


Abstract
Traditionally, multiple sclerosis (MS) has been considered a white matter disease with focal inflammatory lesions. It is, however, becoming clear that significant pathology, such as microglial activation, also takes place outside the plaque areas, that is, in areas of normal-appearing white matter (NAWM) and gray matter (GM). Microglial activation can be detected in vivo using 18-kDa translocator protein (TSPO)–binding radioligands and PET. It is unknown whether fingolimod affects microglial activation in MS. The aim of this study was to investigate whether serial PET can be used to evaluate the effect of fingolimod treatment on microglial activation. Methods: Ten relapsing-remitting MS patients were studied using the TSPO radioligand 11C-(R)-PK11195. Imaging was performed at baseline and after 8 and 24 wk of fingolimod treatment. Eight healthy individuals were imaged for comparison. Microglial activation was evaluated as distribution volume ratio of 11C‐(R)-PK11195. Results: The patients had MS for an average of 7.9 ± 4.3 y (mean ± SD), their total relapses averaged 4 ± 2.4, and their Expanded Disability Status Scale was 2.7 ± 0.5. The patients were switched to fingolimod because of safety reasons or therapy escalation. The mean washout period before the initiation of fingolimod was 2.3 ± 1.1 mo. The patients were clinically stable on fingolimod. At baseline, microglial activation was significantly higher in the combined NAWM and GM areas of MS patients than in healthy controls (P = 0.021). 11C‐(R)-PK11195 binding was reduced (−12.31%) within the combined T2 lesion area after 6 mo of fingolimod treatment (P = 0.040) but not in the areas of NAWM or GM. Conclusion: Fingolimod treatment reduced microglial/macrophage activation at the site of focal inflammatory lesions, presumably by preventing leukocyte trafficking from the periphery. It did not affect the widespread, diffuse microglial activation in the NAWM and GM. The study opens new vistas for designing future therapeutic studies in MS that use the evaluation of microglial activation as an imaging outcome measure.
In multiple sclerosis (MS), neuroinflammation and neurodegeneration are 2 main components in the disease pathogenesis. Usually, the disease initiates as relapsing-remitting MS (RRMS), where focal demyelination of the cerebral white matter (WM) is the hallmark of the disease. At an average of 10 y after disease onset, RRMS shifts into secondary progressive MS and slowly leads to disability (1). Thus far, prevention of worsening of progressive MS has proven a major challenge, whereas great advances have been achieved in developing treatments for RRMS (2). This likely reflects the differences in the pathogenic mechanisms behind the respective disease subtypes. In RRMS, the major determinant driving the onset of relapses and the focal inflammatory lesions in the central nervous system (CNS) is the overreactivity of the adaptive immune system, with resulting inflammatory cell trafficking from the periphery into the CNS (1). Patients with progressive disease, instead, present with chronic lesions with or without microglial activation (i.e., chronic active or chronic inactive lesions), and widespread microglial activation in the normal-appearing WM (NAWM), which colocalizes with signs of neuronal damage (3,4). The microglial activation can be detected in vivo using PET and radioligands binding to an 18-kDa translocator protein (TSPO), which is expressed by activated microglial cells (5–7). It has, however, become clear that activated microglia consist of a wide array of microglial phenotypes, some of which are likely beneficial and contribute to clearing of debris, whereas some are proinflammatory and contribute to the relentless CNS damage related to progressive MS (8,9). Presently, these different phenotypes cannot be distinguished in vivo.
Fingolimod is the first oral disease-modifying therapy (DMT) developed for the treatment of RRMS. It blocks the egress of lymphocytes from secondary lymphoid tissues and thereby prevents their entry into the CNS (10,11). In addition, being strongly lipophilic, fingolimod penetrates into the CNS and binds to sphingosine 1 phosphate receptor on CNS-resident glial and neural cells (12). Importantly, in animal models of MS, fingolimod modulates microglial and astroglial cells (13–15), which can be detected in vivo as a diminished TSPO ligand signal (13). Hence, fingolimod also has the potential to directly alleviate inflammation and neurodegeneration contained within the CNS of MS patients. However, in a recent study of primary progressive MS, fingolimod was proven ineffective in halting the disease progression (16).
Given the suspected role of activated microglial cells in MS, TSPO PET in the NAWM has been suggested to be a potential surrogate outcome measure in trials of progressive MS (5–7). Thus far, the effect of any DMT on microglial activation has been evaluated in only 1 MS study (17). The aim of the present study was to determine the kinetics of microglial activation in various brain areas of a cohort of RRMS patients before and after fingolimod treatment using serial in vivo TSPO imaging. Furthermore, we wanted to evaluate whether in this cohort of RRMS patients with a disease duration averaging 8 y signs of microglial activation could already be seen outside the focal lesions.
MATERIALS AND METHODS
Study Subjects
The study was performed as an academic, investigator-initiated study at Turku PET Centre and Turku University Hospital. The study protocol was approved by the Ethics Committee of the Hospital District of Southwest Finland. The study was registered to ClinicalTrials.gov (identifier NCT02139696). All participants signed a written informed consent form according to the Declaration of Helsinki.
Patients with RRMS diagnosed according to the 2005 revised McDonald criteria (18), who were initiating treatment with oral fingolimod (0.5 mg) daily, were recruited from a single center. Key eligibility criteria were an age of 18–65 y, an RRMS diagnosis for more than 5 y before enrollment, and a neurologist’s treatment decision of switching from previous treatment to fingolimod according to current care guidelines. Disease duration was calculated as the time from the first demyelinating event to the first PET scan. A minimum washout period of 1 mo after previous DMT was required.
Exclusion criteria included corticosteroid treatment within 30 d of evaluation, active neurologic or autoimmune disease other than MS, or another comorbidity considered significant, inability to tolerate PET or MRI, and a current or desired pregnancy in the 6 mo after study enrollment.
Procedures
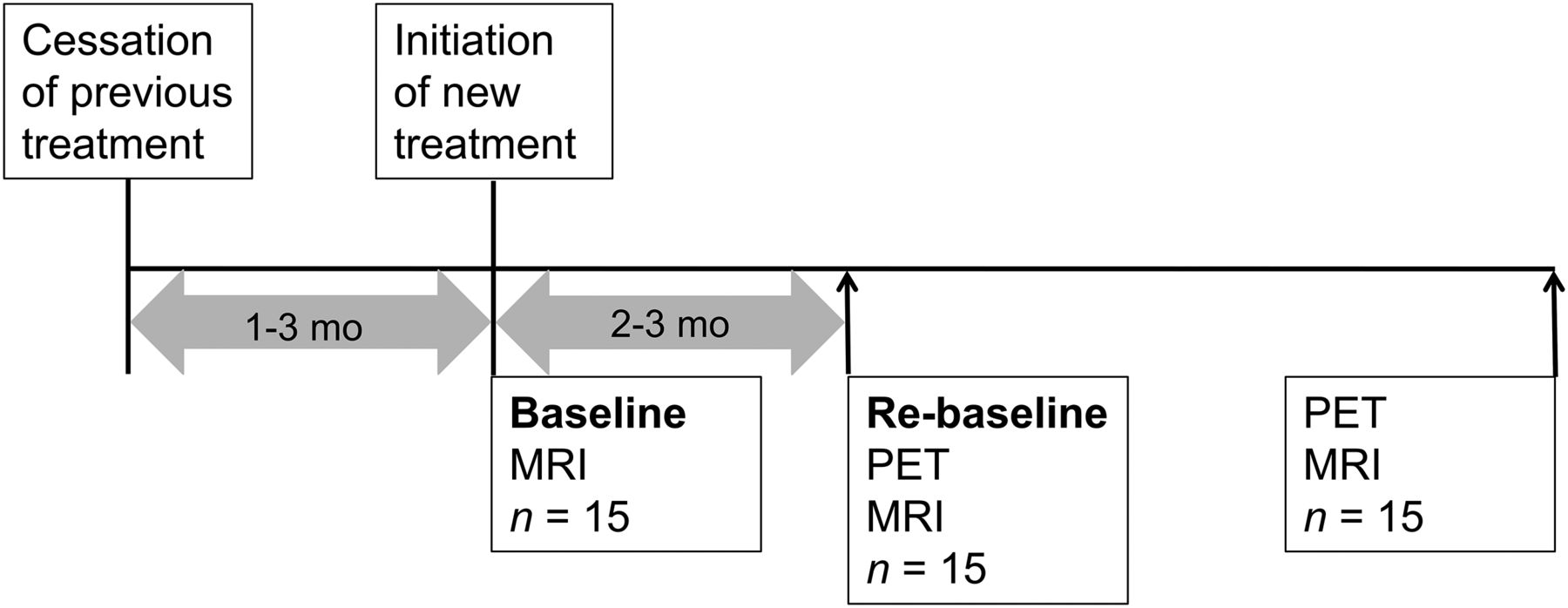
One of 11 recruited patients withdrew from the study after baseline imaging. Ten patients were imaged both at baseline and after 24 wk of fingolimod treatment. They were on average 42 y old (SD, 9.0). Seven patients underwent PET and MRI also after 6−8 wk (range, 53−102 d). Expanded Disability Status Scale, safety evaluation, and recording of relapses were performed at baseline, after 6−8 wk, and after 24 wk of fingolimod treatment (Fig. 1). The baseline PET was compared with historical PET data (with identical imaging methodology) from 8 healthy individuals (6 women, 2 men; mean age ± SD, 49.8 ± 7.9 y; range, 42–61 y).
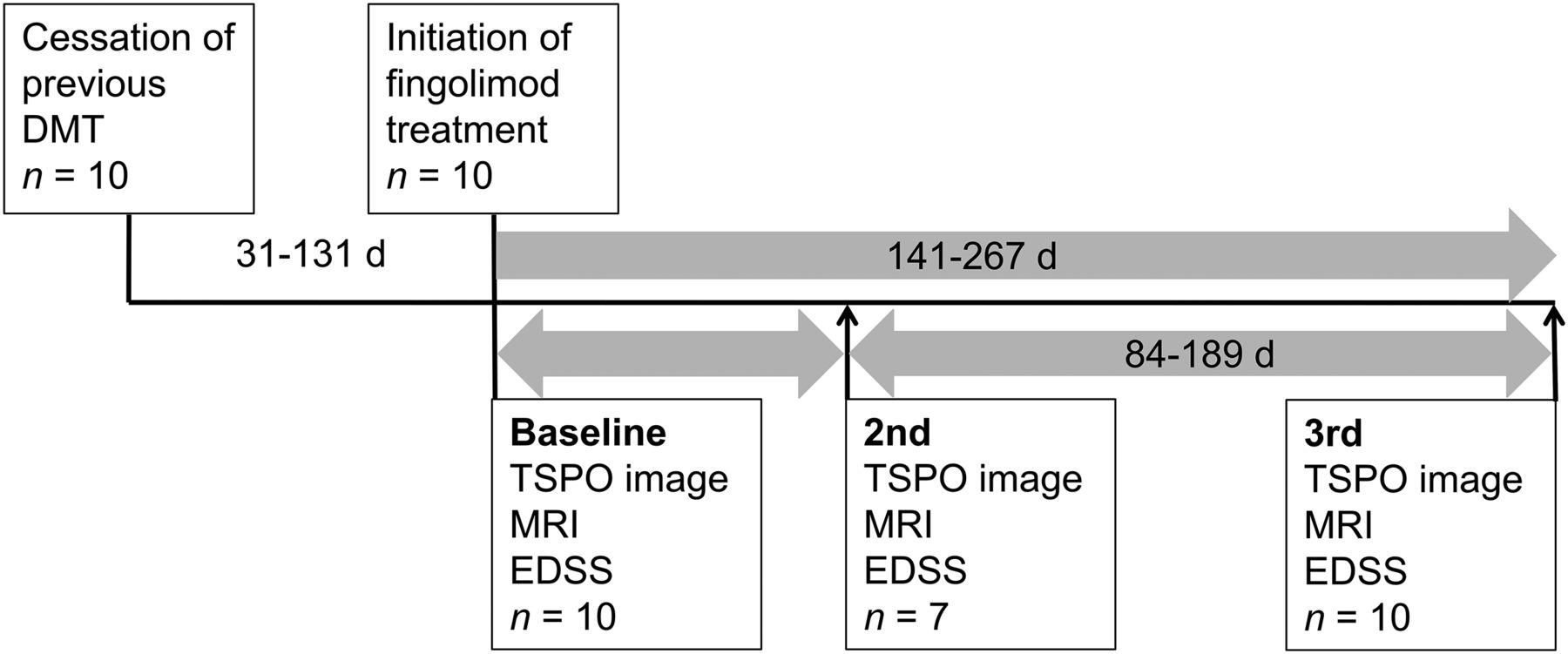
Schematic representation of study timeline. EDSS = Expanded Disability Status Scale.
Outcomes
The primary outcome was the change in microglial activation after 24 wk of fingolimod treatment. In addition, TSPO binding was evaluated after 6−8 wk of treatment (n = 7). We also evaluated the effect of fingolimod treatment on MRI parameters. Finally, we evaluated how microglial activation at baseline correlated with MS duration, age, and washout period before initiation of fingolimod treatment and how microglial activation correlated to duration of treatment.
MRI and Data Analysis
For the evaluation of MS pathology and for the acquisition of anatomic reference for the PET images, conventional MRI was performed with a 3-T Ingenuity TF PET/MR scanner (Philips). MRI of the control group was performed with a Gyroscan Intera 1.5-T Nova Dual scanner (Philips). The routine sequences included axial T2, 3-dimensional (3D) fluid-attenuated inversion recovery, 3D T1, and 3D T1 with gadolinium enhancement.
For each patient, the T1 image at the first time point was coregistered in statistical parametric mapping (SPM8, version 8; Wellcome Trust Centre for Neuroimaging) to the sum image of realigned PET frames of the first session. All the other MR images were then coregistered to the T1 image of the first session. For each time point, the MS lesions were identified using the Lesion Segmentation Tool (a toolbox running in SPM8) (19) as described previously (6). The resulting lesion masks were used to fill the corresponding T1 image with the lesion-filling tool in Lesion Segmentation Tool. The filled T1 was then used for segmenting gray matter (GM) and WM volumes with Freesurfer 5.3 software (http://surfer.nmr.mgh.harvard.edu/).
The volumes of T2 lesion masks acquired with the Lesion Segmentation Tool were used for the T2 lesion load evaluation. Additionally, for each patient the Lesion Segmentation Tool masks at each time point were combined to a unified lesion region of interest (ROI). An average filled T1 image was also calculated from the filled T1 images of all MRI sessions, and it was used for ROI delineation for the cerebellum, striatum, thalamus, WM, and cortical GM with Freesurfer. Finally, a NAWM ROI was created by removing the lesion ROI from the WM ROI. The mixed GM and WM region contains the cortical GM, subcortical GM structures, and the WM (NAWM and WM lesions), thus representing the global brain parenchyma.
11C-(R)-PK11195 Radioligand Production and PET
The radiochemical synthesis of 11C‐(R)-PK11195 was performed as described before (6). The injected doses in each evaluated group did not differ from each other (data not shown). PET was performed with a brain-dedicated ECAT High-Resolution Research Tomograph scanner (CTI/Siemens) with an intrinsic spatial resolution of approximately 2.5 mm (20). First, a 6-min transmission scan for attenuation correction was obtained using a 137Cs point source. Thereafter, 60-min dynamic imaging was started simultaneously with the intravenous, bolus injection of the radioligand. Head movements were minimized using a thermoplastic mask.
PET Analysis
Images were reconstructed using 17 time frames as described previously (6). The dynamic data were then smoothed using a gaussian 2.5-mm postreconstruction filter (6). Possible displacements between frames were corrected using mutual information realignment in SPM8. For each patient, the PET images from the subsequent sessions were coregistered to the PET image of the first session using the sum images of each session. Finally, all images were resliced to match an MR voxel size of 1 mm.
For the estimation of the 11C‐(R)-PK11195 distribution volume ratio (DVR), the time–activity curve corresponding to a reference region devoid of specific TSPO binding was acquired for each PET session using a supervised cluster algorithm with 4 predefined kinetic tissue classes (SuperPK software) as described previously (6). As an additional step, the intersection of the clustered reference region maps within each subject’s PET sessions was calculated, and this region was then used to extract the individual reference region for each PET session. The reference tissue–input Logan method, with a 20- to 60-min time interval, was applied to the regional time–activity curves using the supervised cluster algorithm gray reference input.
To correct for the partial-volume effect caused by differences in 11C‐(R)-PK11195 binding between adjacent ROIs and by increased radioligand binding in the meningeal, vascular, bone, and soft tissue next to cortical GM, a partial-volume correction using the Geometric Transfer Matrix method (21) was performed for all regional time–activity curves. A gaussian function with 2.5 mm full width at half maximum was used to approximate the scanner point-spread function, and for each cortical region a corresponding background ROI was used to correct for the background activity.
Statistical Analysis
The statistical analyses were performed using SPSS (version 23; IBM Statistics). The data distribution was evaluated with the Shapiro–Wilk test. When normally distributed, the group characteristics were reported as mean and SD. Nonnormally distributed variables were reported as medians and interquartile range. The comparisons of the imaging parameters between healthy controls and RRMS patients were performed using analysis of covariance with age as a covariate because of the different, but overlapping, distributions of age. To rule out the possible effect of volumetric changes in the DVR estimates, correlational analyses between the changes in T2 lesional, NAWM, cortical GM, and total cerebral volumes and the changes in respective DVRs in these ROIs were examined using Spearman correlation.
Serial PET data were analyzed with repeated-measures ANOVA with Bonferroni adjustment. Other pairwise comparisons were performed with the nonparametric Mann–Whitney U test or related-samples Wilcoxon signed-rank test, wherein the nonparametric tests were chosen due to the low number of subjects per group. The associations between variables were analyzed with Spearman correlation. A P value of less than 0.05 was considered statistically significant for all analyses unless stated otherwise.
On the basis of earlier 11C‐(R)-PK11195 studies (6,17), we estimated that a sample size of 10 subjects per group would suffice to reveal a 15% change between groups and a 5% change between the time points in the 11C‐(R)-PK11195 DVR within 1 examined ROI. However, because of the pilot nature of this study, a detailed a priori sample size calculation for repeated-measures ANOVA was unfeasible due to the lack of detailed data on the expected effect size and variance with a function of time.
RESULTS
Clinical Characteristics of Subjects
All participants were relapse-free for at least 3 mo before the initial PET scan (median, 9.6 mo; range, 3−77 mo). The mean disease duration was 7.9 y (SD, 4.3), and total relapse number averaged 4 (SD, 2.4). The patients were switched to fingolimod because of safety reasons or therapy escalation. The mean washout period before the initiation of fingolimod treatment was 2.3 mo (SD, 1.1). All patients were clinically and radiologically (MRI) stable during the entire study, there were no gadolinium-enhancing lesions, and no adverse events occurred (Supplemental Tables 1−2; supplemental materials are available at http://jnm.snmjournals.org). The subject demographics are shown in Supplemental Table 3.
11C-(R)-PK11195 Binding at Beginning of Fingolimod Treatment
Microglial activation measured as 11C‐(R)-PK11195 DVR was significantly higher among the MS patients (n = 10) than in healthy controls in the combined NAWM and GM area (P = 0.021) at baseline. Similarly, 11C‐(R)-PK11195 binding in thalami was stronger among MS patients than in controls (P = 0.006). In the separate NAWM and cortical GM ROIs, there was a nonsignificant trend toward higher 11C-(R)-PK11195 DVR in MS patients than in controls (P = 0.096 and P = 0.052, respectively). In other ROIs, no significant differences were found between the groups (Fig. 2A; Supplemental Table 1).
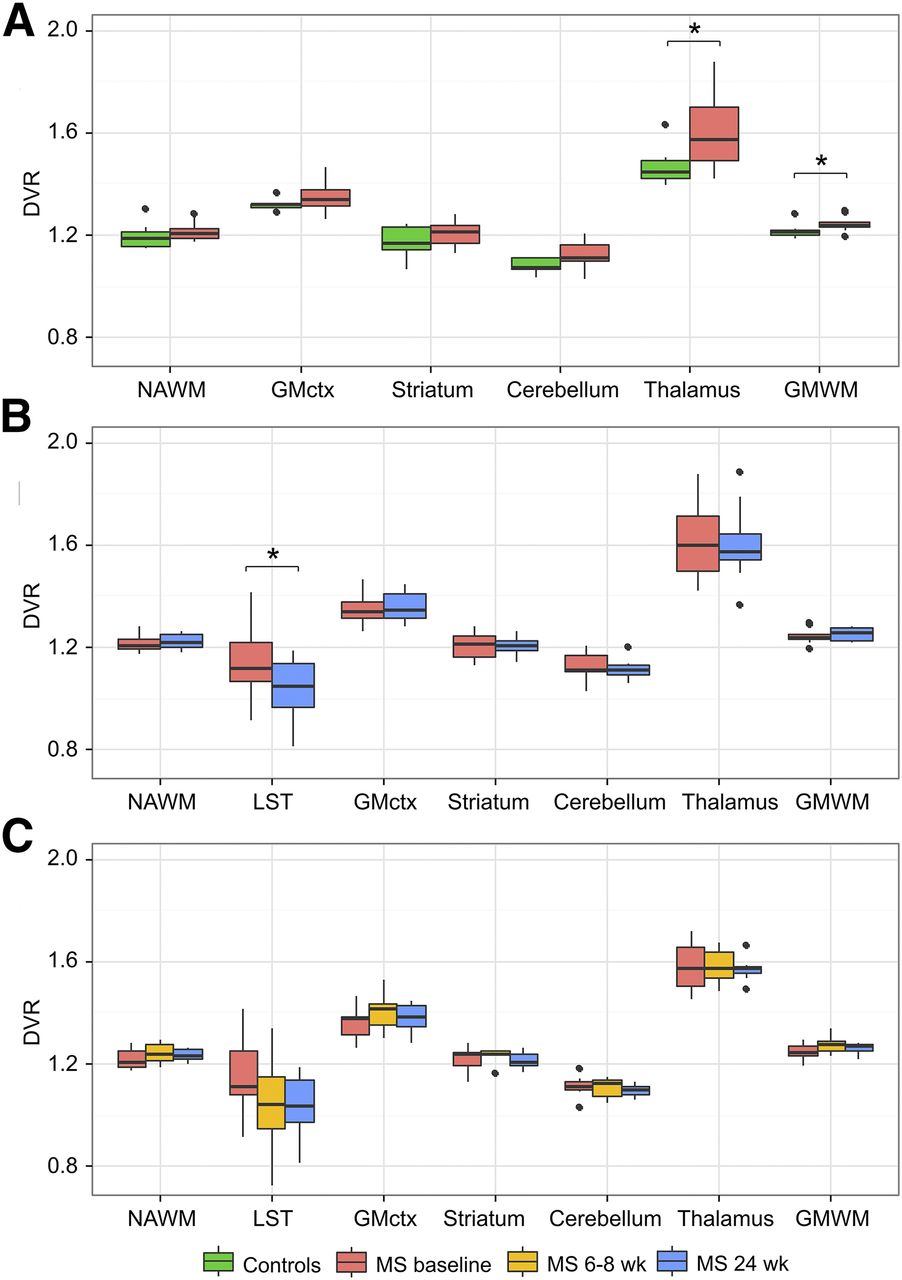
ROI-specific 11C-(R)-PK11195 DVR values by group and imaging time point. Box plot results show median DVR values with first and third quartiles. (A) Healthy controls (n = 8) and RRMS patients (n = 10) at baseline. RRMS patients (n = 10) at baseline and at 6-mo time points (B) and RRMS patients (n = 7) (C) at baseline, 2 mo, and 6 mo. Asterisk denotes statistically significant group difference at level of P < 0.05 in analysis of covariance with age as covariate. Whiskers are calculated using formula 1.5 × interquartile range. Data beyond end of whiskers are outliers and plotted as points. GMctx = cortical GM; GMWM = combined GM and WM areas; LST = area representing lesion mask, which contains all T2 lesion areas in corresponding MRI.
11C-(R)-PK11195 Binding Was Reduced in T2 Lesions After 6 Months of Fingolimod Treatment Compared with Baseline
Ten participants underwent PET and MRI at baseline before initiating fingolimod treatment and again after 6 mo. Fingolimod treatment was initiated within a week of baseline imaging, and the mean time between the PET scans was 190 d (range, 141−267 d; Fig. 1). 11C‐(R)-PK11195 binding was reduced in the combined T2 lesion area after 6 mo of treatment with fingolimod (−12.31%; P = 0.040) but not in other brain areas (Fig. 2B; Supplemental Table 1).
Serial 11C-(R)-PK11195 Imaging During Fingolimod Treatment
Seven participants were scanned 3 times: first at baseline and again 2 and 6 mo after the treatment initiation (Fig. 1). When 3 serial DVRs were evaluated, no statistically significant alterations were observed in any of the ROIs at group level (Fig. 2C; Supplemental Table 2). Interestingly, there was a slight increase in 11C-(R)-PK11195 DVR in 5 of 7 patients in the NAWM and in 6 of 7 patients in cortical GM during the first 2 mo of fingolimod treatment (Fig. 3).
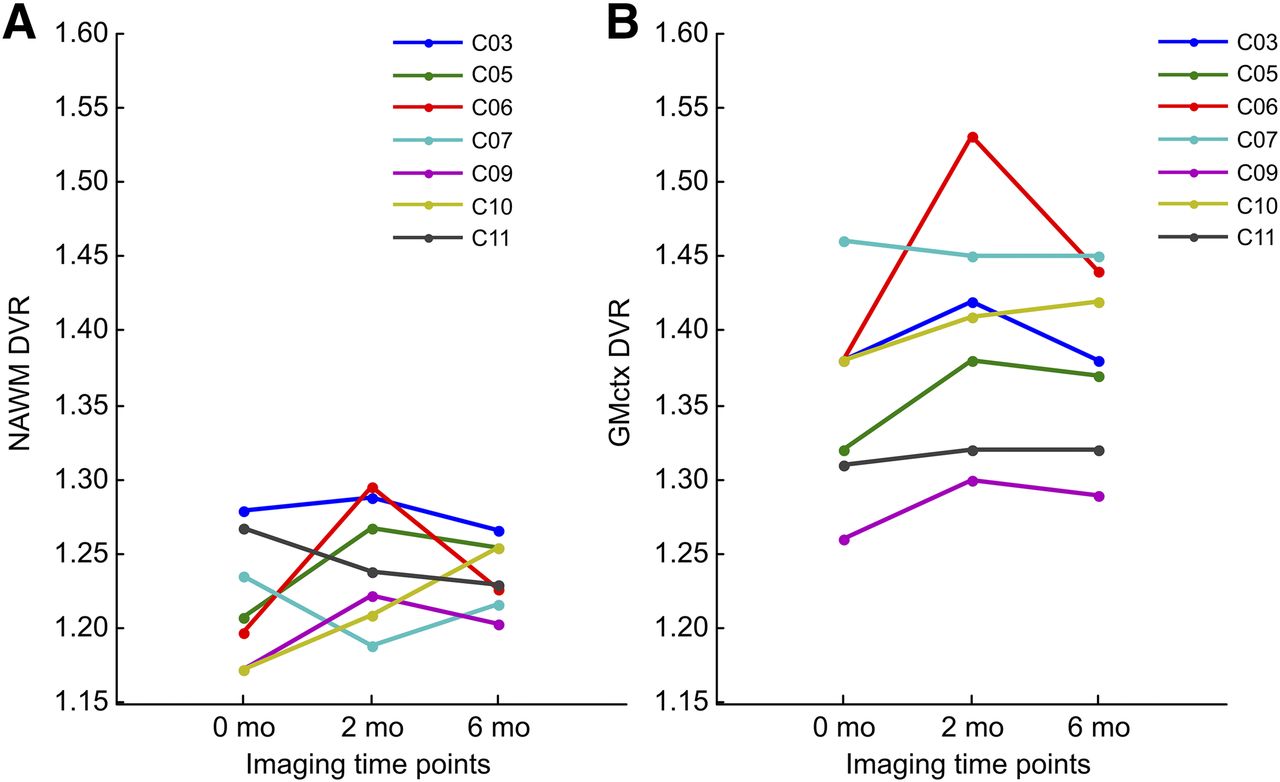
Serial PET imaging results. Individual 11C‐(R)-PK11195 DVR values are shown in NAWM (A) and normal-appearing cortical GM (B) at baseline, 2-mo, and 6-mo time points in 7 RRMS patients with longitudinal PET data.
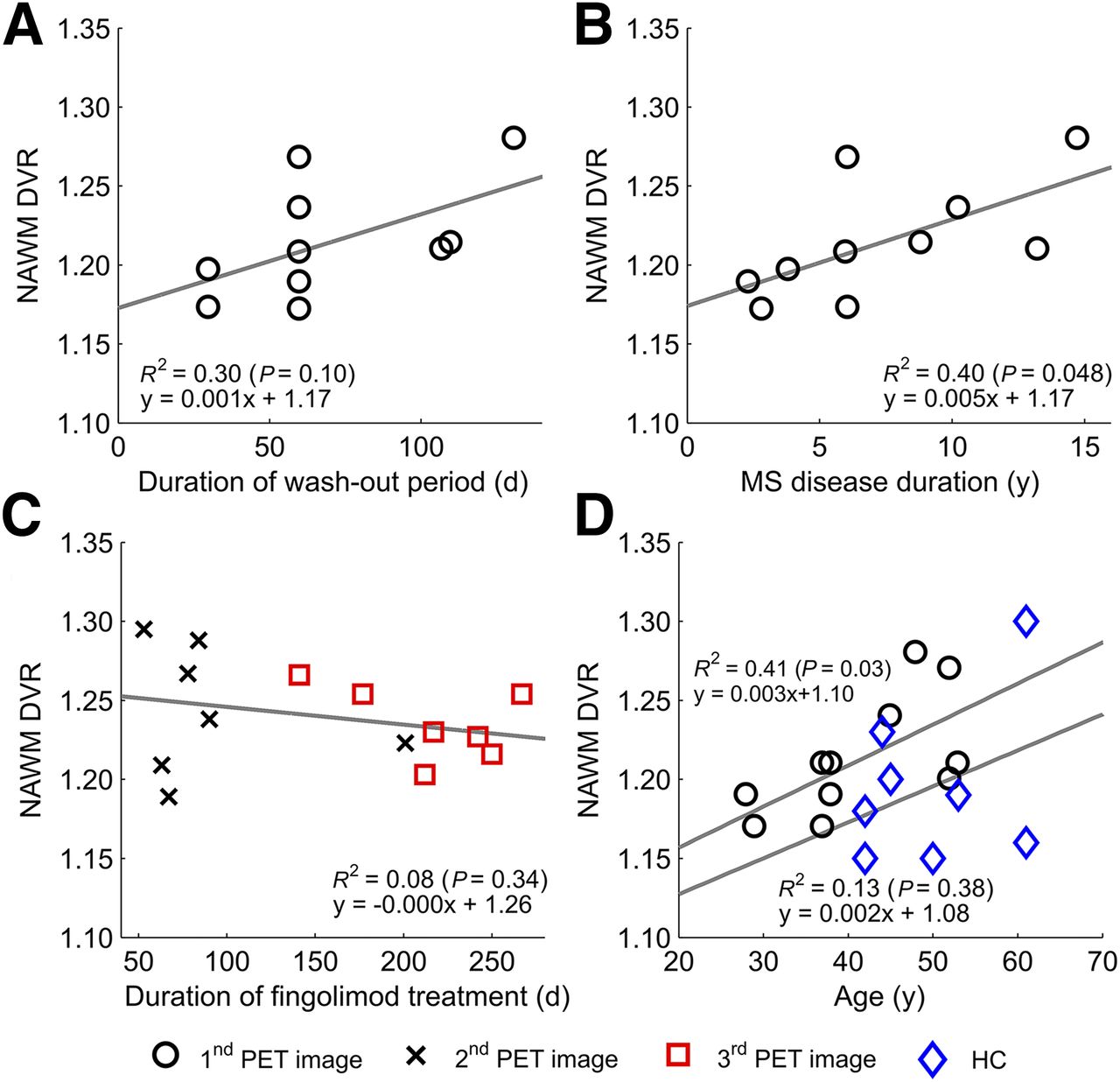
Longer Washout Period, Longer Disease Duration, and Higher Age Associate with Higher Microglial Activation in NAWM
Treatment-free periods before fingolimod initiation varied between 30 and 131 d, and interestingly there was a nonsignificant trend for a correlation between longer washout period and higher NAWM DVR at baseline (n = 10; Spearman correlation, 0.616; P = 0.058; Fig. 4A). Moreover, a higher baseline DVR in the NAWM was associated with longer disease duration (Spearman correlation, 0.754; P = 0.012; Fig. 4B). There was a slight trend between longer fingolimod treatment and lower DVR in the NAWM in the second and third imaging time points, but this was statistically nonsignificant (Spearman correlation, 0.238; P = 0.358; Fig. 4C). Furthermore, higher age correlated with higher DVR in the NAWM among MS patients (Spearman correlation, 0.636; P = 0.048; Fig. 4D).
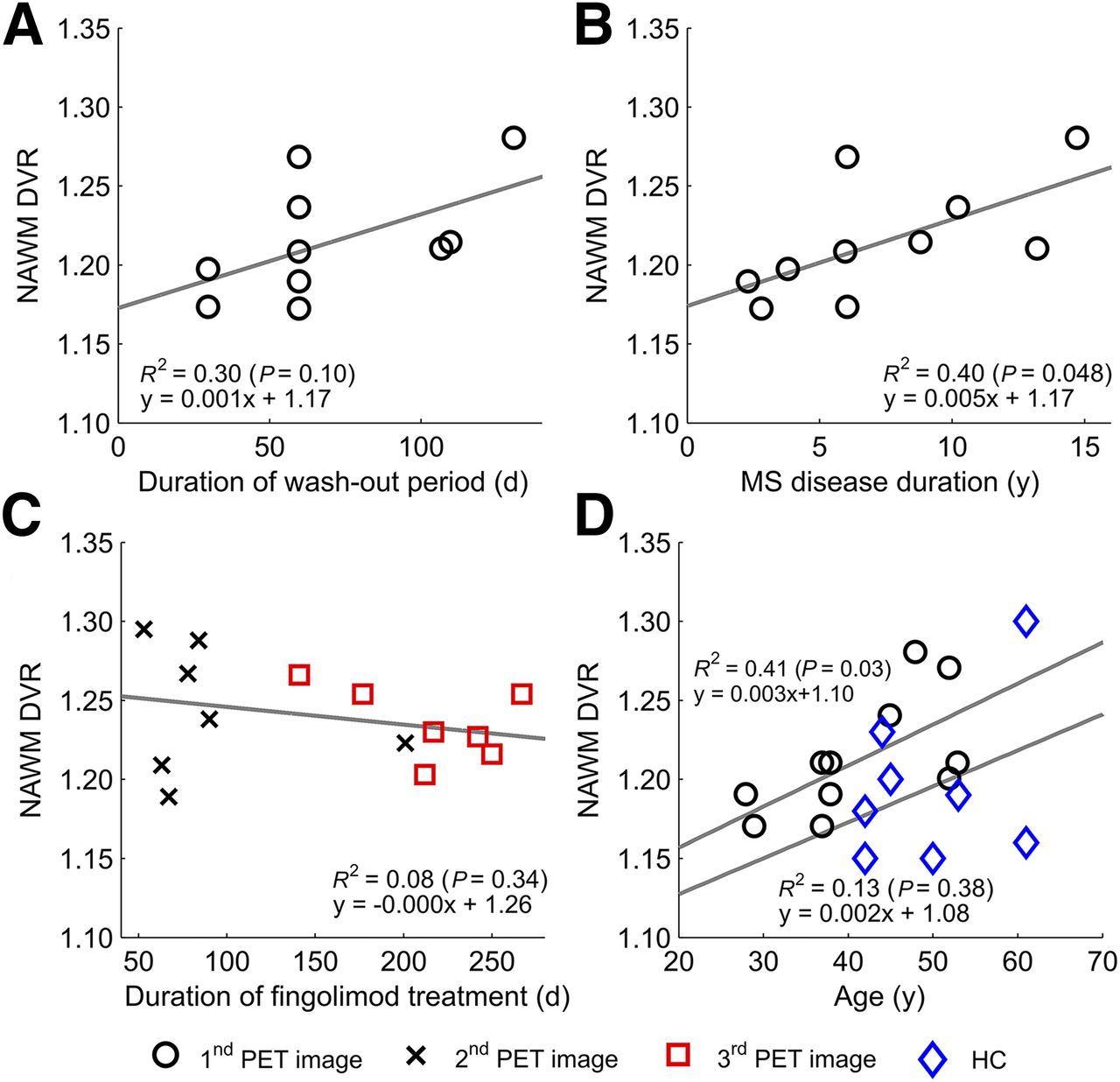
Longer washout period, longer disease duration, and higher age associate with higher microglial activation in NAWM. Correlations are shown between duration of treatment-free period before initiation of fingolimod-treatment and baseline 11C‐(R)-PK11195 binding in NAWM (n = 10) (A), disease duration and 11C‐(R)-PK11195 binding in NAWM at first imaging time point (n = 10) (B), duration from initiation of fingolimod to second and third imaging time points and 11C-(R)-PK11195 binding in NAWM in respective time points (n = 7, pooled n = 14) (C), and age and 11C-(R)-PK11195 binding in NAWM in healthy controls (HC) and RRMS patients at first imaging time point (D). Linear correlations are visualized as regression lines and squared Pearson coefficients in images, and corresponding Spearman correlations are 0.616 (P = 0.058) (A), 0.754 (P = 0.012) (B), 0.238 (P = 0.358) (C), and 0.636 (P = 0.048) (D) for patients and 0.450 (P = 0.313) for HC, statistically significant at level of P < 0.05.
Power and Sample Size Analyses for Future Studies
Using the findings of the present study, we wanted to calculate the sample size needed to demonstrate a statistically significant alteration in microglial activation within the NAWM. We discovered that to reveal a ±5% treatment effect with a statistical significance of P less than 0.05, the sample size and the respective statistical power for a 5% increase between baseline and 2 mo are 13 and 0.907, respectively. For a 5% decrease both in 2- to 6-mo and in 2-mo to 1-y intervals, the sample size is 14 and power 0.913.
DISCUSSION
This study used 11C‐(R)-PK11195 binding to measure the degree of microglial activation in MS patients before and after fingolimod treatment. Microglial activation was reduced after 6 mo of fingolimod treatment in areas corresponding to focal T2 lesions. Microglial activation in the NAWM or GM was not affected. Microglial activation correlates with neurodegenerative changes related to MS progression (3,22), and it is conceivable that modulation of microglial activation might actually alleviate degeneration and decelerate the disease progression. The effects of DMTs on microglial activation in the NAWM in MS have thus far remained unclear. In the 1 previously published study, in which MS treatment effect on microglial activation was evaluated, 11C‐(R)-PK11195 imaging was performed before and after 1 y of treatment with glatiramer acetate (17). In the present study, we opted to image at earlier time points to understand the kinetics of treatment-associated alterations in microglial activation. We used the 11C‐(R)-PK11195 radioligand despite its shortcomings, such as the limited signal-to-noise ratio. It is a widely and successfully used TSPO-binding radioligand in the study of neurodegenerative disease, and particularly the advanced and reliable quantification methods available for this ligand make it useful for clinical assessment of brain pathology (23).
We compared the degree of microglial activation among MS patients at the beginning of fingolimod treatment with a control group of healthy individuals, and in this evaluation, the DVR in the combined NAWM and GM area was higher in the patient group than in the control group. The control group was somewhat older than the patient group, and it is possible that this slightly affected the results of the comparison of the DVRs in individual ROIs in MS versus controls, despite the attempt to statistically control for this by performing the comparison between MS patient and control DVRs using analysis of covariance with age as a covariate. In individual evaluation of NAWM and cortical GM ROIs, there was a strong, although nonsignificant, trend toward higher 11C-(R)-PK11195 DVR in MS patients (P = 0.096 and 0.052, respectively), suggesting that the disease-related pathology had already started to spread outside the focal lesions and manifested as diffuse microglial activation. Moreover, the baseline DVR tended to correlate with the duration of the washout period, which might be relevant to the returning inflammatory activity after a DMT cessation; the longer the washout period, the higher the microglial activation at baseline among MS patients.
On the basis of our results, it seems evident that in the short term, the positive fingolimod treatment effect in MS comes from the prevention of focal inflammatory activity. This result is in line with larger studies using MRI as an outcome measure, which demonstrate a reduction in focal inflammatory activity after fingolimod treatment (24). Our results suggest that there are perhaps no direct effects mediated by fingolimod binding to sphingosine 1 phosphate receptors on microglial cells. Rather, after 2 mo of treatment, 11C‐(R)-PK11195 binding increased slightly in 5 of 7 patients in the NAWM and in 6 of 7 patients in cortical GM. This might be a reflection of the still increasing inflammatory activity emerging from the peripheral immune system after the previous DMT cessation. In line with this, our patients also demonstrated a slight increase in T2 lesion load in the respective time point.
A small number of study subjects is a clear limitation of our study. Being the first study to incorporate serial TSPO PET within a relatively short time frame in MS, the estimation of an appropriate sample size versus statistical power was challenging. The small number of subjects was probably the reason why we found no statistically significant alterations in several of the measured parameters. The reproducibility of 11C-(R)-PK11195 imaging using an older generation HR+ scanner and the supervised clustering algorithm has proven feasible among healthy controls (25), but no data have yet existed on the repeatability of 11C-(R)-PK11195 PET imaging in MS using higher resolution PET scanners, such as HRRT. This has to be taken into consideration when interpreting the results on radioligand binding especially in the nonlesional areas with lower variance. Our results also show that the washout period needs to be considered when designing TSPO PET studies. To catch the pivot point of the highest TSPO binding after a drug-free washout period, we recommend obtaining the first PET image 2−3 mo after the initiation of DMT (Fig. 5). If comparisons are to be done to a baseline image obtained immediately before the initiation of treatment, a longer follow-up should be considered. Our estimates show that with such a design one should be able to reach adequate statistical power with a feasible number of study subjects in terms of radiation safety and imaging costs. On the basis of the power calculations and the results of the present study, 15 patients per study group should be enough to reach sufficient statistical power to demonstrate the effect of fingolimod on microglial activation within the NAWM. The magnitude of the possible treatment effect of other DMTs on TSPO binding cannot, however, be predicted only from these results. Thus, there remains a challenge with power calculations for the future TSPO PET treatment studies with other DMTs in MS. Taking into account the lessons learned from the present study, we suggest a putative design for performing short-term studies evaluating the treatment effects of DMTs on diffuse microglial activation in MS (Fig. 5). Here, the recommended number of study subjects can be applied with confidence only to fingolimod studies. Finally, TSPO imaging has limitations in terms of identifying a particular microglial phenotype, and efforts are continuing for development of better PET ligands to differentiate between microglial cells with different functions.
Putative design for performing short-term PET studies for evaluation of efficacy of DMTs to reduce diffuse microglial activation in MS brain.
CONCLUSION
The present study is the first one to apply serial TSPO PET to evaluate microglial response to a DMT in MS. A reduction in microglial/macrophage activation was demonstrated after 6 mo of fingolimod treatment in brain areas corresponding to T2 lesions, but no alteration in radioligand binding was observed in the NAWM or GM. The study demonstrates the possibilities and challenges of TSPO PET in longitudinal evaluation of microglial activation status in response to treatment and opens new vistas for designing future therapeutic studies that use the evaluation of microglial activation as an imaging outcome measure.
DISCLOSURE
Financial support for this study was provided by the Finnish Academy, the Sigrid Juselius Foundation, The Finnish MS Foundation, The Finnish Medical Foundation, The State Research Funding, Novartis Pharma, and the European Union's Seventh Framework Program (FP7/2007-2013) under grant agreement HEALTH-F2-2011-278850 (INMiND). No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We are thankful to all subjects participating in the study and making it possible.
Footnotes
↵* Contributed equally to this work.
Published online Mar. 23, 2017.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication August 29, 2016.
- Accepted for publication March 13, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Nasal foralumab treatment of PIRA induces regulatory immunity, dampens microglial activation and stabilizes clinical progression in non-active secondary progressive MS
- Integrating TSPO-PET imaging with metabolomics for enhanced prognostic accuracy in multiple sclerosis
- Innate Immune Cell-Related Pathology in the Thalamus Signals a Risk for Disability Progression in Multiple Sclerosis
- Natalizumab treatment reduces microglial activation in the white matter of the MS brain