


Abstract
Inflammation contributes to ventricular remodeling after myocardial ischemia, but its role in nonischemic heart failure is poorly understood. Local tissue inflammation is difficult to assess serially during pathogenesis. Although 18F-FDG accumulates in inflammatory leukocytes and thus may identify inflammation in the myocardial microenvironment, it remains unclear whether this imaging technique can isolate diffuse leukocytes in pressure-overload heart failure. We aimed to evaluate whether inflammation with 18F-FDG can be serially imaged in the early stages of pressure-overload–induced heart failure and to compare the time course with functional impairment assessed by cardiac MRI. Methods: C57Bl6/N mice underwent transverse aortic constriction (TAC) (n = 22), sham surgery (n = 12), or coronary ligation as an inflammation-positive control (n = 5). MRI assessed ventricular geometry and contractile function at 2 and 8 d after TAC. Immunostaining identified the extent of inflammatory leukocyte infiltration early in pressure overload. 18F-FDG PET scans were acquired at 3 and 7 d after TAC, under ketamine-xylazine anesthesia to suppress cardiomyocyte glucose uptake. Results: Pressure overload evoked rapid left ventricular dilation compared with sham (end-systolic volume, day 2: 40.6 ± 10.2 μL vs. 23.8 ± 1.7 μL, P < 0.001). Contractile function was similarly impaired (ejection fraction, day 2: 40.9% ± 9.7% vs. 59.2% ± 4.4%, P < 0.001). The severity of contractile impairment was proportional to histology-defined myocardial macrophage density on day 8 (r = −0.669, P = 0.010). PET imaging identified significantly higher left ventricular 18F-FDG accumulation in TAC mice than in sham mice on day 3 (10.5 ± 4.1 percentage injected dose [%ID]/g vs. 3.8 ± 0.9 %ID/g, P < 0.001) and on day 7 (7.8 ± 3.7 %ID/g vs. 3.0 ± 0.8 %ID/g, P = 0.006), though the efficiency of cardiomyocyte suppression was variable among TAC mice. The 18F-FDG signal correlated with ejection fraction (r = −0.75, P = 0.01) and ventricular volume (r = 0.75, P < 0.01). Western immunoblotting demonstrated a 60% elevation of myocardial glucose transporter 4 expression in the left ventricle at 8 d after TAC, indicating altered glucose metabolism. Conclusion: TAC induces rapid changes in left ventricular geometry and contractile function, with a parallel modest infiltration of inflammatory macrophages. Metabolic remodeling overshadows inflammatory leukocyte signal using 18F-FDG PET imaging. More selective inflammatory tracers are requisite to identify the diffuse local inflammation in pressure overload.
Heart failure (HF) is a multifactorial syndrome and the common endpoint of many cardiovascular diseases. Despite improvements in early treatment strategies, HF remains a prevalent cause of death worldwide and a significant burden on the health-care system (1). Conventional therapeutic approaches combine multiple drugs targeting nonspecific symptoms and common mechanisms of HF, to support cardiac function and improve survival (2). But various specific pathogenetic mechanisms contributing to development and progression of HF remain undertreated, particularly in nonischemic HF (3). Recent evidence suggests that inflammatory leukocytes and macrophages may play a role in nonischemic HF progression (4) and contribute to interstitial fibrosis and contractile dysfunction (4). HF patients exhibit elevated circulating white blood cells, inflammatory cytokines, and biomarkers, regardless of pathogenesis (5,6). As such, inflammation has emerged as a potential therapeutic target for not only ischemic HF but also nonischemic cardiomyopathies (7).
Acute infiltration of inflammatory leukocytes predicts functional decline and progression of HF after ischemic injury (8). Conversely, inflammatory cell infiltration and adverse ventricular remodeling in nonischemic pressure overload are less well characterized, but the temporal dynamics and involved cell population are thought to be distinct from ischemic injury (9). At 7 d after aortic banding, the total macrophages and leukocyte content of the left ventricle are maximal (10–12) and may contribute to ventricular remodeling (13,14). Although systemic inflammation may be quantified through biomarkers or leukocyte count, local myocardial inflammatory activity is more difficult to assess. PET using 18F-FDG robustly accumulates in activated inflammatory leukocytes, including macrophages (15). In the myocardium, however, this accumulation is complicated by physiologic uptake of 18F-FDG into viable myocytes—uptake that is further enhanced by ischemia or other injury (16). Acute pressure overload instigates rapid changes in myocyte glucose metabolism, and such changes are believed to contribute to ventricular hypertrophy (17,18). Nevertheless, inflammation-targeted 18F-FDG imaging, using preparation protocols for suppression of myocyte uptake, has demonstrated prognostic value for outcomes after myocardial infarction in patients and animal models (8,19). But the feasibility of imaging myocardial inflammation in nonischemic HF is undefined.
We hypothesized that 18F-FDG PET imaging would identify infiltrating macrophages and inflammatory leukocytes in the myocardium early in the pathogenesis of nonischemic pressure-overload–induced HF. Accordingly, in mice after transverse aortic constriction (TAC) (using a myocyte uptake suppression protocol), we compared left ventricular uptake of 18F-FDG with progressive changes in left ventricular geometry and contractile function on cardiac MRI. This comparison was complemented by histologic and biochemical tissue workup to determine factors underlying the imaging signal.
MATERIALS AND METHODS
Animals
All animal experiments were approved by the state authority (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit) and the local animal ethics review board. Male C57BL/6N mice (n = 39, 23.7 ± 1.5 g) were purchased from Charles River and housed in groups in a temperature-controlled facility under a 14-h/10-h light/dark cycle with food and water freely available.
TAC
The mice underwent TAC (n = 22) or sham surgery (n = 12) under isoflurane anesthesia, as described previously (20). Briefly, the mice were pretreated with the analgesic carprofen (5 mg/kg subcutaneously) and anesthetized by isoflurane (induction at 3%–4%, 3 L of oxygen per minute; maintained at 1.5%–2% after oral intubation under mechanical ventilation). Atropine sulfate (0.05 mg/kg, subcutaneously) prevented bradycardia and tracheal mucus accumulation. A lateral thoracotomy in the second intercostal space was performed and the thymus gently retracted. The aorta was exposed by blunt dissection and gently separated from surrounding tissue between the brachiocephalic trunk and the left carotid artery. To constrict the transverse aorta, a 6-0 silk suture was secured around the vessel using a blunt 25-gauge needle to define a consistent ligature diameter. After the suture was secured, the needle was removed, the rib cage and skin closed, and the animal allowed to recover before being returned to the home cage. For the sham operation, the identical procedure was followed without securing the suture. After surgery, oral analgesia was maintained with tramadol (2.5 mg/100 mL of drinking water).
Myocardial Infarction (MI)
As a positive control that exhibits robust macrophage infiltration, MI was induced by coronary artery ligation as previously described (21). Briefly, the mice (n = 5), pretreated with butorphanol analgesic (2 mg/kg subcutaneously), were anesthetized with isoflurane (induction at 3%–4%, 3 L of oxygen per minute; maintained at 1.5%–2% after oral intubation under mechanical ventilation). Left thoracotomy was performed, and the left anterior descending coronary artery was permanently ligated.
Study Design
Ventricular geometry and contractile function early in pressure-overload HF were assessed by cardiac MRI at 2 and 8 d after surgery. Inflammation was serially assessed by 18F-FDG PET under ketamine–xylazine (K/X) anesthesia to suppress cardiomyocyte glucose uptake at 3 and 7 d after surgery. These imaging time points were selected on the basis of the expected maximal inflammatory cell infiltration (day 7) based on flow cytometry studies (10,14), with an intermediate time point to allow evaluation of changes over the first week after TAC. One group of mice was killed on day 3 for histologic workup. All remaining mice were killed by cervical dislocation on days 8–9 for ex vivo tissue workup. 18F-FDG images were acquired on day 3 after surgical MI, which evokes robust inflammatory cell infiltration of the infarct territory.
Histopathology
For histologic assessment of inflammation, mice were killed on day 3 (n = 4) after TAC or after MI (n = 1) and on day 8 (n = 8) after TAC or after sham surgery (n = 8). Hearts were removed, cleaned in phosphate-buffered saline, embedded in optimal-cutting-temperature compound (Tissue Tek), and snap-frozen in liquid nitrogen. Adjacent long-axis 10-μm sections were sliced using a cryostat (Shandon) and thaw-mounted onto charged slides (SuperFrost Plus). Immunostaining for CD68 and Ly6G identified macrophages and granulocytes, respectively.
Small-Animal MRI
Mice (TAC, n = 10; sham, n = 4) underwent MRI at 2 and 8 d after surgery using a 7-T small-animal MRI system (Pharmascan 70/16 with ParaVision software, version 6.0.1; Bruker BioSpin). Cardiac function and geometry were evaluated using a navigator-based self-gated cine MRI sequence (IntraGate FLASH; Bruker BioSpin) as described previously (22).
A 72-mm-diameter transmit volume coil (T11070 89/72 Quad to AD; Bruker BioSpin) was used in combination with an anatomically shaped 4-element mouse cardiac phased-array surface receive coil (T20027V3; Bruker BioSpin). The animals were positioned prone, and body temperature was maintained by a water-heated system during examination. All scans were performed with the animals under isoflurane inhalation anesthesia (3% induction, 2% maintenance). Respiration rate was monitored and kept between 50 and 60 breaths per minute.
Cine images were acquired in 4-chamber view to visualize the aortic banding and in a short-axis orientation that covered the whole left ventricle to evaluate ventricular geometry and contractile function. The sequence parameters (short axis) were a 25 × 25 mm field of view, a 196 × 196 matrix, a 0.9-mm slice thickness, a 0.9-mm slice distance, an 84.64-ms repetition time, a 1.84-ms echo time, a 45° flip angle, and 24 reconstructed phases.
MR Image Analysis
Image analysis was performed with CVI (version 5.3.6; Circle Cardiovascular Imaging Inc.) to determine ventricular volumes and to assess heart function. Endocardial and epicardial contours were defined on short-axis sections and used to calculate ventricular mass at end systole and end diastole; ejection fraction was calculated from endocardial volumes.
Small-Animal PET
Mice (TAC, n = 10; sham, n = 4; MI, n = 5) underwent PET imaging using a dedicated small-animal PET camera (Inveon DPET; Siemens) as previously described (21). Briefly, the mice were anesthetized with ketamine (84 mg/kg intraperitoneally)/xylazine (11.2 mg/kg intraperitoneally) to suppress glucose uptake by cardiomyocytes (21) and were positioned prone on the imaging bed with the heart centered in the scanner field of view. 18F-FDG (12.7 ± 0.9 MBq) was administered as a 0.15-mL bolus through a lateral tail-vein catheter. A 60-min dynamic list-mode PET scan was obtained. 18F-FDG was synthesized using standard kits and production methods. One mouse died between the first and second PET acquisitions.
PET Image Analysis
Images were dynamically histogrammed and reconstructed using an iterative ordered-subset expectation-maximization 3-dimensional/maximum-a-posteriori algorithm as described previously (21). Analysis was performed with Inveon Research Workplace (version 4.2; Siemens Medical Solutions USA) to evaluate left ventricular volume and signal. A region of interest was defined on the left ventricular myocardium by interactive thresholding as previously described (21). The size and position of the region of interest were verified by anatomic landmarks in the fused CT images. The signal was evaluated semiquantitatively as percentage injected dose per gram of tissue (%ID/g).
All PET images were reconstructed to a 128 × 128 × 159 image matrix (0.78 × 0.78 × 0.80 mm) using an ordered-subset expectation-maximization 3-dimensional/maximum-a-posteriori algorithm (β = 0.01, 2 ordered-subset expectation-maximization iterations in 16 subsets, 18 maximum-a-posteriori iterations) with scanner-applied corrections for decay and scatter. A 57Co transmission scan was used for attenuation correction (14).
Western Immunoblotting
The hearts from a subset of animals (TAC, n = 4; sham, n = 4) were processed for immunoblotting using lysates of the left and right ventricles. The hearts were excised and dissected to separate the left from the right ventricles and then flash-frozen under liquid nitrogen. Tissue was stored at −80°C until use. Western immunoblotting was performed as described previously (17). Briefly, tissue samples were homogenized in lysis buffer separated by gel electrophoresis. The primary antibodies were mouse monoclonal anti–glucose transporter 1 (GLUT1) (SPM498, 1/5,000) and mouse monoclonal anti–glucose transporter 4 (GLUT4) (ab65267, 1 μg/mL) (Abcam). The secondary antibody was rabbit polyclonal anti–mouse IgG conjugated to horseradish peroxidase (Abcam). Immunoblots were analyzed by chemiluminescence, and integrated density values normalized to β-actin loading control were compared between groups.
Statistics
Statistical analysis was conducted using Prism (version 6; GraphPad). All data are shown as mean ± SD. Serial imaging data obtained for individual animals were compared using the Student paired t test. TAC and sham groups were compared using the Welch t test for unequal variance. Pearson product-moment correlation coefficients described the relationship between pairs of continuous variables. A P value of less than 0.05 was considered statistically significant.
RESULTS
Pressure Overload Evokes Early Left Ventricular Dilation and Cardiac Dysfunction
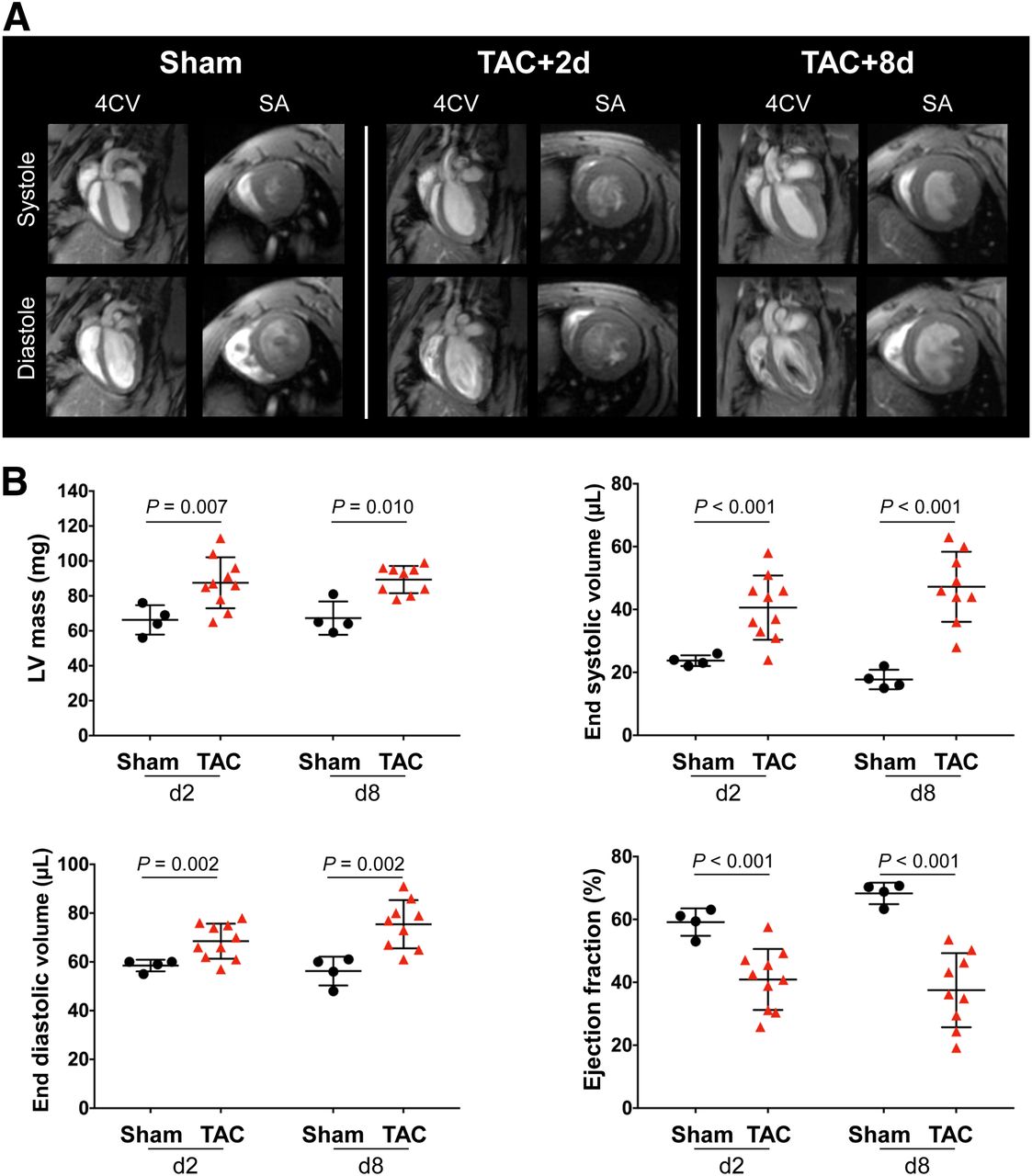
Within 2 d of TAC, cardiac MRI demonstrated a 30% increase in left ventricular mass and a corresponding elevation in ventricular volume compared with sham (Fig. 1A). Ventricular mass was consistently elevated in the first week after TAC surgery (day 2: 87.5 ± 14.6 mg vs. 66.3 ± 8.4 mg, P = 0.007; day 8: 89.3 ± 7.8 mg vs. 67.3 ± 9.5 mg, P = 0.010) (Fig. 1B). Similarly, a progressive increase in end-systolic volumes and end-diastolic volumes was identified from 2 and 8 d after surgery (end-systolic volume, day 2: 40.6 ± 10.2 μL vs. 23.8 ± 1.7 μL, P < 0.001; day 8: 47.2 ± 11.1 μL vs. 17.8 ± 3.1, P < 0.001) (end-diastolic volume, day 2: 68.5 ± 7.2 μL vs. 58.5 ± 2.4 μL, P = 0.002; day 8: 75.4 ± 9.9 vs. 56.3 ± 5.9, P = 0.002) compared with sham (Fig. 1B). TAC animals exhibited a marked reduction in ejection fraction at 2 d (40.9% ± 9.7% vs. 59.2% ± 4.4%, P < 0.001) and 8 d (37.5 ± 11.8 vs. 68.3 ± 3.4, P < 0.001) after surgery compared with sham (Fig. 1B), consistent with early and persistently impaired contractile function after pressure overload.

(A) Representative cardiac 4-chamber view (4CV) and short-axis (SA) images showing elevated heart volume and dilatation of left ventricle after TAC. (B) Quantitative functional analysis of left ventricular mass, end-systolic volume, end-diastolic volume, and ejection fraction.
Left Ventricular Macrophage Infiltration Correlates with Contractile Dysfunction
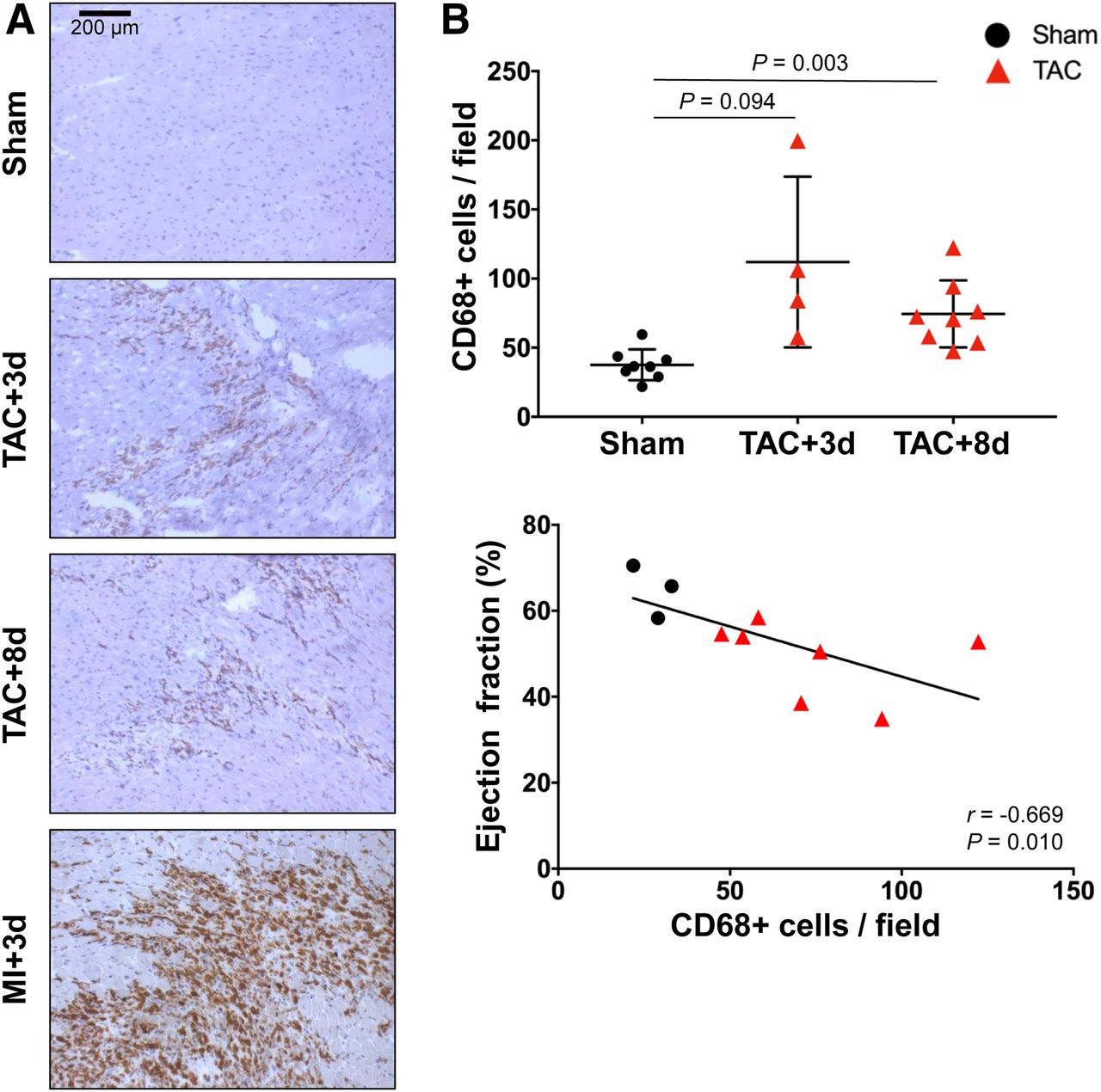
To investigate myocardial inflammation, CD68 and Ly6G immunostaining identified inflammatory leukocyte infiltration. CD68-positive macrophages were noted throughout the left ventricle after TAC, at a diffuse density compared with myocardial infarction (Fig. 2A). Semiquantification of cell infiltration revealed a 3-fold elevation of CD68-positive cells in the left ventricle of TAC mice compared with sham on day 3 and twice as many cells on day 8 (day 3: 111.9 ± 61.8 cells per field, P = 0.094; day 8: 74.4 ± 24.3 cells per field, P = 0.003; vs. sham: 37.6 ± 11.2 cells per field) (Fig. 2B). Macrophage density after TAC was much lower than the extensive staining after MI, for which quantitative calculation is complicated by the large abundance of CD68-positive staining (Fig. 2A). Nevertheless, in TAC and sham mice, the CD68 cell count inversely correlated with ejection fraction (r = −0.669, P = 0.010) (Fig. 2B), suggesting a relationship between inflammatory cell infiltration and functional decline. Ly6G-positive granulocytes were present but at a lower density than macrophages after TAC (Supplemental Fig. 1).
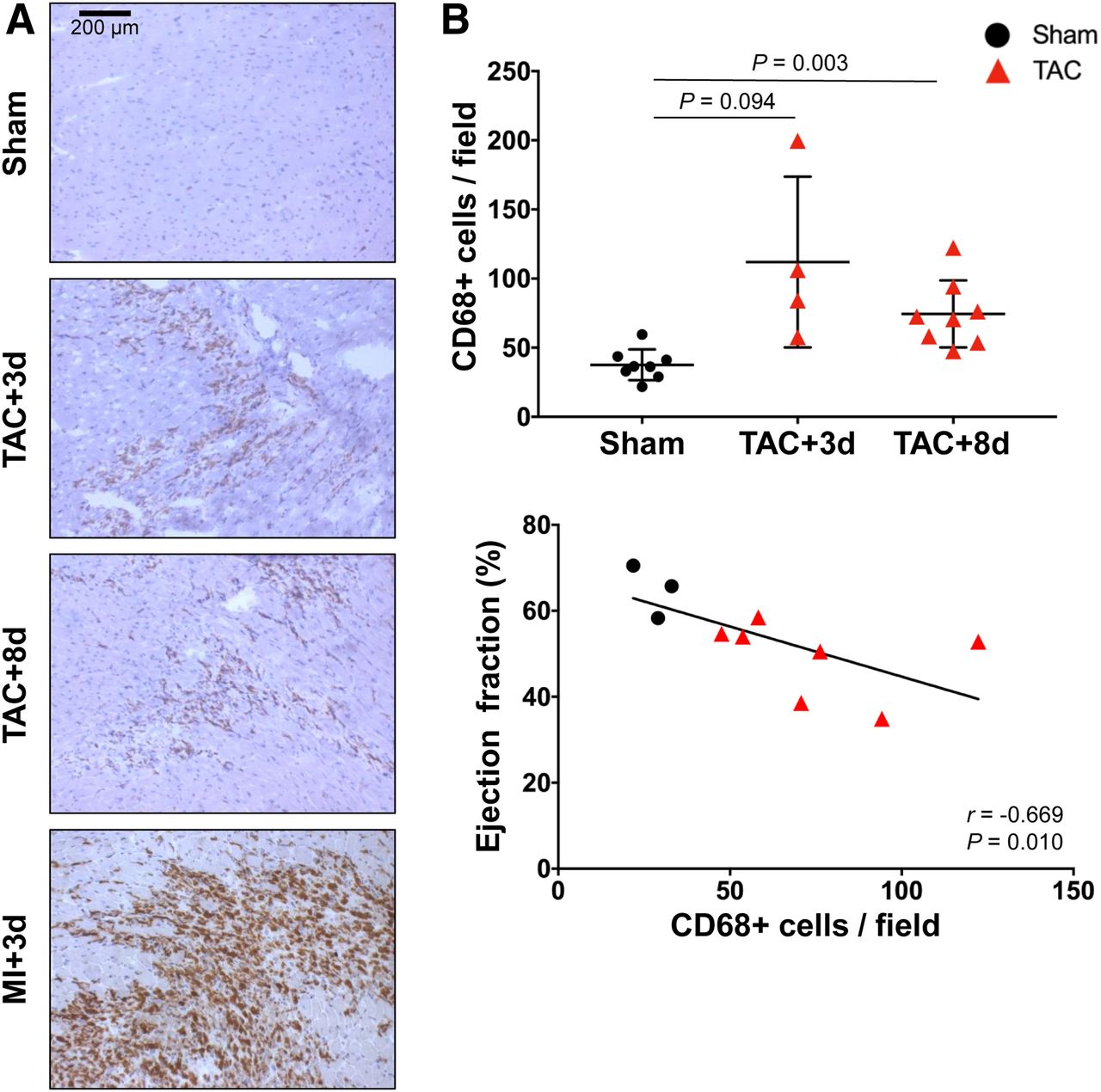
(A) CD68 immunostaining of representative long-axis sections at 3 and 8 d after TAC and 3 d after MI. (B) Quantitative analysis of stained sections showing elevation of CD68-positive cells per field at 3 and 8 d after TAC, which inversely correlates with impaired ejection fraction at 8 d after TAC.
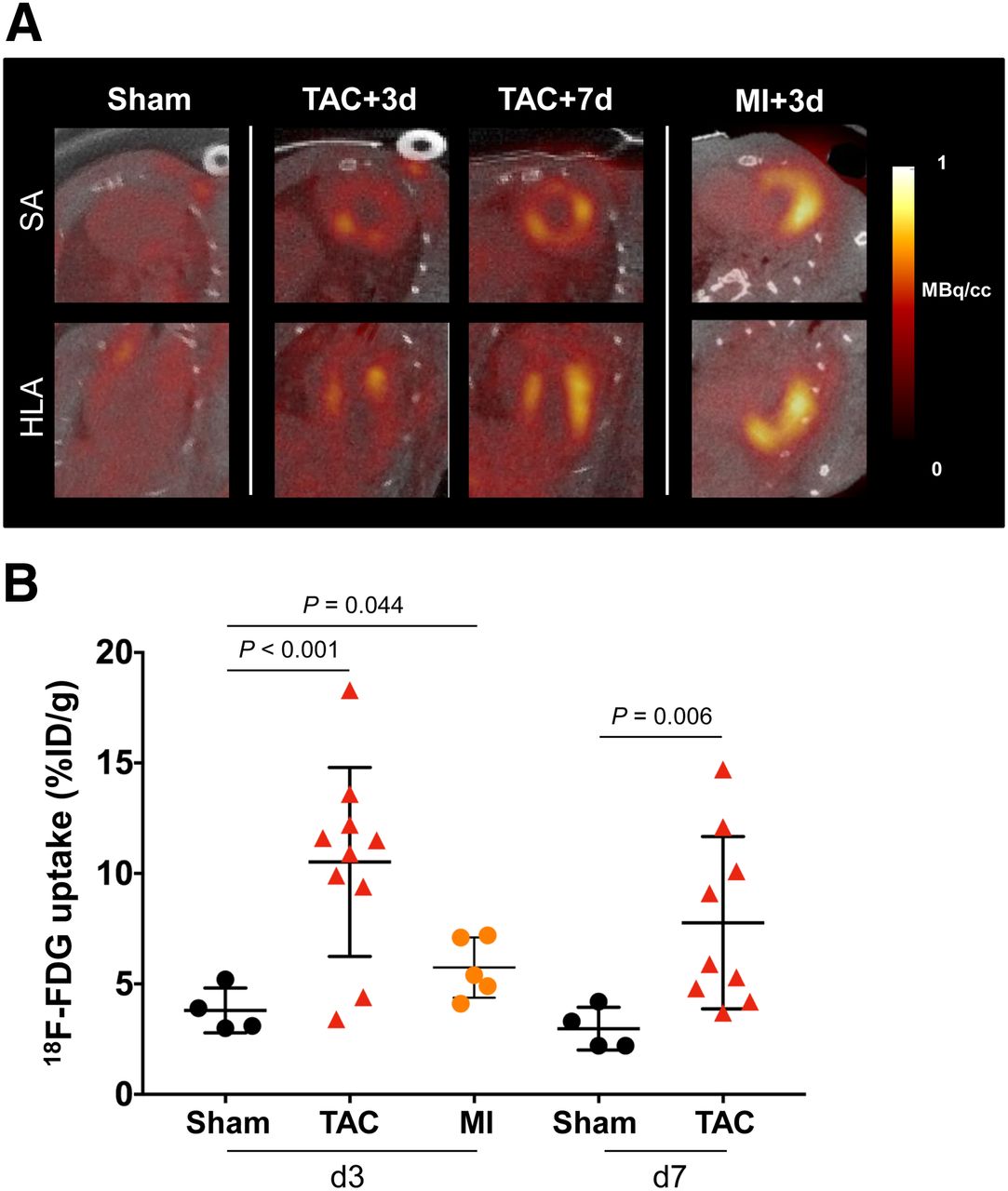
Myocardial 18F-FDG Signal Under K/X Suppression Is Elevated After TAC
K/X anesthesia significantly lowered cardiomyocyte 18F-FDG uptake in sham-operated animals to background levels (<5 %ID/g) (Supplemental Fig. 2). In inflammation-positive control MI mice, cardiomyocyte uptake of 18F-FDG was effectively suppressed, with global myocardial PET signal less than 10 %ID/g, and a hot spot localized to the infarct territory, consistent with histology findings (Fig. 3A). By contrast, TAC animals exhibited a range of modest to robust 18F-FDG accumulation throughout the left ventricle, despite a significantly lower inflammatory cell content. The left ventricular 18F-FDG uptake in TAC mice exceeded that of MI at 3 d (8/10 mice) and at 7 d (4/9 mice), suggesting ineffective cardiomyocyte suppression despite an identical K/X protocol (Supplemental Fig. 3). Semiquantitative analysis confirmed a higher left ventricular 18F-FDG signal in TAC mice than in sham mice on day 3 (+176%, 10.5 ± 4.1 %ID/g vs. 3.8 ± 0.9 %ID/g, P < 0.001) and on day 7 (+160%, 7.8 ± 3.7 %ID/g vs. 3.0 ± 0.8 %ID/g, P = 0.006) (Fig. 3B). The inconsistency of the PET signal localization and intensity necessitated further interrogation of the source of this 18F-FDG uptake.

(A) Representative 18F-FDG short-axis (SA) and horizontal long-axis (HLA) images under effective K/X suppression of cardiomyocyte glucose uptake. Global myocardial inflammation is seen at 3 and 7 d after TAC. Elevated 18F-FDG uptake is localized to infarct territory after MI. (B) Semiquantitative assessment of 18F-FDG mean %ID/g.
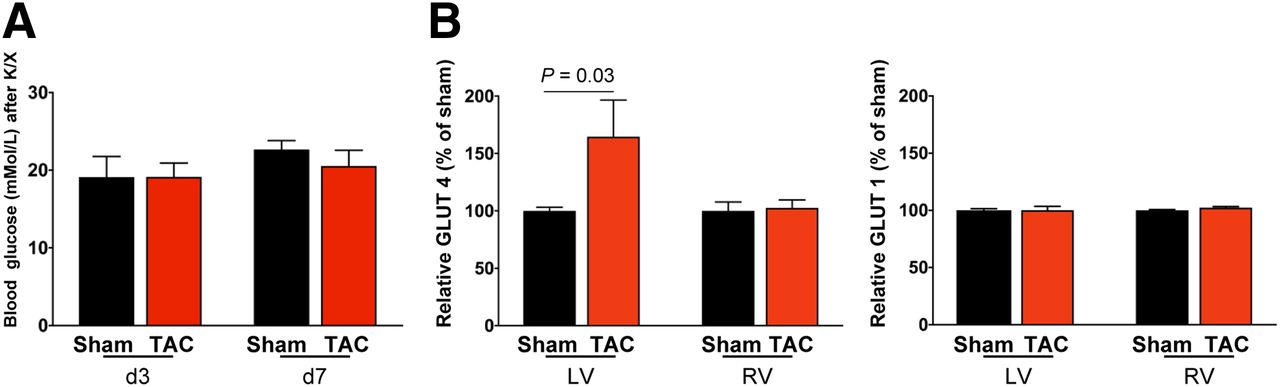
TAC Induces Altered Myocardial Glucose Transporter Expression
Because 18F-FDG uptake was more robust than expected from histologic evidence of leukocyte infiltration, we further investigated the efficacy of K/X to suppress glucose uptake after TAC. Blood glucose measurements at the time of acquisition confirmed hyperglycemia in both TAC and sham-operated animals (Fig. 4A) and were comparable between mice above and below the 10 %ID/g threshold (P = 0.35). Western immunoblotting demonstrated no difference in insulin-independent GLUT1 expression between TAC and sham-operated animals in the right or left ventricle. However, GLUT4 expression was 60% higher in TAC mice than in sham mice, specifically in the left ventricle (Fig. 4B). Accordingly, metabolic changes in substrate use in pressure-overloaded cardiomyocytes are a major contributor to the increased 18F-FDG PET signal after TAC.
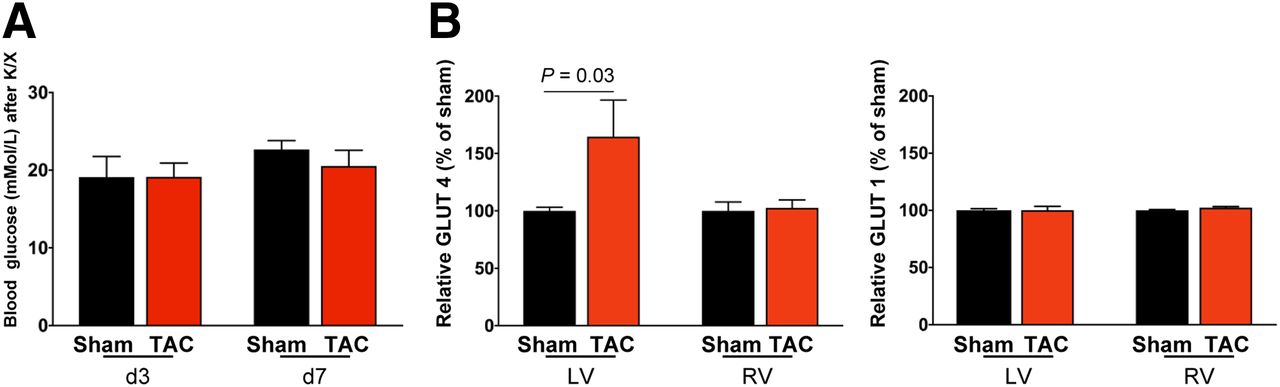
(A) Blood glucose levels are comparably elevated under K/X anesthesia in TAC and sham mice. (B) Quantification of Western immunoblots at 8 d after TAC. GLUT4 expression is elevated in left ventricle (LV) but not right ventricle (RV) after cardiac pressure overload, and there is no change in insulin-independent GLUT1.
Elevated Myocardial 18F-FDG Uptake Is Related to Early Functional Impairment
As both inflammation and metabolic changes may influence ventricular remodeling and cardiac function, we assessed the relationship between left ventricular 18F-FDG signal and cardiac function. At 3 d after TAC, a strong correlation was observed with ejection fraction (r = −0.75, P = 0.01), end-systolic volume (r = 0.75, P < 0.01), and end-diastolic volume (r = −0.67, P < 0.05) (Figs. 5A–5C), suggesting that animals with worse cardiac function exhibit ineffective cardiomyocyte suppression reflecting altered metabolic substrate utilization. This relationship was less prominent for left ventricular mass and stroke volume at 3 d (Fig. 5D) and for all parameters at 7 d (Supplemental Fig. 4).

Correlation of cardiac geometry and function with 18F-FDG accumulation in left ventricle at 3 d after TAC, displaying inverse correlation with ejection fraction (A) and direct correlation with end-systolic volume (B), end-diastolic volume (C), and left ventricular (LV) mass (D).
DISCUSSION
Myocardial inflammation in cardiovascular disease is a powerful prognosticator of functional decline and an emerging therapeutic target. Although strongly implicated in acute myocardial infarction and ischemic HF, the contribution of inflammatory leukocytes to nonischemic pressure-overload HF is less well characterized, owing in part to the challenge of interrogating local inflammation noninvasively. Here, we evaluated the temporal relationship between ventricular geometric change, contractile function, and inflammation, and the capability of 18F-FDG to noninvasively image inflammatory cell content. Histologic analysis revealed mild, diffuse inflammatory cell infiltration in the left ventricle early after pressure overload, but although 18F-FDG uptake was increased, the intensity of the signal was disproportionate to the severity of inflammation. Tissue immunoblotting demonstrated a selective upregulation of GLUT4 in the overloaded left ventricle. The strain incurred by the myocardium during pressure overload renders K/X suppression of cardiomyocyte glucose transport ineffective, such that the 18F-FDG signal early after TAC, although correlating with cardiac dysfunction, may partly reflect inflammation but is probably dominated by metabolically compromised cardiomyocytes.
The role and time course of inflammatory cell invasion in myocardial infarction are well characterized, describing dynamic mobilization of leukocytes (23). By comparison, the role of inflammation in nonischemic HF remains equivocal, though flow cytometry has identified inflammatory cell content in late-stage pressure-overload HF (10). Previous studies demonstrated transient proinflammatory cytokine upregulation and macrophage, but not granulocyte, recruitment, reaching a maximum at 7 d after aortic constriction (10,11). Monocyte-derived cardiac macrophages are considered main contributors to adverse remodeling after TAC (14). In the present study, we observed modest, diffuse CD68-positive macrophage infiltration of the left ventricle beginning 3 d after TAC and persisting to 7 d. Notably, MRI revealed elevated ventricular mass, dilatation, and contractile dysfunction in parallel to or preceding inflammatory cell infiltration. It is difficult to distinguish the role of inflammation in ventricular remodeling, considering the need to sample tissues at multiple time points.
Noninvasive interrogation of inflammatory leukocytes is therefore attractive for monitoring disease progression. In several preclinical and clinical studies, 18F-FDG uptake identified inflammation in cardiovascular diseases, such as acute MI (19), preferentially targeting metabolically active proinflammatory macrophages in cardiac tissue (15,24). However, the robust uptake of 18F-FDG by cardiomyocytes limits the ability to specifically target inflammation in the heart. Previous studies established significantly higher 18F-FDG uptake by the left ventricle within 24 h of aortic constriction, and this uptake was effectively inhibited by β-blocker therapy (18). Elevated glucose utilization is associated with increased glucose-6 phosphate expression (17), which would be consistent with the inability to suppress cardiomyocyte 18F-FDG uptake in the present study. Moreover, the increased rate of glucose transport gradually increases over weeks of pressure overload (17), as may reflect the higher success of K/X cardiomyocyte suppression at the earlier time point (day 3) of the current study. Accordingly, despite comparably elevated blood glucose levels after K/X anesthesia, upregulation of GLUT4 in the overloaded left ventricle prevents the normal suppression pattern. Notably, the chronic stages of pressure overload lead to regional hypometabolism, wherein the extent of myocardial metabolic defects correlates with changes in ventricular volume (25). As such, whereas diffuse inflammatory cells invade the myocardium in pressure overload, enhanced cardiomyocyte glucose utilization obscures the delineation of inflammation using 18F-FDG. Of note, contractile function declined with rising 18F-FDG uptake, consistent with altered metabolic substrate utilization in progressive remodeling. The interplay between inflammation and fibrosis believed to contribute to the progression of pressure-overload HF (26,27) could be more effectively evaluated by specific inflammation agents (28).
Some limitations to the present study should be considered. First, because our aim was to characterize the acute development of inflammation in pressure overload, we did not assess the long-term functional outcome. We were interested primarily in the acute stage of disease, when inflammation is thought to predicate remodeling and fibrosis before terminal HF. The rapid change in ventricular geometry and decline in contractile function in parallel with inflammation and metabolic perturbation suggests we have captured the active stage of disease. Second, because we used separate camera systems, we could not assess cardiac function and 18F-FDG distribution on the same day; such being the case, correlations might be imprecise. Despite a gradual increase in ventricular volumes and mass, ejection fraction remained relatively consistent from 2 to 8 d, supporting the correlation between PET and MRI measurements. Finally, we evaluated only K/X anesthesia as a means of suppressing cardiomyocyte 18F-FDG uptake. Earlier studies indicated that continuous administration of propranolol prevents glucose hypermetabolism (18). Because the sympathetic nervous system is also known to modulate ventricular remodeling and inflammatory leukocyte mobilization (29), and the temporal pattern of leukocyte infiltration is poorly characterized, we focused specifically on K/X suppression.
CONCLUSION
TAC induces rapid changes in left ventricular geometry and contractile function with a parallel modest increase in acute inflammatory cell infiltration. 18F-FDG imaging cannot effectively resolve inflammatory cells in the presence of extensive cardiomyocyte metabolic remodeling, as in pressure-overload HF. A more selective molecular tracer targeting inflammation may provide a clearer illustration of the local inflammatory response and its contribution to pathogenesis.
DISCLOSURE
This study was partly supported by the German Research Foundation (DFG, Clinical Research Group KFO311, Excellence Cluster REBIRTH-2, and research grant TH2161/1-1). No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can 18F-FDG imaging identify inflammatory leukocyte infiltration of the myocardium early in pressure-overload–induced HF?
PERTINENT FINDINGS: Cardiac 18F-FDG uptake is elevated after TAC but is disproportionate to the diffuse infiltration of inflammatory macrophages because of increased glucose metabolism by the overloaded heart.
IMPLICATIONS FOR PATIENT CARE: Assessment of inflammation with 18F-FDG may be imprecise in nonischemic HF, necessitating more specific imaging agents.
Acknowledgments
We thank the Preclinical Molecular Imaging Core Laboratory and the Small Animal MRI Centre for technical assistance with these experiments.
Footnotes
Published online Oct. 25, 2019.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication June 19, 2019.
- Accepted for publication September 25, 2019.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}