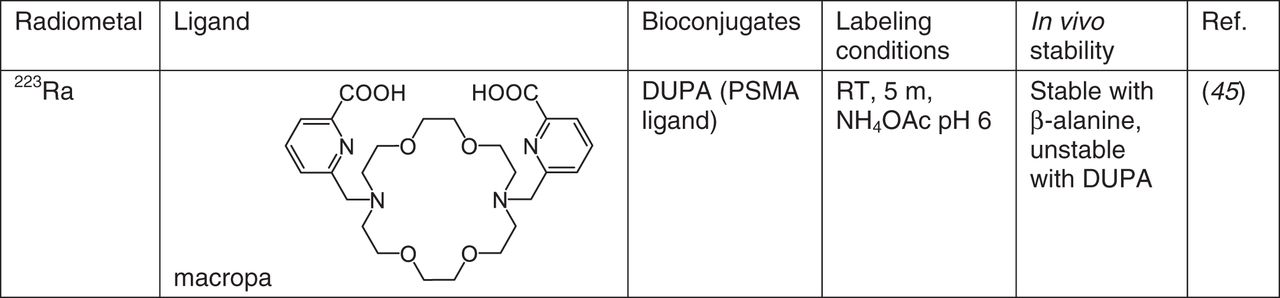
223Ra
So far, the only 223Ra2+ chelator with demonstrated in vivo stability is macropa, manifesting in low bone uptake of [223Ra] [Ra(macropa-β-alanine)] (Fig. 5) (45). However, a PSMA-macropa conjugate, DUPA, showed no difference from [223Ra]RaCl2 in subsequent biodistribution studies, highlighting the difficulty of developing stable chelates for Ra2+. DOTA and Kryptofix 2.2.2 (Merck) bind Ra2+ as well, evidenced by competition extraction experiments against calix[4]arene tetraacetate (46), but the stability of these complexes has yet to be determined. Although calix[4]arene is effective in extracting Ra2+ in neutral pH solution, the stability of the complex is poor (46). A recent review examines the use of nanoparticles to immobilize 223Ra, including liposomes, barium sulphate, lanthanum phosphate, and hydroxyapatite, among others (26).

Chelators for 223Ra. RT = room temperature.
No suitable chelator for in vivo delivery of 223Ra has been identified to date; although macropa shows promise, much work remains to develop a suitable chelate for Ra2+.
225Ac
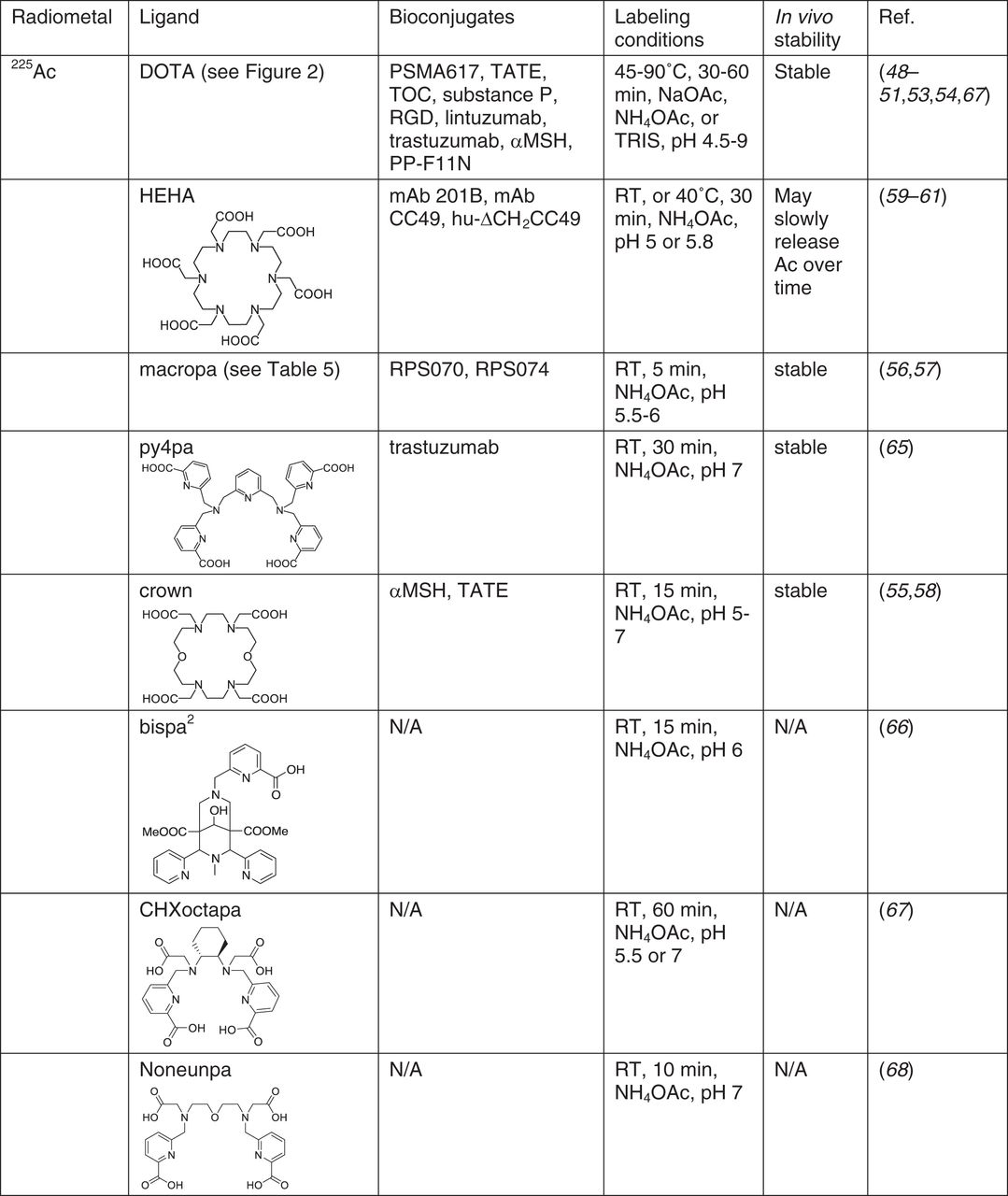
Most 225Ac chelates developed to date are macrocycles; DOTA remains the gold standard for Ac labeling for all clinical work. Examples include225Ac-PSMA617, 225Ac-DOTATOC, and 225Ac-DOTA-HuM195 (Fig. 6) (8). Labeling can be performed using 1-step (47) or 2-step (48) methods, with the latter being used for heat-sensitive targeting biomolecules. On the clinical front, 225Ac-DOTA bioconjugates of 225Ac-PSMA617, 225Ac-DOTATOC, and 225Ac-DOTA-HuM195 (8) have been evaluated. On the preclinical front, 225Ac-DOTATOC (49,50), -F3 (51), -c(RDGyK) (52), -MC1RL (53), -HuM195 (48), -J591 (anti-PSMA) (48), -B4 (anti-CD19) (48), and -trastuzumab (54) are all reported. It is noteworthy that unexpected high liver or spleen uptake observed in some studies may not be related to the in vivo stability of 225Ac-DOTA but rather to radiolysis (55), necessitating further study.

Chelators for 225Ac. N/A = not applicable; RT = room temperature.
Large macrocyclic chelates have been used to ease the steric constraints for large metal ions and to increase the coordination number. 18-membered macrocycles with 6 donor atoms (N or O) have been extensively studied. Macropa showed high specificity toward Ac3+ (56). It has been used to label PSMA-targeted RPS070 (56) and RPS074 (57) and showed good in vivo stability, effective tumor uptake, and therapy efficacy. Crown, another emerging chelate with structural similarity to DOTA, showed high labeling yields with Ac3+ at low concentrations (55,58). It was conjugated to an αMSH peptide targeting melanocortin 1 receptor and showed high tumor uptake and very low uptake in normal tissues and organs (55). HEHA, one of the first chelates developed specifically for Ac3+, showed promise in early studies (59,60), but a conjugate with mAb-B201B slowly released free Ac3+ from the targeting organ (lungs) (61). Other macrocyclic chelates, such as PEPA (59), TETPA (62), TETA (62), DOTP (63), and macropid (Macrobid; Allergan Pharmaceuticals International Ltd.) (64), suffer either low labeling yield or poor in vivo stability, indicating that increasing ring size or adding more donor atoms may not always work.
Acyclic chelates with picolinic acid moieties have shown significant promise as Ac3+ chelates as well. Py4pa demonstrated a high Ac labeling yield at low chelate concentrations (65). The biodistribution of 225Ac-py4pa-trastuzumab showed effective tumor accumulation and relatively low liver uptake. Bispa2, CHXoctapa, and noneunpa all showed an excellent labeling yield at low chelate concentrations (66–68). Those chelates can coordinate 111In as well, giving the advantage of potential theranostic pairs.
Overall, new chelators such as macropa, crown, and py4pa are highly promising because of the improved radiochemical yield, specific activity, and mild labeling conditions. One important area to address is whether the new chelators are capable of binding with an easily accessible imaging isotope, such as 68Ga (imaging is essential for clinical translation of 225Ac-DOTA radiopharmaceuticals). For the chelators with a demonstrated ability to bind with an imaging isotope (Bispa2, CHXoctapa, noneunpa), in vivo studies are necessary to assess the stability and applications.
226/227Th
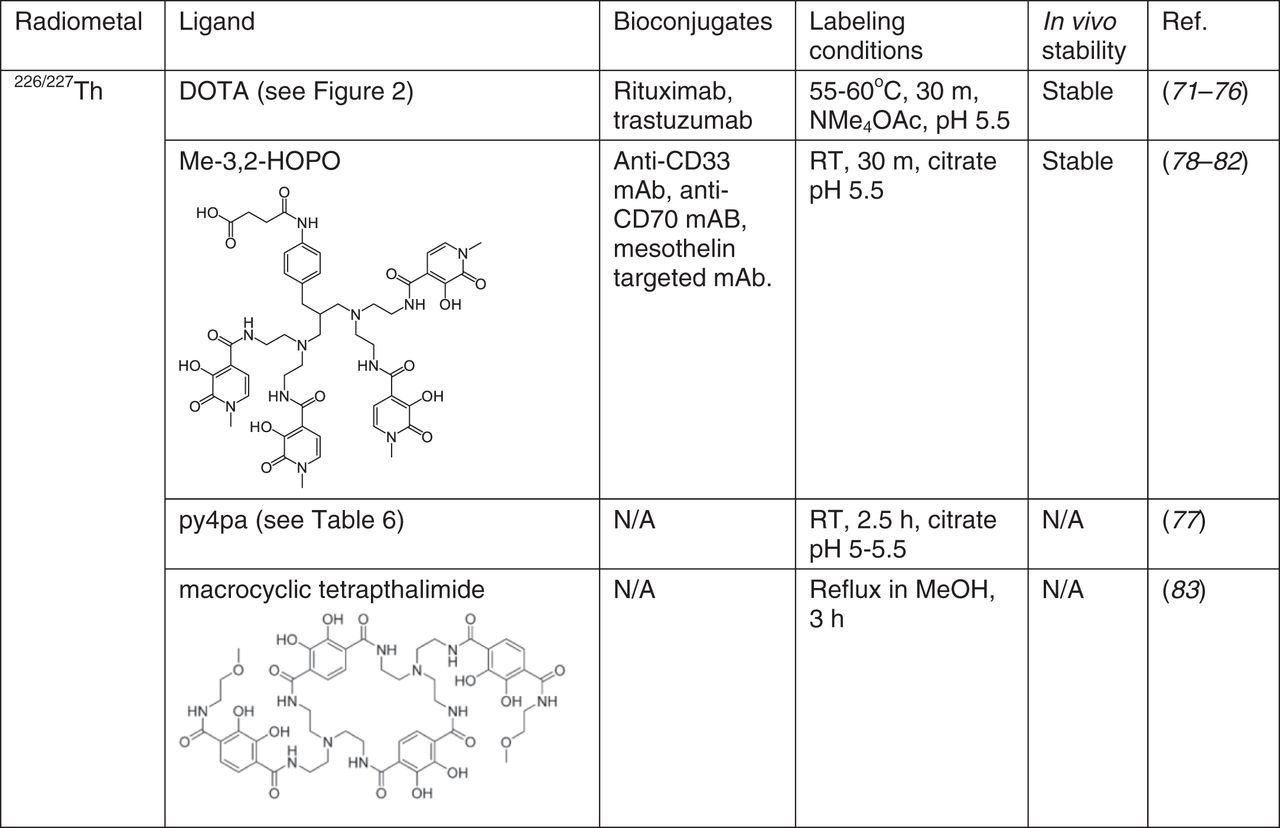
Early studies of 227Th chelation used polyphosphonate-based chelates to target bone. Acyclic chelators ethylenediamine tetra(methylene phosphonic acid) (EDTMP) and diethylenetriamine penta(methylene phosphonic acid) (DTPMP) were evaluated in addition to the macrocyclic ligand (DOTMP) for this purpose (69,70). In animal studies, all 3 complexes exhibited enhanced bone uptake compared with free [227Th]Th4+, indicating that the phosphonate groups can facilitate bone targeting of this radionuclide and that these chelators form stable in vivo complexes with Th4+.
On the basis of its success with many other radiometals, the bifunctional analog of DOTA was studied to chelate 227Th. Early coordination chemistry studies, as well as more recent solid-state structural studies of Th-DOTA complexes (71–73) indicate that they should be stable in aqueous solution (Fig. 7). The most common process involves a low-yielding 2-step reaction; 227Th is initially radiolabeled with bifunctional DOTA-NCS at an elevated temperature and then is conjugated to an antibody (74,75). A single-step radiolabeling reaction with a trastuzumab conjugated to DOTA required very long (2 d) reaction times (76). Despite the poor radiolabeling kinetics, the Th-DOTA complex retains good stability in vivo (75), enabling its use for various TAT applications.

Chelators for 226/227Th. N/A = not applicable; RT = room temperature.
Alternative chelators such as the picolinic acid “pa” chelators octapa, neunpa-p-Bn-NO2, pypa, and py4pa were investigated for their ability to complex 277Th (77). Among those 4 chelates, py4pa gave a high radiochemical yield (87% in 2.5 h), and the complex remained intact in phosphate-buffered saline solution for over 2 weeks. In view of these promising results, py4pa was investigated for chelating the shorter-lived 226Th (77). Microwave irradiation afforded the radiolabeled complex in high yield, marking the first occasion of complexation of the 226Th radionuclide.
Given the oxophilic nature of the Th4+ ion, the use of oxygen-rich chelating agents for these radionuclides is a promising approach. An octadentate hydroxypyridonate-based chelator bearing 4 bidentate 3-hydroxy-N-methyl-2-pyridinones (Me-3,2-HOPO) was validated to be an effective chelator for 227Th (78). This ligand quantitatively incorporated 227Th after 30 min at ambient temperature, marking a significant enhancement over the elevated temperatures required for DOTA; the resulting 227Th complex was stable in vivo, as reflected by a lack of bone uptake. This chelator enabled several tumor-targeting constructs to demonstrate the in vivo therapeutic potential of TAT with 227Th (79–81). Detailed analytic studies on Th4+ chelation with this ligand revealed that it possesses a large (>20 orders of magnitude) thermodynamic selectivity for +4 over +3 ions (82). This large selectivity for +4 ions may explain the high stability of its Th4+ complex in vivo, as biologically common +3 ions such as Fe3+ cannot effectively displace the actinide. The status of the Me-3,2-HOPO chelate as the current gold standard for 227Th chelation has inspired additional studies on ligands of this type. A recently reported macrocyclic tetrapthalimide ligand was found to have the highest thermodynamic affinity for Th4+ discovered to date (log K = 1054), representing a promising candidate for 227Th TAT (83).
Overall, oxygen-rich chelators are effective for stabilizing the hard, oxophilic Th4+ ion in vivo. The HOPO-containing ligands are among the most promising candidates for use with Th-based TAT, but these chelators suffer from challenging chemical syntheses. The development of more readily accessible polydentate oxygen-rich chelators will meet an important need for advancing Th-based TAT.
230U
The chemically hard and oxophilic nature of the [UO2]2+ ion dictates anionic oxygen-donor ligands. These conclusions are supported by many decades of [UO2]2+ coordination chemistry (84). Nevertheless, there have been surprisingly few studies to develop chelators specifically for 230U TAT. The lack of 230U TAT studies is a consequence of the limited availability of the radionuclide and narrow knowledge of its potential importance in nuclear medicine. Researchers have investigated the interactions of the [UO2]2+ ion with the human serum proteins transferrin and albumin, in addition to other small molecules, such as carbonate, that are present in blood in order to assess the needed efficacy of a potential 230U chelator (85). An effective chelator for this radionuclide was predicted to have a stability constant in excess of 1019 (85); there are few chelators with such a high stability constant for [UO2]2+. For example, calixarene ligands were found to form unstable complexes with [238UO2]2+ that dissociated in serum (86). Given the expansive literature and efforts to develop U decorporation agents, however, it is likely that there are promising candidates for 230U chelation extant. For example, the deferoxamine complex of UO22+ has a stability constant within this range (87), as does a class of hexadentate equatorial-spanning terephthalamide(bis-hydroxypyridinone) ligands (88), suggesting that these chelators may be useful for 230U TAT applications.
Although the [UO2]2+ ion predominates in aqueous solution, U4+ is also found. DOTA complexes of U4+ are well characterized and are stable to oxidation in aqueous solution (72,73,89). HOPO-based ligands can facilitate the reduction of [UO2]2+ to U4+ in water (90). HEHA, investigated for 225Ac, also forms a stereochemically rigid and therefore potentially inert complex with U4+ (91). Thus, these results suggest that targeting the chelation of U4+, perhaps by modulating the redox conditions of the radiochemical isolation and separation of 230U, may provide an alternative effective means for delivering this radionuclide to tumor cells. Although 230U has been proposed to be a promising candidate for TAT, there are currently no in vivo or in vitro studies that have demonstrated its efficacy for eliminating malignant cells. As such, chelation efforts for this radionuclide are somewhat underdeveloped; however, the principles of U chelation that have been established for other applications are likely pertinent to future 230U TAT applications.
CHALLENGES AND OPPORTUNITIES
Lutathera, as the first regulatory body–approved (United States and European Union) compound for peptide receptor radionuclide therapy, has stimulated substantial interest in targeted radiotherapy, particularly to treat patients with metastatic disease. α-emitters are highly promising for radiotherapeutic agents, illustrating viability in the treatment of a wide range of malignancies. This surge of interest, particularly in the actinides, however, has outpaced the understanding of the chemistry of each individual element; this understanding remains poorly developed because of the limited supply of the elements and the usually intense radioactivity of all isotopes. Any future coordination chemistry research will depend on reliable availability of α-emitting radionuclides. New production methods coming on-line now mean that supply is approaching levels capable of supporting preclinical as well as clinical studies on a routine basis, and further research into the coordination chemistry of these isotopes is now possible. With half-lives that range from around 1 h (212,213Bi) to weeks (227Th), coupled with the radiologic and biologic considerations needed, the challenges in chelating these metals are considerable.
An important step to advance novel chelates is to find a readily available imaging isotope for those specific chelates for patient stratification and treatment monitoring, which can be challenging because relevant α-emitters typically maintain significantly different chemical properties. The challenge can be addressed either by designing and testing chelates that can accommodate both (68) or by developing imaging radioisotopes, such as 134Ce/134La (92), that are closer in nature to emerging actinide-based α-emitters.
One of the major impediments to using α-emitting radionuclides has been the need for improved chelators or labeling methods to ensure that they remain stable in vivo in order to deliver their payload to the target tissue and minimize the dose that can arise because of dissociation and off-target tissue localization.
Metal-ligand bonding in actinide complexes has been thought to be driven primarily by electrostatic interactions and steric constraints, with limited orbital interaction. Early efforts show that one can match isotopes to chelators, but actinides show significantly more covalency than lanthanides (68). Further studies of fundamental actinide coordination and organometallic chemistry will enlighten more specific chelate designs.
α-recoil will cause the progenies to leave the chelates and free the daughters, which may carry significant or even most of the energy, as in the case of 225Ac decay. Fast pharmacokinetics and sufficient internalization are highly desirable to limit off-target toxicity and maximize the tumor dose. Other methods to mitigate the risk include encapsulating in nanoparticles or localized administration (10).
The tumor microenvironment includes, as 3 major cell types, immune, stromal, and vascular cells, and the potential to target each will become more exact using unique combinations of elements/isotopes with chelators and more specific targeting vectors. The incorporation of α-emitters into nanomaterials (with or without chelation) presents another front of interest to TAT.
In summary, a focus on new tightly binding, selective chelating ligands that can yield high specific activity radiopharmaceuticals will soon be necessary. Reproducibly versatile and subsequently translatable chelation chemistry is required. Other areas important for realizing the full potential of α-emitting radiopharmaceuticals include, but are not limited to: modular and automated (smart) technologies for high-level synthesis, kit-formulation for ease of preparation, as well as a better understanding of the biology of daughter isotopes released via recoil, as this could be a limiting factor in the efficacy of emerging radiopharmaceuticals due to their toxicity. All of these challenges represent opportunities for research in the expanding field of TAT.
DISCLOSURE
TRIUMF receives funding via a contribution agreement with the National Research Council of Canada. Justin Wilson receives support through the U.S. National Institutes of Health under awards R21EB027282 and R01EB029259. Chris Orvig has received many years of support from NSERC and CIHR. Paul Schaffer is a consultant for ARTMS, Inc., as chief technology officer. Justin Wilson holds equity in Ratio Therapeutics, which has licensed macropa for chelation of α-emitters. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online September 9, 2021.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- Received for publication June 11, 2021.
- Revision received August 5, 2021.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}