


Abstract
The purpose of this study was to evaluate the ability of 21-18F-fluoro-16α,17α-[(R)-(1′-α-furylmethylidene)dioxy]-19-norpregn-4-ene-3,20-dione (18F-FFNP) to measure alterations in progesterone receptor (PR) protein level and isoform expression in response to 17β-estradiol (E2) challenge. Methods: T47D human breast cancer cells and female mice bearing T47D tumor xenografts were treated with E2 to increase PR expression. 18F-FFNP uptake was measured using cell uptake and tissue biodistribution assays. MDA-MB-231 breast cancer clonal cell lines were generated that express the A or B isoform of human PR. PR protein levels, transcriptional function, and subcellular localization were determined. In vitro 18F-FFNP binding was measured via saturation and competitive binding curves. In vivo 18F-FFNP uptake was measured using tumor xenografts and PET. Statistical significance was determined using ANOVA and t tests. Results: After 48 and 72 h of E2, 18F-FFNP uptake in T47D cells was maximally increased compared with both vehicle and 24 h of E2 treatment (P < 0.0001 vs. ethanol; P = 0.02 and P = 0.0002 vs. 24 h for 48 and 72 h, respectively). T47D tumor xenografts in mice treated with 72 h of E2 had maximal 18F-FFNP uptake compared with ethanol-treated mice (percentage injected dose per gram: 11.3 ± 1.4 vs. 5.2 ± 0.81, P = 0.002). Corresponding tumor-to-muscle uptake ratios were 4.1 ± 0.6, 3.9 ± 0.5, and 2.3 ± 0.4 for 48 h of E2, 72 h of E2, and ethanol-treated mice, respectively. There was no significant preferential 18F-FFNP binding or uptake by PR-A versus PR-B in the PR isoform–specific cell lines and tumor xenografts. Conclusion: 18F-FFNP is capable of measuring estrogen-induced shifts in total PR expression in human breast cancer cells and tumor xenografts, with equivalent isoform binding.
See an invited perspective on this article on page 218.
Estrogen receptor α (ERα)–positive breast cancer comprises the largest class of breast cancer and the largest number of breast cancer deaths each year (1). More than half of recurrences in patients with ERα-positive breast cancer occur 6 or more years after initial diagnosis and completion of adjuvant treatment (2,3). Impactful information on endocrine sensitivity may be revealed by monitoring changes in estrogen-regulated target genes as a surrogate measure of upstream ERα inhibition.
A classic ERα target is progesterone receptor (PR), which is upregulated in the presence of estrogen (4,5). Through separate promoters, the PGR gene encodes 2 functionally distinct ligand-activated transcription factors, PR-A and PR-B (6–8). In normal breast tissue, PR-A and PR-B are present in equimolar amounts; however, in breast cancer a higher ratio of PR-A to PR-B has been observed (9,10). Imbalanced PR isoform expression may influence prognosis and benefit from endocrine therapy (11,12).
A molecular imaging agent for quantitative assessment of PR expression in vivo using PET is 21-18F-fluoro-16α,17α-[(R)-(1′-α-furylmethylidene)dioxy]-19-norpregn-4-ene-3,20-dione (18F-FFNP). This radioligand has high relative binding affinity to PR and low nonspecific binding (13,14). The binding specificity of 18F-FFNP for PR has been demonstrated using tissue biodistribution assays with blocking treatments (13,15). One study of 20 patients has been published, to date, demonstrating the safety, dosimetry, and ability of 18F-FFNP PET imaging for identifying PR-positive breast cancer (16).
Endocrine sensitivity can be investigated by measuring physiologic responses to short-course, low-dose estrogen treatment (estradiol challenge) (17,18). This approach is being investigated in a clinical trial testing whether a change in 18F-FFNP uptake after 1 d of administration of low-dose estradiol is predictive of endocrine therapy response in patients with ERα-positive metastatic breast cancer (NCT02455453, Clinical trials.gov). The purpose of this study was to evaluate the ability of 18F-FFNP to measure alterations in PR protein level and isoform expression in response to estradiol challenge in breast cancer cells and tumor xenografts.
MATERIALS AND METHODS
Cell Culture
Experiments were performed under a protocol approved by the Office of Biologic Safety. T47D cells were obtained from the American Type Culture Collection. MDA-MB-231 cells were obtained from the Mallinckrodt Institute of Radiology Pre-Clinical PET/CT Imaging Facility. Cell lines were authenticated using short tandem repeat analysis and tested negative for murine pathogens and Mycoplasma contamination (IDEXX BioResearch). MDA-MB-231 and T47D cells were cultured in Dulbecco modified Eagle medium (Corning) or in Roswell Park Memorial Institute medium (Corning), respectively, with 10% fetal bovine serum (VWR) and 1% penicillin and streptomycin (Gibco) at 37°C and 10% or 5% CO2.
Expression plasmids were created containing either human PR-A or PR-B complementary DNA (6,19). These plasmids were stably transfected into PR-negative MDA-MB-231 cells, resulting in the stable clonal cell lines 231 PR-A and 231 PR-B. Cells were grown with a 200 μg/mL concentration of hygromycin B (Life Technologies) to maintain selection.
Transcriptional Reporter Gene Assay
Cells were grown in steroid hormone–depleted medium (10% charcoal/dextran-stripped fetal bovine serum in phenol red–free medium with 1% penicillin and streptomycin and 2% l-glutamine) for 3 d and then seeded in a 6-well plate (5 × 105 cells per well). The following day, cells were cotransfected with glucocorticoid/progesterone response element luciferase (0.75 μg) and CMV-β-galactosidase (0.25 μg) reporter plasmids using Lipofectamine 3000 (Life Technologies). After 5 h, the medium was changed, and cells were incubated overnight and then treated with ethanol or 100 nM promegestone (R5020; Perkin Elmer) for 24 h. Luciferase activity (Promega) and β-galactosidase activity (Tropix) were assayed following manufacturer protocols.
Immunofluorescence
Cells were grown in chamber slides (Sarstedt) and then fixed with 3.7% formaldehyde, permeabilized with 0.5% Triton X-100 (Sigma), blocked with 10% goat serum for 1 h at 37°C, and incubated with anti-PR antibody (1:100, AB-52; Santa Cruz) overnight at 4°C. Slides were then probed with AlexaFluor 488 antimouse antibody (1:100, Life Technologies) for 1 h at room temperature. The slides were then mounted with ProLong Gold Antifade with 4′,6-diamidino-2-phenylindole (Life Technologies) and imaged using confocal microscopy (Leica SP8 STED).
Cell Proliferation Assay
Cells grown in steroid hormone–depleted medium for 3 d were seeded in 96-well plates (2,500 cells per well) and then treated with 10 nM 17β-estradiol (E2; Sigma) or ethanol for 3, 7, and 12 d. Proliferation was analyzed by measuring DNA content using CyQUANT (Invitrogen) and a DNA standard curve (R2 = 0.96–0.99).
Western Blot
Whole-cell lysates were prepared from cell pellets or pulverized flash-frozen tumors using radioimmunoprecipitation assay buffer containing 2 mM sodium orthovanadate, protease (1:500), and phosphatase (1:100) inhibitor cocktails (Sigma). Protein was quantified with a Bradford assay, and equal amounts were run on 7.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Millipore). Antibodies were used at saturating concentrations for PR (1:1,000, NCL-L-PGR-312; Leica Biosystems), β-actin (1:10,000; Sigma), and horseradish peroxidase-conjugated IgG (1:3,000; GE Healthcare). Blots were incubated with Clarity Western enhanced chemiluminescence substrate (Bio-Rad), and chemiluminescence was imaged using GeneSys GBox:XX6 (Syngene). Bands for PR-B and PR-A protein were quantified using ImageJ software and normalized to the β-actin loading control.
Tumor Xenografts
Experiments were performed under an approved protocol from our Institutional Animal Care and Use Committee. Twelve adult female NOD-scid-γ-mice (Jackson Laboratory) were ovariectomized and supplemented with E2 (10 μg/mL) in the drinking water. After 1 wk, the mice were orthotopically injected bilaterally in the third mammary fat pads with 3.0 × 106 T47D cells at a 1:1 ratio with Matrigel (BD Biosciences). Four days before the tissue biodistribution assay, E2 was removed from the drinking water. Ethanol or E2 (20 μg/mouse) diluted in sunflower seed oil (100 μL/mouse) was administered subcutaneously daily for 48 or 72 h. Fifteen female athymic nude mice (Charles River), aged 8 wk, were orthotopically injected with 3.0 × 106 MDA-MB-231, 231 PR-A, or 231 PR-B cells with Matrigel into the third mammary fat pad. Tumor size was measured by calipers, and volume was calculated by (length × width2)/2.
Histology
Excised tumors were fixed in 10% formalin and then paraffin-embedded. Deparaffinization of slides was performed followed by epitope retrieval in citrate buffer (pH 6.5) for 60 min at 95°C. Immunostaining was performed for PR (1:100 NCL-L-PGR-312, clone 16; Leica) or Ki-67 (1:100, clone SP6; Biocare Medical) using the Mouse-on-Mouse Elite Peroxidase Kit or Vectastain ABC HRP Kit (Vector Laboratories). Staining with hematoxylin and eosin was also performed. Using a whole-slide, bright-field imaging system (Aperio Image Scope software), slides were scanned at ×40 magnification. A clinical pathologist with subspecialty training in breast histopathology visually quantified the percentage of cells with nuclear staining.
18F-FFNP Cell Uptake and Tissue Biodistribution Assays
18F-FFNP was synthesized by the University of Wisconsin–Madison Radiopharmaceutical Production Facility using previously described methods (20). Molar activity at the end of synthesis ranged from 74 to 480 GBq/μmol.
Cell uptake assays were performed using previously described methods (20). Briefly, cells were grown in 24-well plates containing steroid hormone–depleted medium and incubated with increasing doses of R5020 (10−12–10−7 M) and 0.074 MBq (2 μCi) of 18F-FFNP for competitive binding curves or with 2 × 10−8 M R5020 with increasing doses of 18F-FFNP (0.004–0.42 MBq; 0.11–11.4 μCi) for 1 h at 37°C for saturation binding curves.
For biodistribution assays, tissues (blood, muscle, tumors, and uterus) were harvested 2 h after tail vein injection of 10 MBq (270 μCi) of 18F-FFNP in an 80- to 180-μL total volume. Uteri were included as a positive control given their high expression of PR and 18F-FFNP uptake, as shown previously (15). Harvested tissues were wet-weighed and radioactivity measured with a γ-counter. Data were background-corrected and decay-corrected and used to calculate the percentage injected dose per gram of tissue (%ID/g) by comparison with prepared standards representing the same injected dose per mouse.
PET/CT Imaging
Nonfasting mice were injected in the tail vein with 10 MBq (270 μCi) of 18F-FFNP. One hour after injection, the mice were anesthetized with 1.5%–2.0% isoflurane and scanned supine in a small-animal PET/CT scanner (Inveon; Siemens Preclinical Solutions). CT imaging lasted 12 min (2 bed positions × 6 min) and was followed by PET image acquisition (40 million counts per mouse; typically <10 min) using 1 PET bed position. PET data were histogrammed into 1 static frame and reconstructed using ordered-subset expectation maximization of 3 dimensions followed by the maximum a posteriori algorithm (18 iterations, 16 subsets). CT attenuation and scatter correction were applied. PET/CT images were automatically coregistered and analyzed using Inveon Research Workplace, version 3.0 (Siemens Medical Solutions). Regions of interest were drawn around the tumor and triceps muscle to determine mean %ID/g. Tumor-to-muscle uptake was calculated as the ratio of mean %ID/g of tumor to that of muscle.
Statistics
Unpaired t tests were used to compare the means between ethanol- and R5020-treated cells for the PR transcriptional reporter gene assay. One-way ANOVA with Tukey posttesting was used when comparing 3 or more experimental groups (GraphPad Prism, version 6.05). Half-maximal inhibitory concentration (IC50) was determined using nonlinear-regression dose-response inhibition (log[inhibitor] vs. normalized response). Dissociation constant (Kd) was determined using nonlinear-regression binding saturation (total and nonspecific binding). Results are presented as mean ± SE. All tests are 2-sided, with a P value of less than 0.05 considered significant.
RESULTS
18F-FFNP Uptake by T47D Cells After Estradiol Challenge
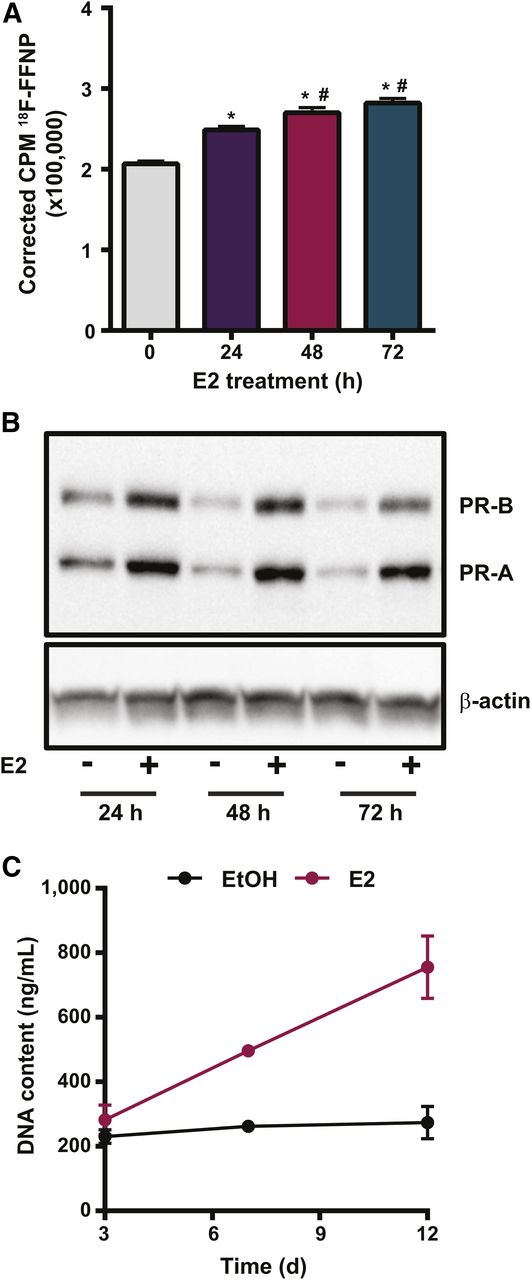
18F-FFNP uptake increased in T47D cells treated with E2 for 24 h (P < 0.0001), 48 h (P < 0.0001), and 72 h (P < 0.0001), compared with ethanol (Fig. 1A). Maximal 18F-FFNP uptake was observed in cells treated with E2 for 48 and 72 h (P = 0.02 and P = 0.0002, respectively), compared with 24 h. Total PR protein increased by 1.9 ± 0.4-, 2.7 ± 0.44-, and 2.3 ± 0.6-fold after 24, 48, and 72 h of E2, respectively, compared with ethanol-treated cells (Fig. 1B). E2 increased both the higher-molecular-weight PR-B isoform (∼120 kDa) and the lower-molecular-weight PR-A isoform (∼94 kDa) (Fig. 1B) and also stimulated cell proliferation beyond 3 d of treatment (Fig. 1C).

18F-FFNP uptake in T47D cells after E2 challenge. (A) Total 18F-FFNP uptake after 1 h in cells treated with 10 nM E2 for 24, 48, or 72 h or ethanol control (0 h). Values represent mean ± SEM background- and decay-corrected counts per minute (CPM) from 3 independent experiments. *P < 0.05 compared with 0 h. #P < 0.05 compared with 24 h E2. (B) Western blot for PR protein after treatment with 10 nM E2 or ethanol for 24, 48, and 72 h. Images are representative of 3 independent experiments. (C) Proliferation assay for cells treated with 10 nM E2 or ethanol for up to 12 d.
18F-FFNP Uptake by T47D Tumor Xenografts After Estradiol Challenge
Because maximal cellular 18F-FFNP uptake occurred at 48 and 72 h, we used these time points for testing whether 18F-FFNP is sensitive for measuring dynamic changes in PR protein expression in a more complex in vivo environment. 18F-FFNP uptake was higher in T47D tumor xenografts than in nonspecific muscle, and blood was higher in all treatment groups (0, 48, and 72 h) compared with nonspecific muscle and with blood (Fig. 2A). Mice treated with 72 h of E2 had maximal tumoral 18F-FFNP uptake, compared with ethanol-treated mice (11.3 ± 1.4 vs. 5.2 ± 0.81 %ID/g, P = 0.002). Corresponding tumor-to-muscle ratios were 4.1 ± 0.6, 3.9 ± 0.5, and 2.3 ± 0.4 for 48 h of E2, 72 h of E2, and ethanol-treated mice, respectively. As expected, strong uterine 18F-FFNP uptake was observed (18.7 ± 2.2 %ID/g) because the uterus is a PR-rich target organ.
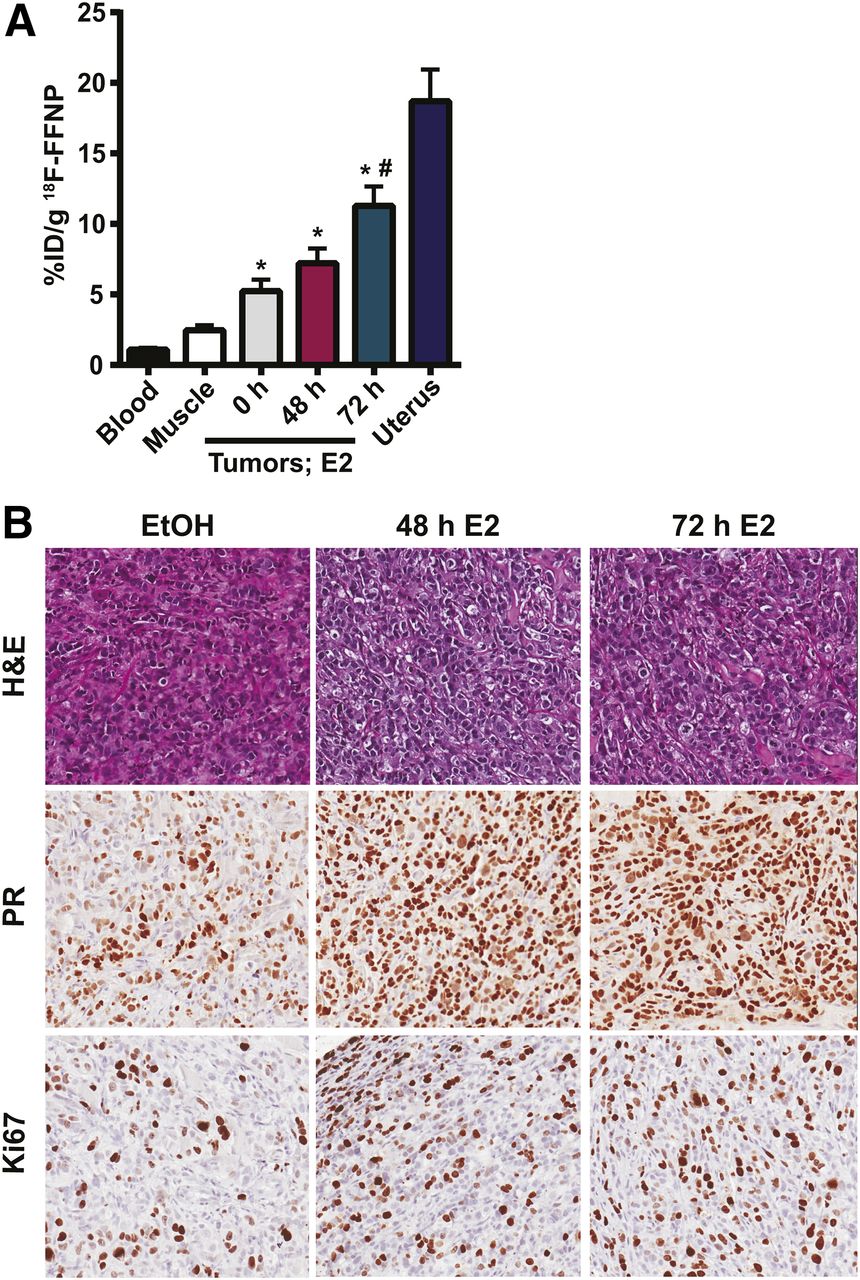
18F-FFNP uptake in T47D tumor xenografts after E2 challenge. Twelve mice with bilateral T47D tumor xenografts were treated with ethanol vehicle (0 h) or for 48 or 72 h with 20 μg of E2 39 d after implantation (n = 8 tumors per treatment). (A) Tissue biodistribution assay performed 2 h after 10 MBq (270 μCi) 18F-FFNP injection. *P < 0.05 compared with muscle. #P < 0.05 compared with vehicle control. (B) Representative hematoxylin and eosin (H&E) and immunohistochemistry (PR and Ki-67) of excised tumors.
Tumor xenografts in mice treated with 48 and 72 h of E2 had 91.7% ± 1.7% and 96.7% ± 1.7% positive nuclear staining for PR, which was more than the ethanol-treated controls (70.0% ± 5.8% positive nuclear staining) (P = 0.01 and 0.005, respectively; Fig. 2B). There was also a trend toward increased tumor Ki-67 staining in mice treated with 48 or 72 h of E2, compared with ethanol controls (41.7% ± 7.3% and 46.7% ± 3.3% vs. 31.7% ± 9.3%, respectively); however, this difference was not statistically significant (Fig. 2B). There was also no significant difference in tumor volume in mice treated with 48 or 72 h of E2 and mice treated with ethanol (50.6 ± 9.2 and 57.9 ± 7.5 vs. 47.1 ± 4.0 mm3).
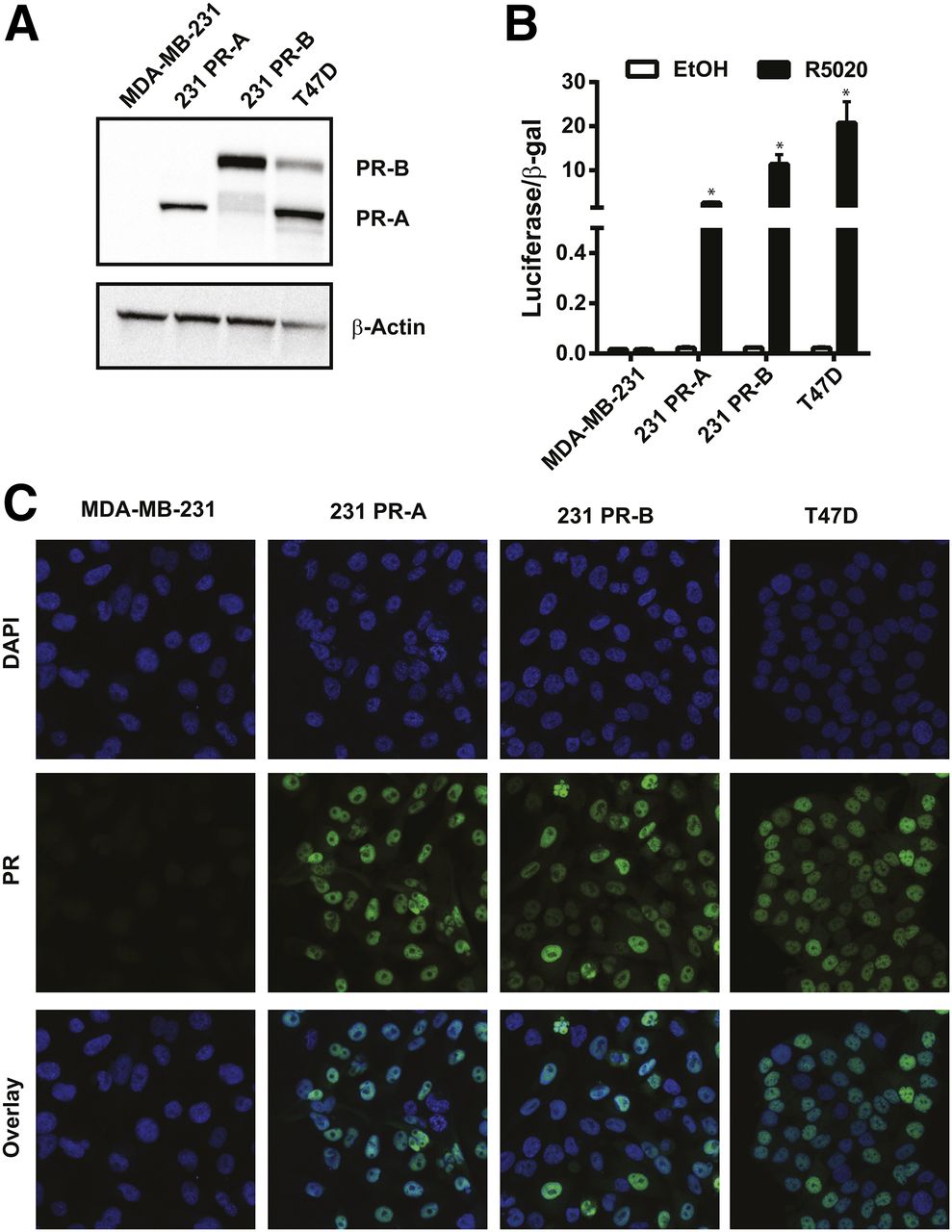
PR Isoform Expression, Subcellular Localization, and Functional Activity in Engineered Cells
To investigate the separate role of each isoform on overall 18F-FFNP binding, PR-negative MDA-MB-231 breast cancer cells were stably transfected with plasmids containing either PR-A or PR-B (Fig. 3A). PR transcriptional activity was markedly increased in response to R5020, compared with ethanol treatment, in 231 PR-A (P = 0.0003) and 231 PR-B cells (P = 0.0063) (Fig. 3B). As expected, PR transcriptional activity was increased in T47D cells after R5020 treatment, compared with ethanol (P = 0.0128), and there was no induction of PR activity in the parental MDA-MB-231 cells (P = 0.95). The engineered 231 PR-A and PR-B cells had nuclear localization of PR isoform protein similar to that of T47D cells when grown in cell culture medium containing steroid hormones (Fig. 3C).

PR isoform protein expression, subcellular localization, and functional activity. (A) Western blot for PR protein in MDA-MB-231, 231 PR-A, 231 PR-B, and T47D cell lines (representative of 3 independent experiments). (B) Steroid hormone–deprived cells treated with either ethanol (EtOH) or R5020 for 24 h. Luciferase reporter gene activity was measured and then normalized to β-galactosidase activity. Values represent mean ± SEM of 3 independent experiments. *P < 0.05 compared with ethanol for each cell line. (C) Immunofluorescence of PR localization and 4′,6-diamidino-2-phenylindole (DAPI) nuclear staining (×100 magnification). Images are representatives of 3 individual experiments.
Comparison of 18F-FFNP Uptake in Breast Cancer Cells Expressing PR-A Versus PR-B
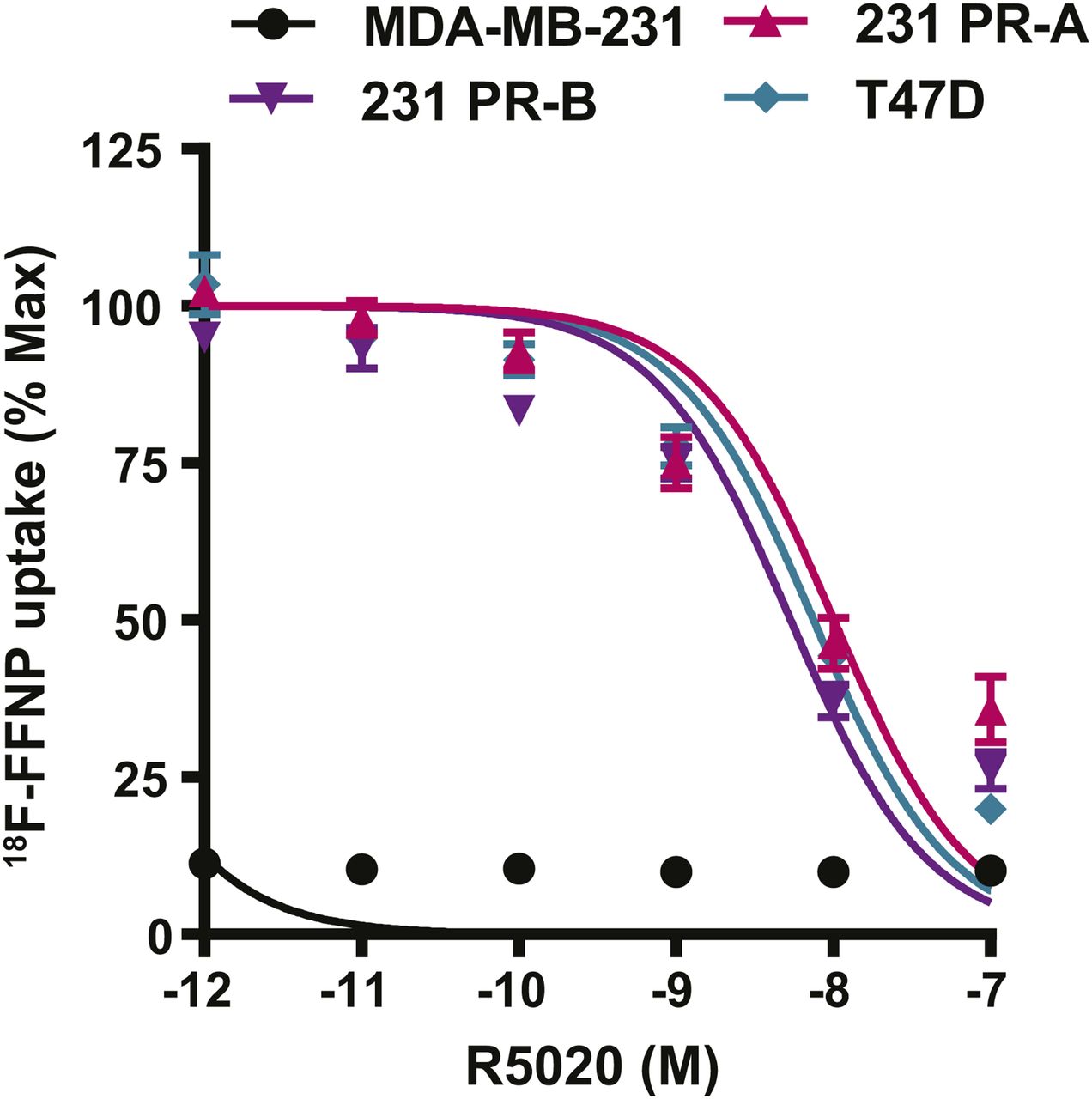
Competitive binding curves were performed to determine whether 18F-FFNP has preferential binding for either PR-A or PR-B (Fig. 4). 231 PR-A cells had an IC50 of 10 nM (95% confidence interval [CI], 6.7–15 nM). 231 PR-B cells had an IC50 of 5.4 nM (95% CI, 3.8–7.6 nM). T47D cells, which express both isoforms, had an IC50 of 7.5 nM (95% CI, 5.7–9.9 nM). The IC50 values did not significantly differ across the 3 cell lines. The parental MDA-MB-231 cells had no specific 18F-FFNP uptake.
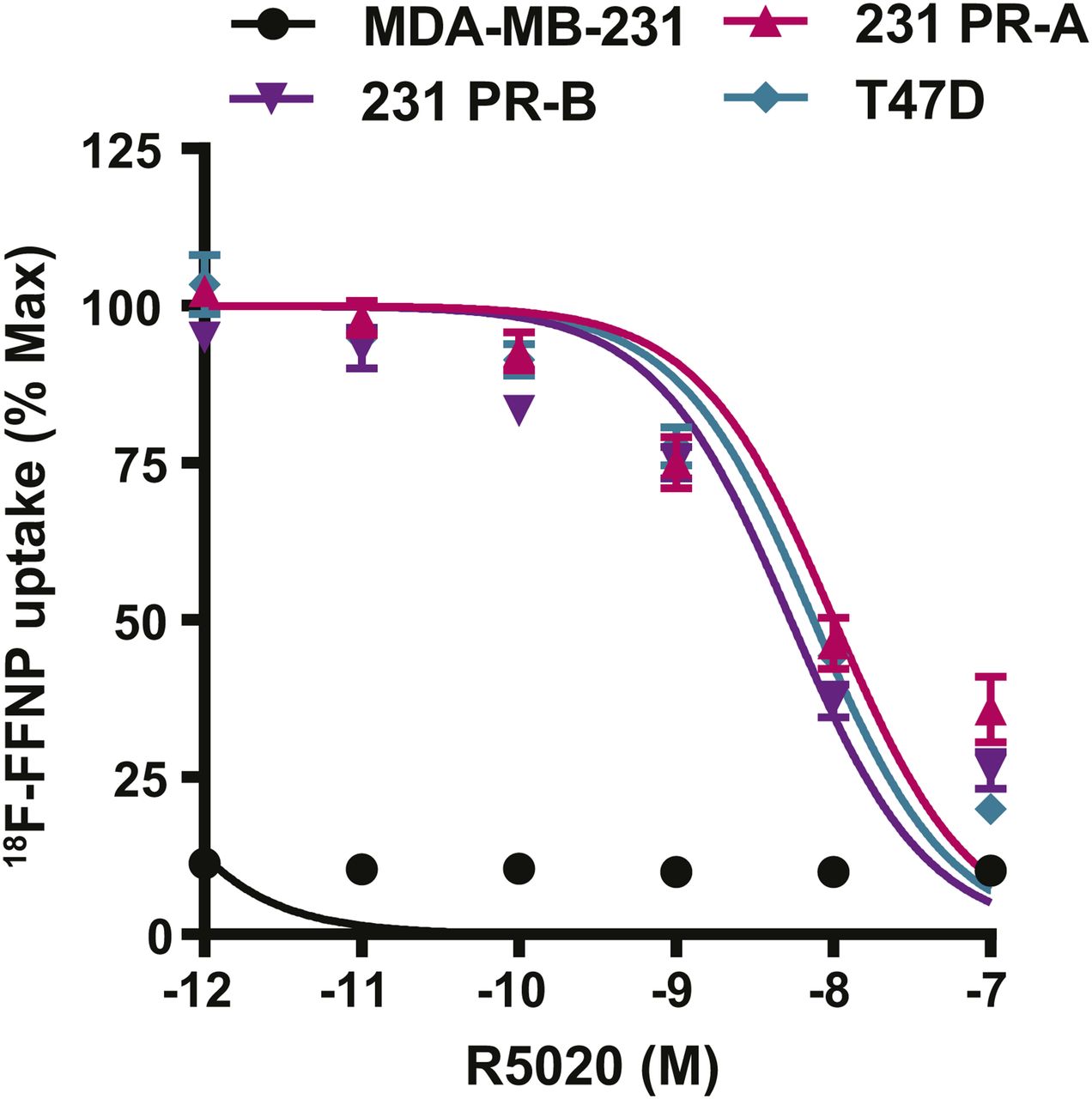
18F-FFNP uptake in cells expressing PR-A or PR-B. MDA-MB-231, 231 PR-A, 231 PR-B, and T47D cells were steroid hormone–deprived for 24 h and then treated with increasing amounts of R5020 (10−7–10−12 M) and incubated with 0.074 MBq (2 μCi) of 18F-FFNP for 1 h at 37°C. Decay-corrected counts per minute were normalized to wells containing 18F-FFNP without R5020 to calculate percentage maximum uptake values. MDA-MB-231 cell values were expressed relative to T47D. Values represent mean ± SEM of 3 independent experiments.
Saturation binding curves were also performed to quantify 18F-FFNP binding affinity, the equilibrium Kd, and total receptor density. 231 PR-A cells had lower receptor density than 231 PR-B and T47D cells (970 ± 62 vs. 2,621 ± 189 and 2,934 ± 97 fmol/mg, respectively; P < 0.0001; Table 1). The Kd was 0.82 nM (95% CI, 0.44–1.20) for 231 PR-A cells and 0.35 nM (95% CI, 0.14–0.56 nM) for 231 PR-B cells (Table 1). The Kd for T47D cells was 0.52 nM (95% CI, 0.39–0.66 nM). The Kd values did not significantly differ across the 3 cell lines.
Quantitative 18F-FFNP Binding Parameters
Comparison of 18F-FFNP Uptake in Tumor Xenografts Expressing PR-A Versus PR-B
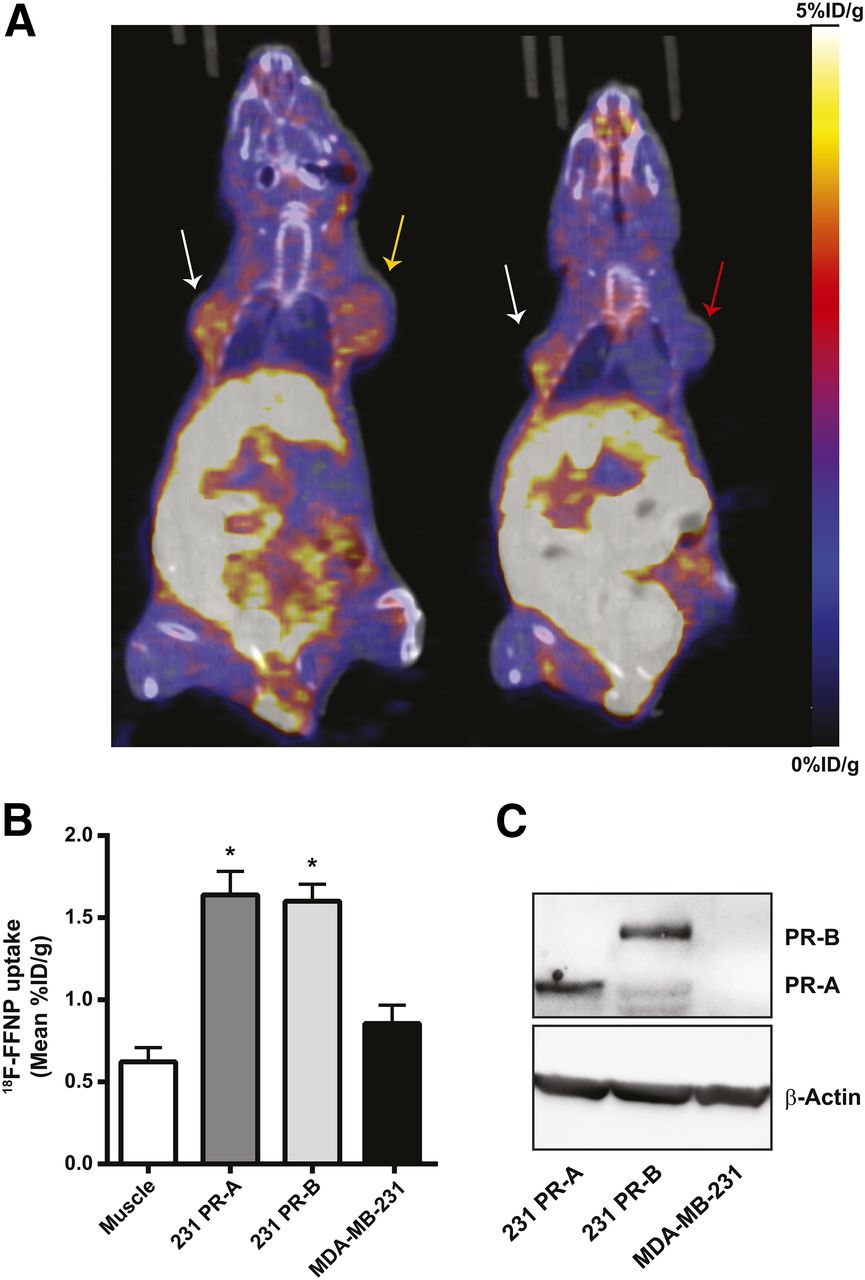
To test whether 18F-FFNP has preferential binding to PR-A or PR-B in vivo, tumor xenografts were generated using the engineered cell lines and PET imaging was performed (Fig. 5A). 18F-FFNP uptake (Fig. 5B) was greater in PR-A xenografts (1.64 ± 0.14 mean %ID/g, P = 0.0014) and PR-B xenografts (1.60 ± 0.10, P = 0.0030) than in MDA-MB-231 tumors (0.86 ± 0.11). However, 18F-FFNP uptake did not significantly differ between the PR-A and PR-B xenografts (P = 0.99). Corresponding tumor-to-muscle ratios were 3.3 ± 0.6, 3.0 ± 0.5, and 1.6 ± 0.4 for 231 PR-A, 231 PR-B, and MDA-MB-231 xenografts, respectively. Western blot analysis confirmed the appropriate expression of PR isoforms in the excised tumors (Fig. 5C).
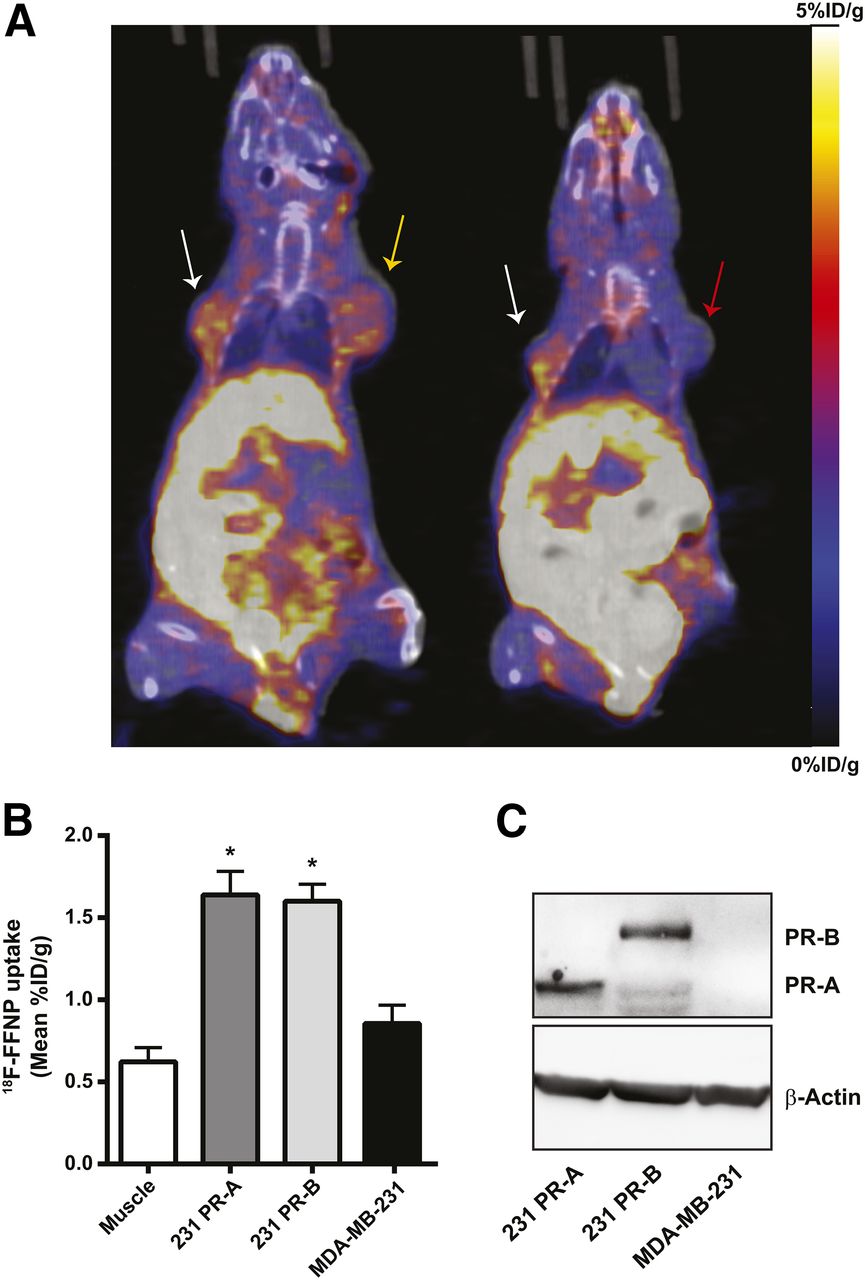
18F-FFNP uptake in tumor xenografts expressing PR-A or PR-B. (A) Representative coronal 18F-FFNP PET/CT images of MDA-MB-231 (red arrow), 231 PR-A (white arrows), and 231 PR-B (yellow arrow) tumor xenografts (19 d after implantation) from 12 mice imaged 1 h after injection with 10 MBq (270 μCi) of 18F-FFNP. Physiologic uptake from hepatobiliary clearance is also visualized. (B) Quantification of 18F-FFNP uptake (mean %ID/g) in muscle and tumor xenografts (231 PR-A, n = 10; 231 PR-B, n = 9; MDA-MB-231, n = 5). Values represent mean ± SEM, *P < 0.05 compared with muscle. (C) Western blot for PR isoform expression in excised tumors.
DISCUSSION
The purpose of this study was to evaluate the ability of 18F-FFNP to measure dynamic changes in PR protein levels and isoform expression in response to E2 challenge. We demonstrated that 18F-FFNP uptake increases in T47D cells after E2 treatment, with maximal uptake by 48 h, and in T47D tumor xenografts in mice treated with E2 for 48 and 72 h. In PR-isoform–specific cell lines and tumor xenografts, there was no significant preferential 18F-FFNP uptake by PR-A versus PR-B. Detecting early changes in PR protein expression with 18F-FFNP may be an earlier indicator of endocrine sensitivity than proliferation biomarkers and can be done noninvasively.
Current practice guidelines use the immunohistochemical presence (>1%) of ER protein as an indication for endocrine therapy. ER expression can also be assessed noninvasively using a radiolabeled estrogen, 18F-fluoroestradiol, and PET imaging to predict endocrine therapy response. Combined analyses of single-center studies demonstrated that patients with a maximum SUV below 1.5 showed no clinical benefit from endocrine therapy (negative predictive value, 88%) (21). However, the positive predictive value was only 65%, indicating that not all patients with positive tumoral 18F-fluoroestradiol uptake will benefit (21). Imaging the expression of PR, a classically regulated downstream ER target gene, using 18F-FFNP, may therefore be useful as an endogenous readout of ER functional activity and for early response assessment to endocrine therapy. An advantage of 18F-FFNP over 18F-fluoroestradiol is that the patient can be taking an ER-blocking agent, such as tamoxifen, fulvestrant, or other emerging oral selective ER degraders, and still have 18F-FFNP measure PR while ER is blocked.
One potential factor that could affect 18F-FFNP binding and the accuracy of PET imaging is the variable expression of PR isoforms observed in breast cancer, which may influence prognosis and therapeutic efficacy (11,12). Current clinical immunohistochemistry assays for PR do not distinguish between isoforms. Although PR-A and PR-B share identical ligand-binding domains in the C terminus, PR-A lacks the first 164 amino acids in the N terminus of PR-B, which changes its function as a transcription factor (6,7). Altered interdomain interactions can occur in the tertiary protein structure and differ depending on the ligand bound, as shown for other related steroid hormone receptors (22–24). Our results showed equivalent PR isoform binding affinity in cells and tumor uptake by 18F-FFNP. Although PR-A expression was 2.7-fold less than PR-B in the engineered cell lines on the basis of total receptor density values, there was comparable protein isoform expression in vivo measured with Western blots of the excised tumors—a finding that is congruent with the comparable tumor uptake of 18F-FFNP measured by imaging. Inherent differences between the in vitro and the more complex in vivo environment most likely account for this observation. For example, extracellular signaling through growth factor pathways may influence relative PR isoform expression in vivo, which may not be present in cell culture, and may have contributed to the equalization we observed in the excised tumors (25). On the basis of our results, we would expect similar diagnostic performance for 18F-FFNP PET imaging in breast cancer patients with variable PR isoform expression patterns.
Methods for demonstrating endocrine sensitivity in breast cancer patients using a short-window treatment continue to be an active area of interest. For example, changes in PR protein levels have been seen on repeat biopsy within a few weeks after initiating tamoxifen therapy in patients with locally advanced or recurrent breast cancer and have correlated with prolonged time to progression and improved survival (26). This technique is being used in an ongoing clinical trial testing whether a change in 18F-FFNP uptake after 1 d of administration of 6 mg of oral estradiol is predictive of endocrine therapy response in patients with ERα-positive metastatic breast cancer. The short-term diagnostic use of low-dose estradiol paired with imaging to demonstrate endocrine sensitivity is distinct from the long-term therapeutic use of high-dose estradiol (30 mg), which may be used clinically after failure of standard endocrine therapy agents (17,18). On the basis of our preclinical results, we would expect to see a similar increase in tumoral 18F-FFNP uptake after estradiol challenge and that this response will predict a benefit from endocrine therapy. However, because we focused on the time points demonstrating maximal 18F-FFNP uptake in cells (48 h and 72 h) for the xenograft experiments, we do not have a 24-h time-point for direct comparison with the clinical trial—a limitation of this study. Another approach measures increased tumor glucose metabolism with 18F-FDG PET imaging after a brief, low-dose estradiol challenge (17,18).
There are few published studies using 18F-FFNP with which to compare our results. The first-in-humans study of 18F-FFNP PET imaging for patients with PR-positive and PR-negative breast cancer consisted of a single baseline imaging session (16). Longitudinal imaging with 18F-FFNP PET before and during endocrine therapy, published by us previously, demonstrated that decreases in 18F-FFNP uptake and PR protein expression in response to 2 types of endocrine therapy (ovariectomy or fulvestrant) occurred in endocrine-sensitive but not in endocrine-resistant STAT1-deficient mouse mammary carcinomas (15,27). Thus, our results contribute to a growing amount of evidence supporting further investigation of 18F-FFNP PET imaging as a potential pharmacodynamic biomarker of endocrine sensitivity and a decision-making tool for tailoring therapy.
CONCLUSION
18F-FFNP is capable of measuring estrogen-induced shifts in total PR expression levels in human breast cancer cells and tumor xenografts, with equivalent isoform binding in vivo.
DISCLOSURE
This work was supported by the University of Wisconsin Institute of Clinical and Translational Research KL2 Scholar Award (KL2TR000428), the Clinical and Translational Science Award (CTSA) program through the NIH National Center for Advancing Translational Sciences (NCATS) grant UL1TR002373, the School of Medicine and Public Health, and the Department of Radiology to Amy Fowler. The University of Wisconsin Translational Research Initiatives in Pathology laboratory is in part supported by the University of Wisconsin Department of Pathology and Laboratory Medicine and UWCCC grant P30 CA014520, and the Experimental Pathology Laboratory is supported by P30 CA014520. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank the University of Wisconsin–Madison Cyclotron Laboratory for 18F production, the Radiopharmaceutical Production Facility for 18F-FFNP synthesis, and the Small Animal Imaging Facility for technical support and work space. We also thank the University of Wisconsin Optical Imaging Core for confocal microscopy and image collection, and we thank the University of Wisconsin Translational Research Initiatives in Pathology laboratory and the Experimental Pathology Laboratory for use of their facilities and services. Lastly, we thank Kirsti Walker for technical assistance and Dr. John Katzenellenbogen for thoughtful discussions and critical reading of the manuscript.
Footnotes
Published online Jul. 20, 2018.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication March 19, 2018.
- Accepted for publication July 13, 2018.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Longitudinal Molecular Imaging of Progesterone Receptor Reveals Early Differential Response to Endocrine Therapy in Breast Cancer with an Activating ESR1 Mutation
- Recent Advances in Imaging Steroid Hormone Receptors in Breast Cancer
- 18F-Fluoroestradiol PET Imaging of Activating Estrogen Receptor-{alpha} Mutations in Breast Cancer
- Measuring Estrogen Receptor Functionality Using Progesterone Receptor PET Imaging: Rising to the (Estradiol) Challenge!