


Abstract
Therapies targeting reductive/oxidative (redox) metabolism hold potential in cancers resistant to chemotherapy and radiation. A redox imaging marker would help identify cancers susceptible to redox-directed therapies. Copper(II)-diacetyl-bis(4-methylthiosemicarbazonato) (Cu-ATSM) is a PET tracer developed for hypoxia imaging that could potentially be used for this purpose. We aimed to demonstrate that Cu-ATSM signal is dependent on cellular redox state, irrespective of hypoxia. Methods: We investigated the relationship between 64Cu-ATSM signal and redox state in human cervical and colon cancer cells. We altered redox state using drug strategies and single-gene mutations in isocitrate dehydrogenases (IDH1/2). Concentrations of reducing molecules were determined by spectrophotometry and liquid chromatography–mass spectrometry and compared with 64Cu-ATSM signal in vitro. Mouse models of cervical cancer were used to evaluate the relationship between 64Cu-ATSM signal and levels of reducing molecules in vivo, as well as to evaluate the change in 64Cu-ATSM signal after redox-active drug treatment. Results: A correlation exists between baseline 64Cu-ATSM signal and cellular concentration of glutathione, nicotinamide adenine dinucleotide phosphate (NADPH), and nicotinamide adenine dinucleotide (NADH). Altering NADH and NADPH metabolism using drug strategies and IDH1 mutations resulted in significant changes in 64Cu-ATSM signal under normoxic conditions. Hypoxia likewise changed 64Cu-ATSM signal, but treatment of hypoxic cells with redox-active drugs resulted in a more dramatic change than hypoxia alone. A significant difference in NADPH was seen between cervical tumor orthotopic implants in vivo, without a corresponding difference in 64Cu-ATSM signal. After treatment with β-lapachone, there was a change in 64Cu-ATSM signal in xenograft tumors smaller than 50 mg but not in larger tumors. Conclusion: 64Cu-ATSM signal reflects redox state, and altering redox state impacts 64Cu-ATSM metabolism. Our animal data suggest there are other modulating factors in vivo. These findings have implications for the use of 64Cu-ATSM as a predictive marker for redox therapies, though further in vivo work is needed.
Therapies targeting reductive/oxidative (redox) metabolism hold promise in cancers resistant to conventional therapies. Many cancer cells upregulate antioxidant systems, specifically the glutathione and thioredoxin pathways, to manage excess reactive oxygen species produced by altered metabolism (1). Mutations in enzymes that impact redox metabolism, such as isocitrate dehydrogenases (IDH1/2), are also seen in many cancers (2,3). Targeting redox metabolism has been shown to be toxic to several different cancers (4–8). In addition, quinolone drugs metabolized by the enzyme NQO1, such as β-lapachone, that increase reactive oxygen species and consume reducing molecules such as nicotinamide adenine dinucleotide phosphate (NADPH) and nicotinamide adenine dinucleotide (NADH) have shown promise and are being evaluated in humans (9–11).
Biomarkers to identify cancers most susceptible to such a strategy are lacking. Metabolic imaging tracers whose mechanism is dependent on redox metabolism are excellent candidates. Copper(II)-diacetyl-bis(4-methylthiosemicarbazonato) (Cu-ATSM) is a PET tracer developed for hypoxia imaging that could serve this purpose. There are data that suggest retention of Cu-ATSM is dependent on the redox state of the cell (12–15). Its relationship to hypoxia may therefore be secondary. For example, Cu-ATSM uptake does not always correlate with traditional markers of hypoxia, such as pimonidazole and carbonic anhydrase 9 staining (16–18). Indeed, the formative works with Cu-ATSM demonstrated the importance of cellular reducing potential, reducing species such as NADH and acid/base balance in the uptake of Cu-ATSM (14,19,20)
We hypothesize that Cu-ATSM retention is primarily dependent on a cell’s ability to reduce Cu-ATSM and that Cu-ATSM therefore holds potential as a predictive imaging marker for therapies directed at redox metabolism. We specifically focus on cervical cancer, a disease in which the standard-of-care treatment for locally advanced disease (chemoradiation therapy) fails in nearly 40% of patients (21). Redox-directed therapies are one potential novel treatment strategy, and we have demonstrated this approach with preclinical data (22). Cervical cancer is also a disease in which Cu-ATSM has been investigated as a prognostic imaging marker in patients (23). The use of Cu-ATSM as a predictive marker for redox therapies therefore holds immediate translational potential.
In this work, we investigate how altering redox state impacts Cu-ATSM signal using drug strategies targeting glutathione, NADH, and NADPH metabolism. We also investigate genetic strategies to alter redox metabolism, as well as the effect of the combination of hypoxia and metabolically directed therapies. Finally, we explore the relationship between redox state and Cu-ATSM signal in clinically relevant in vivo models of cervical cancer.
MATERIALS AND METHODS
Cell Culture, Reagents, and Drug Treatment
Four human cervical cancer cell lines obtained through the ATCC were evaluated: SiHa, ME180, CaSki, and C33A. We also studied HCT116 human colon cancer cells without and with IDH1 (R132H) and IDH2 (R172K) mutations (Horizon). With the exception of the hypoxia experiments, all experiments were performed under standard tissue culture conditions at 37°C and 5% CO2, with cells maintained in Iscove modified Dulbecco medium with 10% fetal bovine serum and a 0.1 mg/mL concentration of gentamycin. HCT116 cells were maintained in McCoy 5A medium with 10% fetal bovine serum and no antibiotic.
Drugs used to alter redox state in vitro included buthionine sulfoximine (BSO; Sigma), β-lapachone (Sigma), rotenone (Sigma), and 6-aminonicotinamide (6-AN; Santa Cruz Biotechnology). All drugs were dissolved in dimethyl sulfoxide diluted to 0.1% in medium, with the exception of 6-AN, which was dissolved in 1.67% dimethyl sulfoxide.
Drugs were diluted in the appropriate medium to their desired concentration. Prior medium was suctioned off cells and replaced with the medium containing drugs or control medium.
Hypoxia Experiments
Hypoxia experiments were performed in a benchtop hypoxia incubator (Coy Laboratory Products) kept at 37°C, with 5% CO2 and 1% O2. Cells were kept in the hypoxia chamber for 24 h before experiments. Media and buffer solutions were equilibrated in the hypoxia chamber before use.
64Cu-ATSM Radioactivity Assays
64Cu-ATSM was synthesized in-house as described in the supplemental methods (supplemental materials are available at http://jnm.snmjournals.org). Cells were plated in 12-well plates such that they were 70%–90% confluent at the time of tracer labeling. Cells were labeled with 0.74 MBq of 64Cu-ATSM per well and incubated with radiotracer for 1 h at 37°C under room oxygen with 5% CO2. Cells were then washed with cold phosphate-buffered saline (4°C) 3 times and lysed with 1% sodium dodecyl sulfate in 10 mM sodium tetraborate decahydrate (Sigma). Cell lysate was counted in a γ-counter. Duplicate 12-well plates were cultured in parallel and subjected to the same conditions. Cells from these plates were counted on a Vi-cell automated cell counter (Beckman Coulter). Measured radioactivity was normalized to number of viable cells for each condition.
For hypoxia experiments, cells were plated in 100-mm-diameter dishes to achieve approximately 70% confluence the following day. Cells to be treated with hypoxia were placed in the hypoxic chamber for 24 h. In the hypoxic chamber, cells were then scraped from the plates, placed into Falcon tubes, spun down, resuspended, and transferred into sealed silica-coated blood collection vials. Experimental drugs were added to these vials through a sealed cap using insulin syringes with a 27-gauge needle, and vials were gently stirred in a vortex mixer and incubated at 37°C with constant agitation on a rocking platform. 64Cu-ATSM was likewise added through the sealed cap and cells were stirred in a vortex mixer and incubated at 37°C for 1 h on the rocking platform. Cells and medium were then transferred to Eppendorf tubes and spun down at 1,500 rpm, medium was suctioned off, and cells were washed with cold phosphate-buffered saline 3 times, repeating the process of spinning down and suctioning off the rinsing solution. Cells were then counted in the γ-counter. Normoxic cells used in these experiments were treated in the same way but under room air. Cells plated and treated in parallel were used to normalize radioactivity to number of viable cells.
64Cu-ATSM Uptake In Vivo
All in vivo studies were conducted according to protocols approved by the Washington University Division of Comparative Medicine and the Institutional Animal Care and Use Committee. The experimental methods for the in vivo experiments are described in detail in the supplemental methods. Briefly, for orthotopic tumors, luciferase-expressing SiHa and ME180 cells were orthotopically injected into the cervix of 6- to 8-wk-old female nude mice (n = 4 for SiHa, n = 5 for ME180). Bioluminescent imaging was performed 1 and 2 wk after injection on a PerkinElmer IVIS50 to confirm implantation. For xenograft tumors, SiHa cells were injected subcutaneously into the flank of 6- to 8-wk-old female nude mice using a serum-free medium/Matrigel (Corning) mixture.
The animals with orthotopic tumors were imaged with MRI and PET/CT. MRI experiments were performed 3, 4, and 5 wk after implantation, and 64Cu-ATSM PET/CT data were collected 4 wk after implantation for 4 mice with ME180 tumors and 3 mice with SiHa tumors. One mouse with a SiHa tumor died immediately before PET/CT imaging; its tumor was immediately dissected and flash-frozen for mass spectrometry. One ME180 tumor was not imaged, but the tumor was dissected and flash-frozen for mass spectrometry. MRI (T1-, T2-, and diffusion-weighted sequences) was performed on an Agilent/Varian DirectDrive 4.7-T small-animal MRI scanner, with a 2.5-cm quadrature birdcage radiofrequency coil. All CT scans were performed on an Inveon small-animal CT scanner (Siemens Healthcare), and PET scanning was performed on either an Inveon small-animal PET scanner (Siemens Healthcare) or on a Focus 220 (Siemens Healthcare). PET imaging was performed from 0 to 60 min after injection of 64Cu-ATSM (3.92 ± 0.11 MBq). After the final MRI, tumors were dissected and flash-frozen.
Image analysis was performed using the Inveon Research Workplace software (Siemens Healthcare). Tumor volumes of interest were drawn on the PET images using the combination of the MRI and fused PET/CT images (supplemental methods). The SUVmean was measured 40–60 min after injection.
Animals with xenograft tumors were used to evaluate biodistribution of 64Cu-ATSM after β-lapachone treatment. This was done on 2 cohorts of mice (n = 10 and n = 12), with animals in each cohort split equally between treatment and control groups. β-lapachone was administered via intraperitoneal injection (30 mg/kg in 0.1% dimethyl sulfoxide), given on 5 consecutive days before and including the day of the biodistribution study. For biodistribution, animals were anesthetized using isoflurane and room air, injected with approximately 0.74 MBq (20 μCi) of 64Cu-ATSM, and sacrificed after a 90-min uptake period. Tumors were washed in 4°C phosphate-buffered saline and flash-frozen, and radioactivity was counted in a Beckman 8000 γ-counter.
Measurement of Redox Metabolites by Spectrophotometry and Liquid Chromatography–Mass Spectrometry–Based Metabolomics
Intracellular levels of reduced and oxidized glutathione (GSH and GSSG) were determined spectrophotometrically using an NADPH recycling assay (8). Briefly, cells were grown in 100-mm dishes, treated with or without drugs, harvested by scraping into 150 μL of 5% 5-sulfosalicylic acid, and then measured with the spectrophotometric assay.
Intracellular levels of NADH and NAD+ (the oxidized form of NADH), and NADPH and NADP+ (the oxidized form of NADPH) were determined using mass spectrometry for both cell samples and animal tumors. These methods are further outlined in the supplemental methods. Briefly, after being washed, scraped (for cells grown in culture), or pulverized (for tumors), cells were extracted as previously described using a 2:2:1 acetonitrile:methanol:water solution with 0.1 formic acid and 2 M ammonium bicarbonate (24). Samples then underwent multiple freeze–thaw cycles, were stirred in a vortex mixer, were sonicated, and finally were centrifuged at 15,000g for 30 min at 4°C. Supernatant (50 μL) was transferred to liquid chromatography–mass spectrometry vials for analysis on an Agilent 6460 triple-quadruple mass spectrometer. Quantitation was accomplished using a standard curve and commercial standards obtained from Sigma. Samples were normalized by extraction volume and tumor weight or by cell number as appropriate.
Statistics
All in vitro experiments were performed in triplicate. All data points displayed represent the mean, and error bars represent SD. Propagation of error was used for data points with multiple sources of error. Correlations displayed represent the Pearson correlation coefficient. Groups were compared using the 2-tailed Student t test assuming normally distributed data with unequal variances. Groups from the in vivo studies were compared with the Mann–Whitney test.
RESULTS
Relationship of Baseline 64Cu-ATSM Uptake to Cellular Reducing Species
There was a significant difference in baseline 64Cu-ATSM signal between the cervical cancer cell lines studied in vitro under standard tissue culture conditions (Supplemental Fig. 1, P < 0.001 by ANOVA). With the hypothesis that 64Cu-ATSM signal reflects the reducing potential of cells, we compared baseline 64Cu-ATSM signal to the concentrations of major reducing species, namely NADH (Fig. 1A), NADPH (Fig. 1B), and GSH (Fig. 1C), and found a positive correlation in each case.
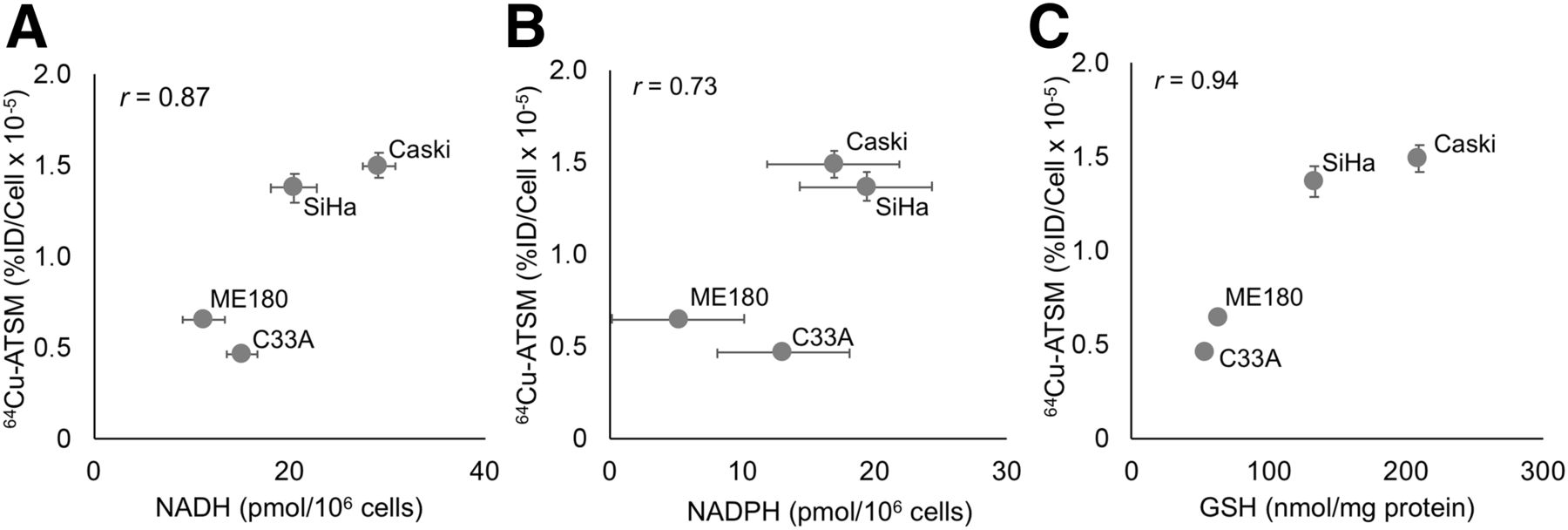
Relationship of baseline 64Cu-ATSM signal to intracellular reducing species. Baseline uptake of 64Cu-ATSM correlates with baseline cellular levels of NADH (A), NADPH (B), and GSH (C). %ID = percentage injected dose.
Impact of Redox-Altering Drugs on 64Cu-ATSM Signal
We then altered metabolism of these molecules using drug strategies and evaluated the impact on 64Cu-ATSM signal. To interfere with NADH-dependent metabolic pathways, cells were treated with rotenone (50 μM for 2 h), a mitochondrial complex I inhibitor that increases NADH, or β-lapachone (10 μM for 15 min), a quinolone that is metabolized by the enzyme NQO1, consuming NADH and NADPH in the process. We tested these drugs on SiHa and ME180 cells, as these cell lines showed significant differences in baseline levels of redox-active molecules and 64Cu-ATSM signal (Fig. 1). They have also shown different sensitivities to redox-directed therapies in our prior work (22). We then evaluated how these drugs change levels of NADH, NADPH, and glutathione in SiHa cells; SiHa cells were selected as they showed the greatest change in 64Cu-ATSM signal with drug treatment, and they are particularly sensitive to redox therapies (22). They therefore represent a relevant model for evaluating Cu-ATSM as a redox marker. In both SiHa and ME180 cells, rotenone significantly increased 64Cu-ATSM signal, and β-lapachone significantly decreased 64Cu-ATSM signal (Fig. 2A). In SiHa cells, treatment with rotenone increased NADH concentration, and treatment with β-lapachone decreased NADH and NAD+ concentrations (Fig. 2B).
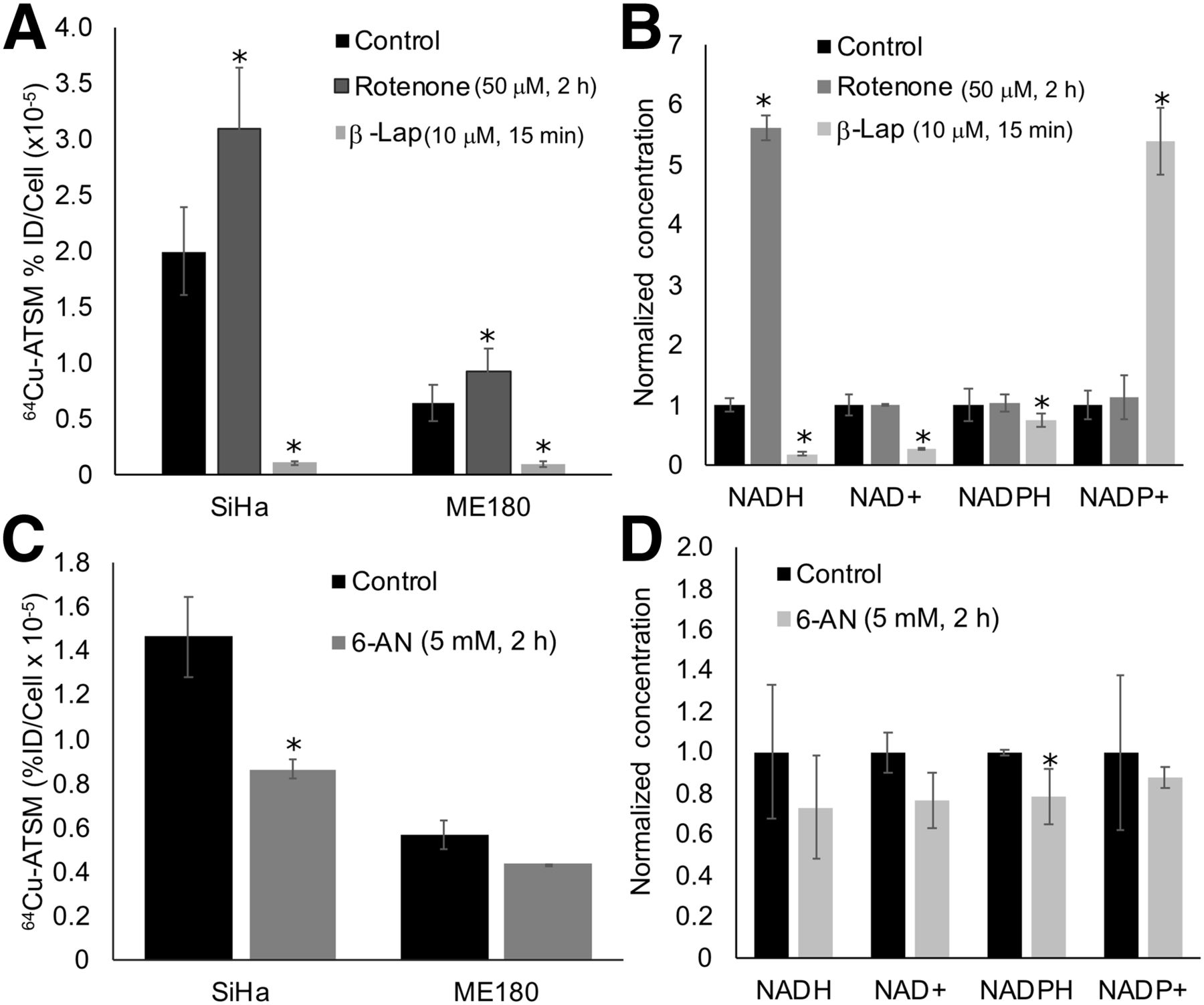
Impact of altering redox metabolism on 64Cu-ATSM signal. (A and B) Altering NADH metabolism with rotenone and β-lapachone significantly changed 64Cu-ATSM signal (A) and likewise resulted in significant changes in NADH, NAD+, and NADPH in SiHa cells (B). (C and D) Altering NADPH metabolism with 6-AN significantly changed 64Cu-ATSM signal (C) and significantly decreased NADPH concentration in SiHa cells (D). *P < 0.05, compared with control. %ID = percentage injected dose; β-lap = β-lapachone.
The decrease in 64Cu-ATSM signal seen with β-lapachone could also be due to interference with NADPH metabolism (Fig. 2B). To better isolate the impact of NADPH metabolism on 64Cu-ATSM signal, cells were treated with 6-AN (5 mM for 2–3 h), an inhibitor of glucose-6-phosphate dehydrogenase that prevents the production of NADPH. SiHa cells treated with 6-AN showed a significant decrease in 64Cu-ATSM signal, and ME180 cells showed a nonsignificant decrease in signal (Fig. 2C). Treatment with 6-AN resulted in a corresponding decrease in NADPH in SiHa cells (Fig. 2D).
To alter the glutathione pathway, cells were treated with BSO (1 mM for 20–24 h), an inhibitor of γ-glutamylcysteine synthetase that blocks glutathione production. Treatment with BSO resulted in a significant decrease in 64Cu-ATSM binding only in SiHa cells. However, BSO significantly decreased GSH and increased the percentage of GSSG across all cell lines (Supplemental Fig. 2).
Impact of Altering Redox Metabolism with IDH Mutants on 64Cu-ATSM Signal
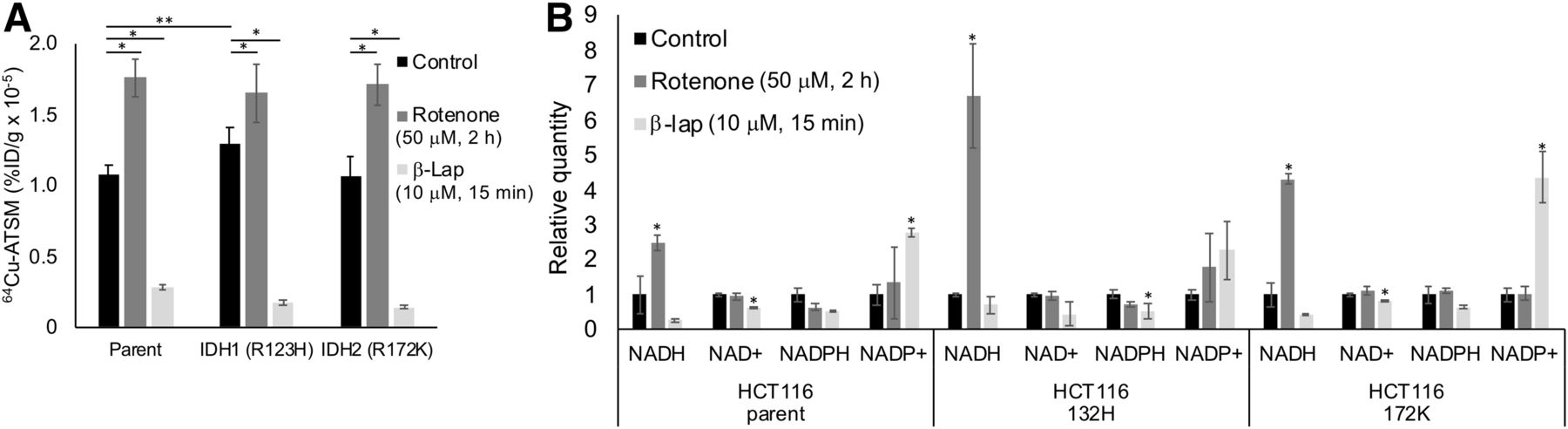
We also investigated the impact of single-gene mutations in redox-active enzymes (IDH1/2) on 64Cu-ATSM signal. In addition, we treated IDH-mutated HCT116 cells with rotenone and β-lapachone to determine the relative impact of single-gene mutations versus drugs on 64Cu-ATSM signal. Untreated cells with an IDH1 mutation showed a significant increase in 64Cu-ATSM signal compared with the parent (Fig. 3A). This increase corresponds to the previously published decrease in NADPH in HCT116 IDH1-mutant cells (25). Both parent and mutant HCT116 cells showed a significant increase in 64Cu-ATSM with rotenone and a decrease with β-lapachone (Fig. 3A). Notably, the effect of the drugs on 64Cu-ATSM signal was greater than the effect of the IDH1 mutation. These changes corresponded to changes in NADH/NAD+ and NADPH/NADP+ similar to those seen in the cervical cancer cells (Fig. 3B). HCT116 parent and mutant cell lines also showed a decrease in GSH levels after treatment with BSO (Supplemental Fig. 3).

Impact of IDH mutations on 64Cu-ATSM signal. (A) In untreated HCT116 cells, IDH1 mutation significantly changed 64Cu-ATSM signal (**P < 0.05, compared with parent cell line). Parent and mutant cell lines showed increase in 64Cu-ATSM signal in response to rotenone and decrease in signal in response to β-lapachone (*P < 0.05, compared with untreated cells). (B) Treatment with rotenone and β-lapachone altered NADH/NAD+ and NADPH/NADP+ levels similarly to what was seen in cervical cancer cells in parent and IDH mutant HCT116 cells (*P < 0.05, compared with untreated controls). %ID = percentage injected dose; β-lap = β-lapachone.
Impact of Hypoxia on 64Cu-ATSM Signal
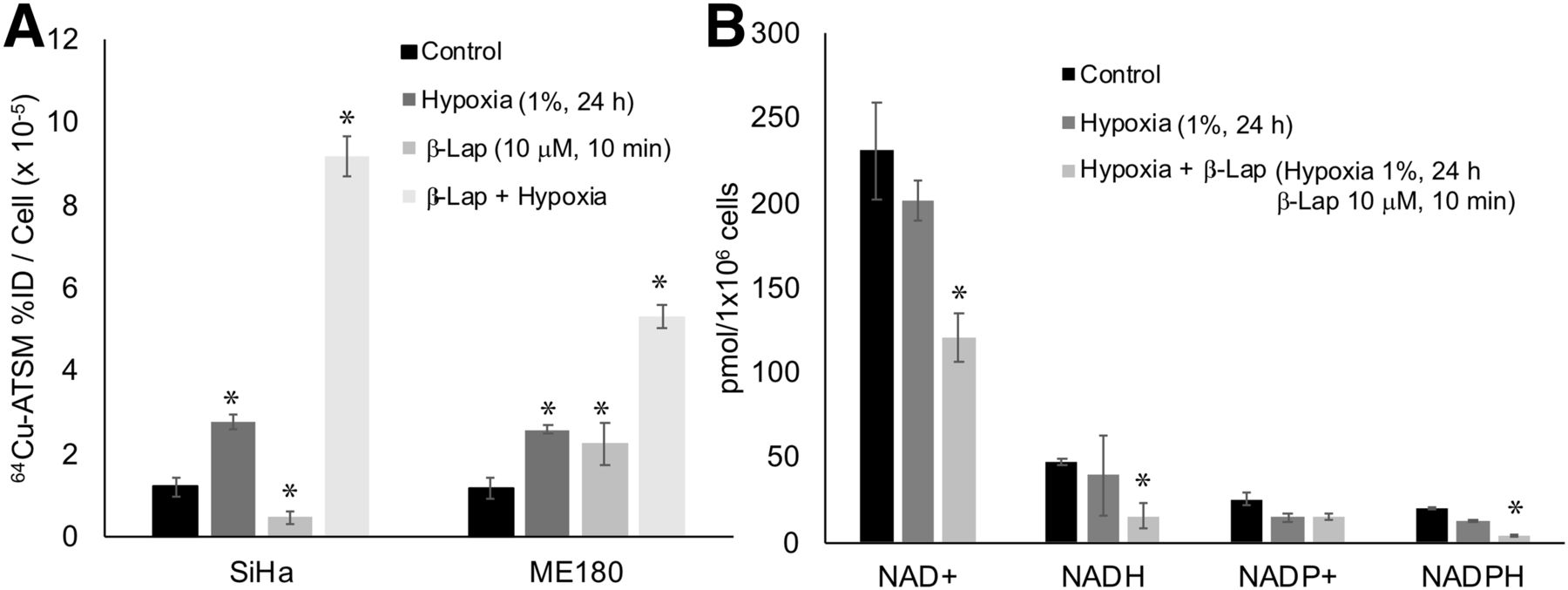
We also investigated the impact of hypoxia on 64Cu-ATSM signal in SiHa and ME180 cells treated with β-lapachone. Hypoxia significantly increased 64Cu-ATSM signal in both cell lines. In these experiments, treatment with β-lapachone significantly decreased 64Cu-ATSM signal in SiHa, but not ME180, cells. Treatment with β-lapachone in hypoxic cells dramatically increased 64Cu-ATSM signal in both cell lines (Fig. 4A). Although the change in 64Cu-ATSM signal seen when β-lapachone was combined with hypoxia was opposite that seen under normoxic conditions, the impact of β-lapachone on NADH and NADPH remained the same; both decreased in hypoxic cells treated with β-lapachone (Fig. 4B).
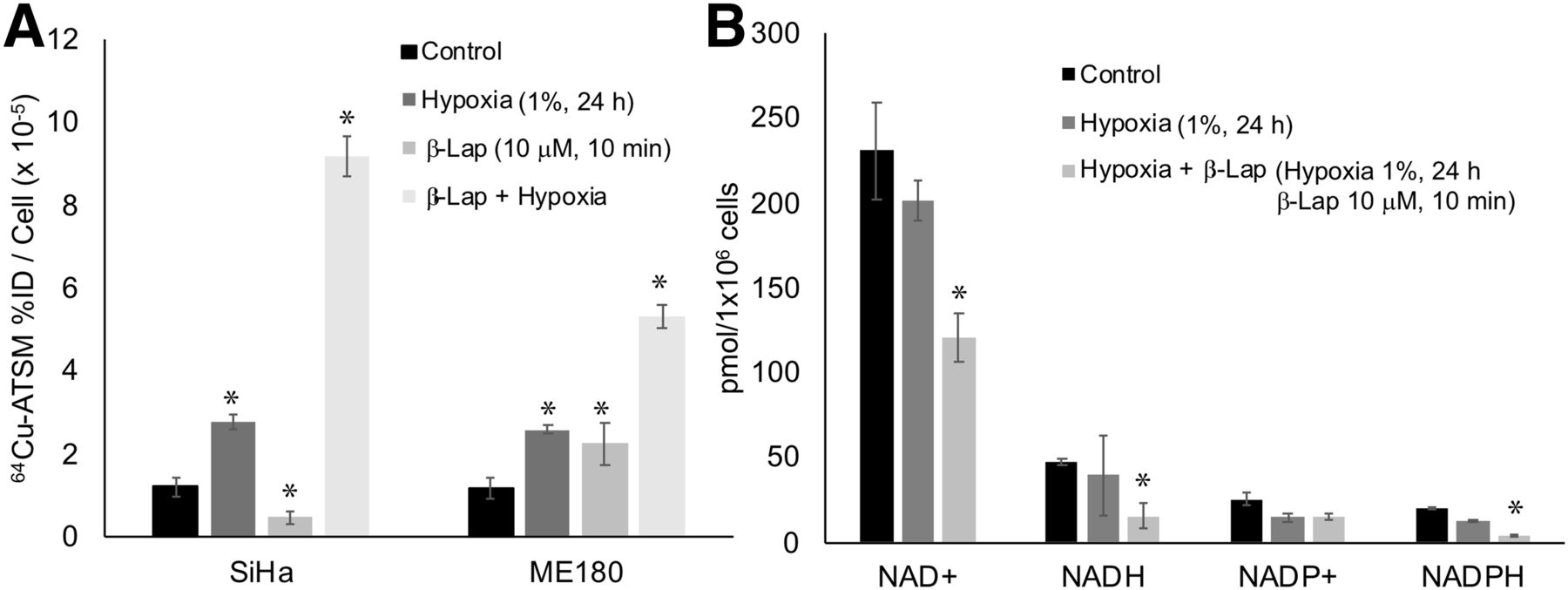
Impact of hypoxia on effects of redox drugs on 64Cu-ATSM signal. (A) Hypoxia increased 64Cu-ATSM signal in both SiHa and ME180 cells. Combination of β-lapachone and hypoxia further increased 64Cu-ATSM signal. (B) Treatment with hypoxia+β-lapachone significantly changed NAD+, NADH, and NADPH. Treatment with hypoxia alone did not significantly change these molecules. %ID = percentage injected dose; β-lap = β-lapachone.
64Cu-ATSM Signal In Vivo
Baseline 64Cu-ATSM signal was studied in vivo in untreated SiHa (n = 3) and ME180 (n = 4) orthotopic tumors to determine whether the differences between cell lines in vitro are likewise seen in vivo (Figs. 5A). No significant difference was seen between SiHa and ME180 tumors in the SUVmean averaged over 40–60 min after injection (Fig. 5B). We also measured NADH/NAD+ and NADPH/NADP+ in all tumors. ME180 tumors showed significantly less NADPH than SiHa tumors, similar to what was seen in vitro. There was no significant difference in NADH, NAD+, or NADP+ (Fig. 5C).

Relationship between 64Cu-ATSM and redox state in vivo. (A) We investigated 64Cu-ATSM signal in orthotopic mouse model of cervical cancer using SiHa (n = 3) and ME180 (n = 4) cells (tumors circled in red). (B) There was no significant difference in average SUVmean between the 2 tumor types. (C) There was significant difference in NADPH concentration between SiHa (n = 4) and ME180 (n = 5) tumors.
64Cu-ATSM signal after treatment with β-lapachone was evaluated in vivo in a biodistribution study using xenograft SiHa tumors. Across 2 cohorts of animals, a significant increase in 64Cu-ATSM signal was seen between controls and animals treated with β-lapachone in tumors weighing less than 50 mg (Table 1). This is similar to the change in 64Cu-ATSM signal seen in vitro with the combination of β-lapachone and hypoxia. This difference was not seen in larger tumors (n = 11).
Biodistribution of 64Cu-ATSM in Xenograft SiHa Tumors with Mass < 50 mg Treated with β-Lapachone vs. Controls
DISCUSSION
We have demonstrated that 64Cu-ATSM signal can be altered in human cancer cells by changes in redox metabolism—particularly by changes in metabolic pathways that utilize NADH/NAD+ and NADPH/NADP+—using both drug strategies and single gene mutations (Figs. 2 and 3). In this work, the drug strategies studied had a greater impact than the gene mutations, suggesting that the drug strategies had a greater impact on the overall redox state of the cells. We have further shown that redox metabolism impacts 64Cu-ATSM signal in the absence of hypoxia, though hypoxia has an important modulating effect (Fig. 4).
Our findings are supported by prior work investigating the mechanism of Cu-ATSM (12–15). Some of the initial work with Cu-ATSM demonstrated that reduction of Cu(II)-ATSM depended on NADH (19). Other in vitro data have supported the importance of NADH- and NADPH-dependent enzymes in the reduction of Cu(II)-ATSM (12,26,27). There are likewise basic chemistry data supporting the importance of reducing molecules in the reduction of copper(II)-bis(thiosemicarbazonato) complexes but demonstrating that some reducing molecules, including GSH, do not have sufficient reduction potential to reduce Cu(II)ATSM (28). These data support our finding that altering NADH and NADPH metabolism significantly impacts Cu-ATSM signal but that altering glutathione does not (Fig. 2; Supplemental Fig. 2).
Although our data show NADH and NADPH metabolism to be important in Cu-ATSM binding, binding is not directly dependent on their concentration. For example, although mutations in IDH1 alter 64Cu-ATSM signal and NADH/NADPH concentrations in vitro, there is no concordance between them (Fig. 3 (25)). In addition, although β-lapachone decreases NADH concentration under both normoxic and hypoxic conditions, its effect on 64Cu-ATSM signal under hypoxia is opposite that under normoxia (Fig. 4). The in vivo data show that although there is a difference in NADPH concentration between SiHa and ME180 tumors, there is no difference in 64Cu-ATSM signal. The in vivo data also show that although a change in 64Cu-ATSM signal was seen after β-lapachone treatment in smaller xenograft tumors, such a change was not seen in larger tumors. Other factors are therefore important to 64Cu-ATSM binding in vivo, some of which have previously been elucidated (e.g., hypoxia and pH (12,16,18,20)). Thus, 64Cu-ATSM signal more likely reflects overall reducing potential, or the flux through redox pathways using NADH/NAD+ or NADPH/NADP+, rather than the concentration of a specific reducing molecule.
A PET tracer that reflects redox metabolism would be valuable. NADH metabolism has been shown to be important in cancer progression and metastatic potential (29). NADPH metabolism is also critical in cancer metabolism and provides reducing equivalents that can be used in reactive-oxygen-species–scavenging pathways to help manage excessive oxidative stress (7). Our work suggests that Cu-ATSM could serve as an imaging marker for drugs targeting these pathways. For example, β-lapachone is a potent redox-active drug that is toxic to cancer cells that overexpress NQO1. Our data demonstrate changes in Cu-ATSM signal after treatment with β-lapachone, and monitoring changes in Cu-ATSM signal from before therapy to after therapy represents a potential means of identifying cancers that will benefit from β-lapachone.
This study has some important limitations. Although we have demonstrated the importance of redox balance in 64Cu-ATSM metabolism, further study of the specific redox chemistry in cells that contribute to Cu-ATSM binding, fully accounting for pH, hypoxia, and other known factors that impact Cu-ATSM chemistry, would be valuable. Our animal data also support the complexity of Cu-ATSM binding in vivo, which we could not fully elucidate here. For example, although differences in redox molecules exist between cell lines at baseline, 64Cu-ATSM signal is the same. In addition, whereas a change in 64Cu-ATSM signal after β-lapachone treatment was seen in small xenograft tumors, such a change was not seen in larger tumors. Further in vivo studies are needed to fully define the physiologic factors that influence Cu-ATSM binding in vivo and their interplay with the redox pathways we focused on here. In addition, further study is needed to validate that changes in 64Cu-ATSM signal after redox-active drug treatment are predictive of response to the therapy in vivo.
CONCLUSION
In this work, we have demonstrated that Cu-ATSM signal is dependent on the redox state of a cell in the absence of hypoxia, using drug strategies and single-gene mutations in IDH1/2 to alter redox state. The baseline level of 64Cu-ATSM signal, or the change in signal after treatment with redox-active drugs, may have value as a predictive marker for response to these redox-directed therapies. Further study is needed to validate 64Cu-ATSM as a predictive marker for redox-directed therapies in vivo.
DISCLOSURE
This work was supported by NIH R01CA181745 to Julie K. Schwarz and by ASTRO Resident Research Seed Grant 531448 and an RSNA Resident Research Grant to John M. Floberg. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Is 64Cu-ATSM dependent on intracellular redox state, irrespective of hypoxia?
PERTINENT FINDINGS: In human cervical cancer and colon cancer cell lines, 64Cu-ATSM signal correlated with intracellular levels of key reducing molecules, including NADPH and NADH. Altering redox state with drug strategies or single germline mutations alters 64Cu-ATSM signal under normoxic and hypoxic conditions.
IMPLICATIONS FOR PATIENT CARE: Intracellular redox state must be considered when using 64Cu-ATSM. 64Cu-ATSM could also be used as an imaging marker for redox-active drugs.
Acknowledgments
We thank Doug Spitz and Collin Heer of the University of Iowa for helpful discussions. We thank Nikki Fettig, Amanda Klaas, Margaret Morris, and John Englebach in Mallinckrodt Institute of Radiology’s Small Animal PET and MR Imaging Facilities for assisting with the mouse imaging experiments. We thank Susan Gelman of the Patti Lab for providing the HCT116 cells.
Footnotes
Published online Oct. 4, 2019.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication May 10, 2019.
- Accepted for publication September 4, 2019.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.