


Abstract
The development of molecular therapies for cancer treatment has created a need to image biochemical and molecular processes to appropriately select tumors that express the drug target, thereby predicting a positive response to therapy. Biomarker-driven molecular imaging is complementary to pathologic analysis and offers a more direct measure of drug efficacy and treatment response, potentially providing early insight into therapeutic futility and allowing response-adapted treatment strategies. Imaging also allows a unique means of assessing the heterogeneity of both intra- and intertumoral targets as well as a mixed response to therapy; this information is important in the setting of metastatic disease. Here we review the development of novel molecular imaging probes and combinations of probes to guide therapy for two new targets and associated therapeutic agents: cyclin-dependent kinase inhibitors and poly(adenosine diphosphate–ribose) polymerase inhibitors.
- novel radiotracers
- PET imaging
- breast cancer
- proliferative status
- proliferation rate
- PARP
- targeted therapies
The expanding array of therapeutic targets for breast cancer has created exciting opportunities to improve treatment, as well as new challenges. Specifically, given the heterogeneity of this disease, how does the breast cancer physician choose the optimal therapy or combination of therapies from an ever-expanding array of options? Equally important, how can imaging help guide the development and testing of drugs designed to target new agents? Molecular imaging is ideally equipped to address both tasks by providing a means to directly measure target expression and drug efficacy that can allow early response-adapted treatment strategies in the case of therapeutic futility. Imaging is complementary to pathology by assessing the heterogeneity of both intra- and intertumoral targets, which provides a window into a mixed response to therapy—especially important in the setting of metastatic disease.
In this review, we highlight the development of molecular imaging probes and new combinations of molecular imaging probes as a key step in developing imaging biomarkers to guide early drug testing and clinical use. We chose two emerging breast cancer treatment strategies: poly(adenosine diphosphate–ribose) polymerase (PARP) and cell cycle inhibition. The former illustrates the development of new probes for a new target, and the latter illustrates how combinations of probes—sometimes designed for similar targets—can guide a new therapeutic strategy.
CELL CYCLE–TARGETED THERAPIES AND NEED TO IMAGE CELL PROLIFERATION
New drugs targeting proteins expressed in different phases of the cell cycle (1–3) motivate the need for more sophisticated methods of assessing the cell cycle in breast cancer and other tumor types. Researchers have noted that “it is of pivotal importance to identify which subgroup of patients would mostly benefit from [cyclin-dependent kinase] CDK4/6 inhibition with biomarker-driven clinical trials” (4). Cell cycle regulators, such as the recently approved palbociclib, act by preventing cell cycle transition to the S phase and halting proliferation. During proliferation, cyclin D1–CDK4/6 complexes sequester the cyclin kinase inhibitors p21Cip1 and p27Kip1, allowing the activation of cyclin E1–CDK2 complexes and cell cycle progression (5). p21Cip1-dependent inhibition of cyclin D1–CDK4/6 in breast cancer cells causes the arrest of cells in a state with characteristics of G0 (quiescence) (Fig. 1) (6). Furthermore, inhibition of CDK4 function with the CDK4/6 inhibitor palbociclib or inducible shutdown of cyclin D1 in mice with HER2/neu-driven breast tumors results in tumor cell senescence, with a markedly reduced fraction of cells expressing the proliferation biomarker Ki-67 (7).
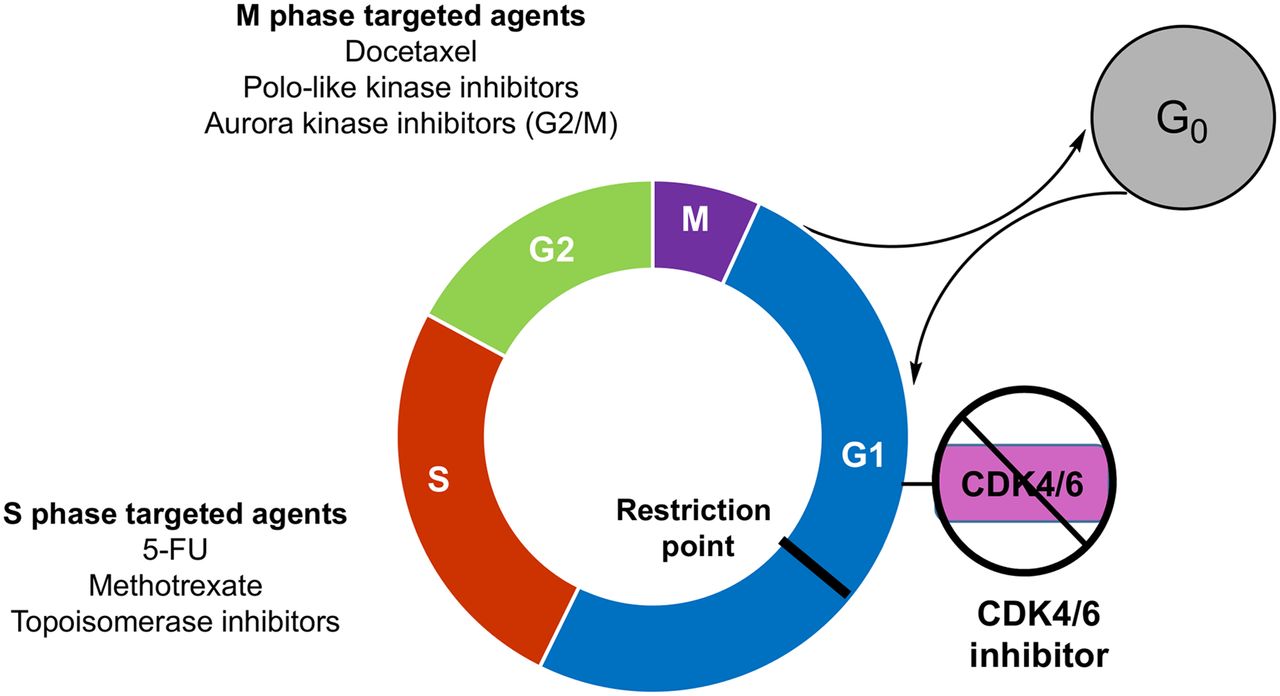
Illustration of proliferating and quiescent tumor cells and representative cell cycle–specific chemotherapeutics. 5-FU = 5-fluorouracil.
Traditional imaging methods, such as MRI, CT, and bone scanning, have a limited ability to predict or monitor treatments that specifically target proliferation and produce a cytostatic response (8). 18F-FDG measures of cancer metabolism allow early insight into the response of breast cancer metastatic disease to cytotoxic and endocrine agents but do not measure proliferation (9) and do not correlate with tumor proliferative status or growth rate (10). Imaging strategies that can measure cell proliferation and cell cycle arrest in G1/G0 have the potential to identify patients likely to respond to cell cycle targeted therapeutics such as CDK, chk-1, and polo kinase inhibitors.
TWO COMPONENTS OF CELL PROLIFERATION: PROLIFERATION RATE AND PROLIFERATIVE STATUS
Proliferation Rate
To provide an understanding of the measures required to monitor cell cycle–targeted agents, we briefly highlight an underlying concept related to cell proliferation. Proliferation rate refers to the time it takes a proliferating cell to pass through the 4 different phases of the cell cycle: G1, S, G2, and M. An alternative term is the tumor doubling time (TD) because the end of the M phase is marked by the division of the tumor cell into two daughter cells. In cell cultures, there are two different ways to measure proliferation rate: counting the number of cells as a function of time to calculate the TD or measuring the S-phase fraction of the tumor with either 3H-thymidine or the thymidine analog 5-bromodeoxyuridine (BrdU) to determine the salvage pathway of DNA synthesis. The key step in the salvage pathway involves phosphorylation of the 5′-hydroxy position of thymidine by the enzyme thymidine kinase 1.
Because the lengths of S, G2, and M are relatively constant, the difference between a rapidly growing tumor (TD, ∼24 h) and a slowly growing tumor (TD, approximately several days) is the length of G1. Tumors having a high proliferation rate (and a rapid TD) have a short G1 phase and a high S-phase fraction (Fig. 2). For example, a tumor with a TD of 48 h has an S-phase fraction of 17%, whereas a tumor with a TD of 240 h has an S-phase fraction of 3%. Radiolabeled thymidine analogs are predicted to have higher uptake in tumors with a higher S-phase fraction (high proliferation rate) than in slower-growing tumors. This prediction was confirmed in an elegant study by Nishii et al. (11), who demonstrated that the uptake of both 2′-deoxy-2′-18F-fluoro-5-methyl-1-β-l-arabinofuranosyluracil (18F-FMAU) and 3′-deoxy-3′-18F-fluorothymidine (18F-FLT) was much higher in a rapidly growing tumor than in a slowly growing tumor (Fig. 3A). A review of clinical PET studies with radiolabeled thymidine analogs is presented in this supplement by Kenny (12).

(Top) Illustration of proliferating (P) and quiescent (Q) tumor cells. TD of left tumor is 48 h, resulting in S-phase fraction (SPF) of 17%. TD of right tumor is 240 h, resulting in SPF of 3% and slower proliferation rate. (Bottom) Proliferative status, measuring ratio of number of P cells to number of Q cells. P/Q ratio is 2 because there are 2 P cells and 1 Q cell. Proliferative status is independent of proliferation rate.
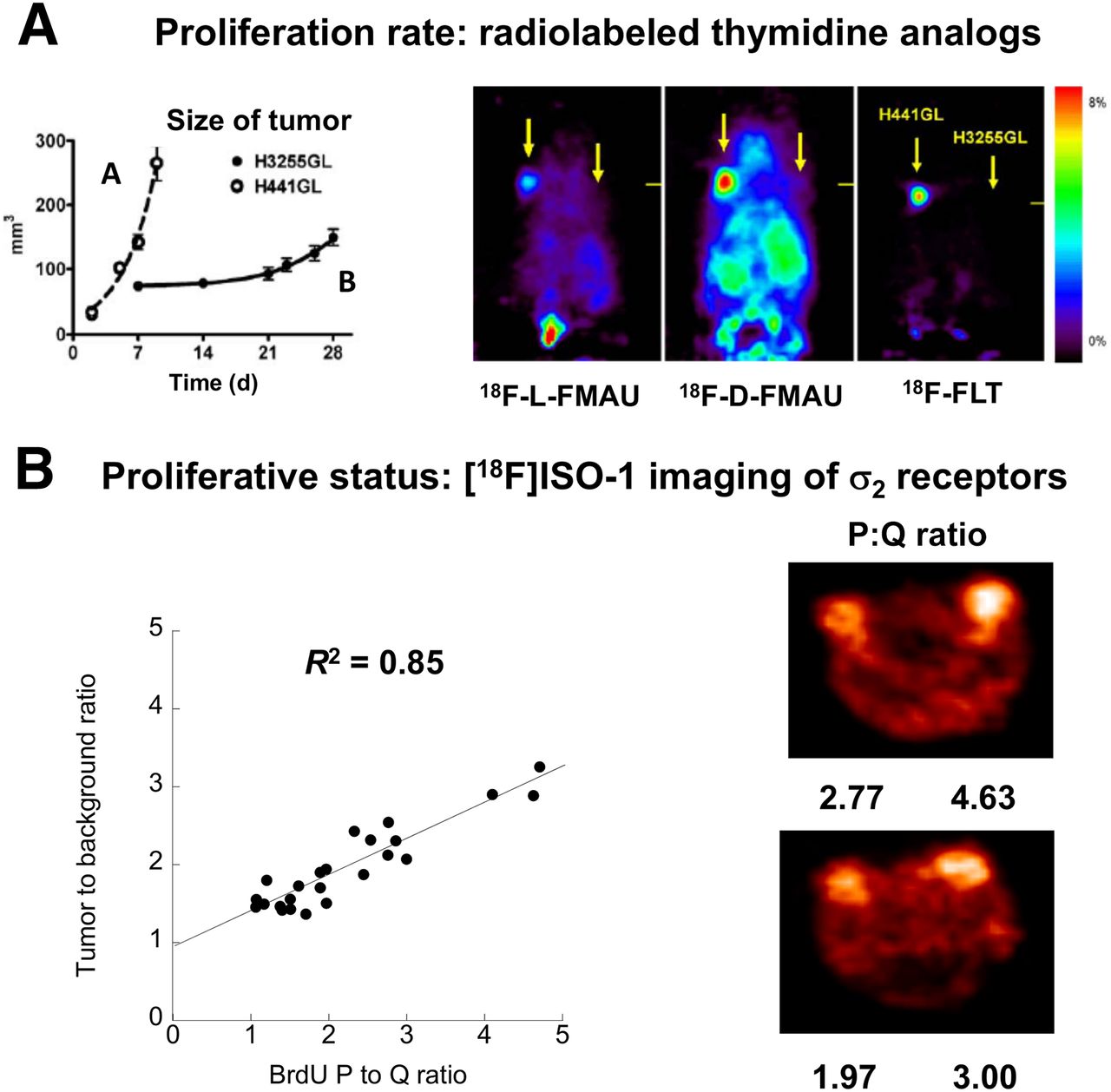
(A) Imaging of proliferation rate with radiolabeled thymidine analogs. Note higher uptake of radiotracers in H441GL tumor, which had faster doubling time (and higher S-phase fraction), than in H3255GL tumor. (B). Imaging of proliferative status with σ2-receptors in a mouse model of breast cancer. Note linear relationship between tumor-to-background ratio of 18F-ISO-1 and P:Q ratio of breast tumor.
Another property of tumor cells that has recently gained increased recognition is tumor quiescence (13–16). It is well known that when tumor cells are deprived of the basic nutrients needed to support cell proliferation or are under the stress of tumor growth and anticancer therapy, they can exit the cell cycle and enter prolonged quiescence (G0) (13–16). All solid tumors, even those as small as 2–5 mm, can contain cancer cells that are proliferating and cancer cells that are quiescent. Determination of the ratio of proliferating cells to quiescent cells (P:Q ratio) is another method for measuring cell proliferation; this property is known as the proliferative status of the tumor (17–19). Several molecular markers have been used to distinguish proliferating and quiescent cell populations in a solid tumor; these include Ki-67 (20–22), proliferating cell nuclear antigen (22), ribonucleotide reductase M1 subunit (23), and chromatin assembly factor 1 (24). Ki-67, which is expressed at high levels in proliferating cells and at lower levels in quiescent cells (25), is generally considered the gold standard for measuring the proliferative status of a broad spectrum of solid tumors (20).
A tumor with a proliferative status of 2 has twice as many proliferating cells as quiescent cells (Fig. 2). Proliferative status is also independent of proliferation rate because proliferation rate measures only the number of cells in S-phase and does not consider the total length of time for tumor progression through the cell cycle. Because quiescent tumor cells are not cycling, their proliferation rate is 0. Quiescent tumor cells are biologically different from senescent normal cells; quiescent tumor cells are undifferentiated cells that can be transformed into cycling, proliferating cells once the conditions of hypoxia or nutrient deprivation are eliminated (18,19,26,27). Senescent cells have undergone terminal differentiation and do not possess the ability to reenter the cell cycle. Quiescent cells can evade therapy and are increasingly being recognized as a primary factor in tumor recurrence (28,29).
Imaging of Proliferative Status: σ2-Receptor as Biomarker for Imaging of Proliferative Status of Breast Cancer
Although Ki-67 is the gold standard for measuring cell proliferation in histology studies and would be an excellent molecular marker for imaging of the proliferative status of breast tumors with PET, there are no small molecules with a high affinity for Ki-67 to serve as lead compounds for PET radiotracer development. Because Ki-67 is a nuclear protein and cell permeabilization methods are needed to label it under cell culture or tissue staining conditions, radiolabeled antibodies for Ki-67 are not expected to work well for the imaging of solid tumors.
Another protein that behaves in a manner similar to that of Ki-67 and has small molecules that bind to the protein with high affinity is the σ2-receptor. The σ2-receptor was initially identified as a biomarker of tumor cells by Vilner et al. (30), who reported a high density of σ2-receptors in many human and murine tumor cells grown under cell culture conditions. Subsequent studies by Mach et al. (17) and Wheeler et al. (19) demonstrated that σ2-receptors were expressed at a 10-fold-higher density in proliferating mouse adenocarcinoma (line 66) cells (66P cells) than in quiescent line 66 cells (66Q cells), both under cell culture conditions (17) and in solid tumor xenografts (19). Furthermore, upregulation of σ2-receptors for the transition from 66Q cells to 66P cells and downregulation from 66P cells to 66Q cells took approximately 3 d (18); these behaviors were similar to those of other membrane-bound receptors.
These observations resulted in extensive efforts to synthesize σ2-receptor–selective radioligands for in vivo imaging studies (3,12). The most promising radiotracer reported to date—and the only σ2-receptor–selective PET radiotracer to be used in human studies—is the conformationally flexible benzamide analog 2-(2-[18F]fluoroethoxy)-N-(4-(3,4-dihydro-6,7-dimethoxyisoquinolin-2(1H)-yl)butyl)-5-methyl-benzamide (18F-ISO-1). In vivo small-animal PET studies demonstrated that 18F-ISO-1 imaged breast tumors in a rodent model of breast cancer (31). However, a key step in the validation of this radiotracer for imaging of the proliferative status of breast tumors was the demonstration of a good correlation between 18F-ISO-1 uptake and the P:Q ratio of a panel of breast tumors. This finding was confirmed in a study by Shoghi et al., who conducted small-animal PET imaging studies with 18F-ISO-1 in nude mice implanted with mouse mammary carcinoma cells (10). After imaging, the tumors were removed and dissociated into cells, and the P:Q ratios of individual tumors were determined with flow cytometry. There was a high correlation between the tumor-to-background ratio of 18F-ISO-1 and the P:Q ratio of the panel of tumors (Fig. 3B). These data confirmed that 18F-ISO-1 is a useful radiotracer for measuring the proliferative status of breast tumors with PET and set the stage for subsequent clinical imaging studies with this radiotracer.
A first-in-human study of 18F-ISO-1 was completed in patients with breast, lymphoma, and head and neck cancers (32). The results of PET/CT studies indicated that 18F-ISO-1 was capable of specifically imaging these tumors (Fig. 4A). Analysis of the dynamic imaging data in breast cancer patients indicated that uptake in tumors peaked early after injection and remained constant through the 60-min scan. The blood activity cleared to a stable level within a few minutes. Metabolite analysis indicated that approximately 90% of the parent compound was present in blood throughout the study, with the remaining 10% being localized in red blood cells. The uptake of 18F-ISO-1 in blood, muscle, and tumors indicated that equilibrium was achieved in these compartments rapidly (10–20 min after intravenous injection), with subsequent stability. Because of these properties of 18F-ISO-1 distribution, a simple tumor-to-muscle ratio was adequate for analyzing the clinical imaging data. It was found that a Ki-67 score of 35% was the best cutoff for distinguishing tumors having high versus low Ki-67 scores (Fig. 4B).

(A) PET/CT imaging studies of 18F-ISO-1 in breast cancer, head and neck cancer, and lymphoma patients. (B) Correlation between Ki-67 score and tumor-to-muscle (T:M) ratio. Red line with arrows demonstrates that T/M ratio for tumors with high Ki-67 scores was above mean for tumors with low Ki-67 scores. (C) Whole-body distribution of 18F-ISO-1 at 2 time points. Lack of uptake of 18F-ISO-1 in bone will permit imaging of bone metastases. GB = gallbladder; GI = gastrointestinal.
One of the potential limitations of using σ2-receptor radiotracers for imaging of the proliferative status of solid tumors is the high density of σ2-receptors in the liver. Whole-body images of 18F-ISO-1 at 30 and 150 min after injection of the tracer indicated some initial uptake of 18F-ISO-1 in the liver (Fig. 4C). However, the tracer was metabolized and cleared through the hepatobiliary system, so the uptake was distributed to the gallbladder by 150 min (32). There was also high uptake of 18F-ISO-1 in the pancreas, which also has a relatively high density of σ2-receptors. The uptake of 18F-ISO-1 in bone was very low, in contrast to what has been reported for 18F-FLT (33,34).
These early human studies suggested that 18F-ISO-1 is capable of imaging bone metastases, and this finding was demonstrated in a phase 0 clinical trial of 18F-ISO-1 in a patient diagnosed with lymphoma. Taken together, these data indicate the promise of 18F-ISO-1 for imaging the proliferative status of breast tumors and predicting response to current and emerging breast cancer treatments. An imaging probe that characterizes proliferating and quiescent cells and that can also image bone metastases may be highly complementary to 18F-FLT PET, especially for evaluating cell cycle–targeted therapies in metastatic disease.
Imaging of Proliferation Rate and Proliferative Status to Guide Cell Cycle–Targeted Therapy
Measurement of the proliferation rate and determination of the proliferative status are complementary methods for imaging cell proliferation. Measurement of the S-phase fraction of tumor cells is the most reliable way to measure the proliferation rate, and with PET this measurement is accomplished with radiolabeled thymidine (11C-thymidine) or thymidine analogs (18F-FLT and 18F-FMAU). Ki-67 is used in histology and flow cytometry studies to measure the pathologic proliferative status of a tumor. The only molecular imaging method that can measure the proliferative status of a tumor involves the use of the σ2-receptor probe 18F-ISO-1. The complementary nature of these two imaging techniques is clearly demonstrated in the small-animal PET imaging studies shown in Figure 3. There was much higher uptake of the radiolabeled thymidine analogs in the faster-growing tumor H441GL tumor (TD, ∼48 h) than in the H3551GL tumor, which had a TD of 240 h. With the σ2-receptor probe 18F-ISO-1, higher uptake was observed in breast tumors with a high P:Q ratio. This example illustrates the complementary nature of the data provided by the two classes of agents, namely, thymidine analogs and σ2-receptor probes. The combination of markers that indicate proliferative status and proliferation rate (as shown in Fig. 2) more fully characterizes the cell cycle status of cancer cells, akin to flow cytometry, with noninvasive quantitative imaging methods. This property is likely to be of increasing importance because targeted therapeutic agents now under development affect cells in a specific phase of the cycle, requiring a more comprehensive knowledge of tumor cell cycle distribution to guide therapy selection and evaluate efficacy.
PARP INHIBITORS AND THE NEED TO IMAGE PARP1 ACTIVITY
PARP is a family of enzymes involved in base excision repair (repair of DNA single-strand breaks) and alternative end joining (repair of DNA double-strand breaks). The molecular basis of PARP1 inhibitor function may depend on the dual roles of PARP1: as a modulator of gene transcription and in DNA damage repair (35). Inhibition of PARP results in persistent single-strand DNA breaks, which are subsequently converted to double-strand breaks. Double-strand breaks can be repaired through homologous recombination, nonhomologous end joining, and alternative end joining. In BRCA-mutated tumors and other tumors with defects in DNA repair, PARP inhibition can lead to genetic errors and instability, with subsequent cell death, because homologous recombination is not active and nonhomologous end joining is error-prone (36).
Preclinical Studies of PARP Inhibitors
Combining PARP inhibition synergistically with radiation for cellular killing has been investigated since the 1980s (37,38). The recent development of more specific and potent PARP inhibitors has spurred human trials based on provocative preliminary data. In 2005, various investigators published the observation that the inhibition of PARP activity in BRCA1- and BRCA2-mutated cells resulted in chromosomal instability, cell cycle arrest, and apoptosis (39–41). Without a viable mechanism of repair, BRCA-mutated cells would selectively undergo cellular lethality. Subsequent in vivo studies demonstrated that PARP inhibition selectively blocked the growth of BRCA2-deficient tumors (39,40). Thus, it was hypothesized that targeting PARP in BRCA-mutated human tumors might be a selective therapeutic strategy.
Early Clinical Trials
On the basis of strong preclinical data, selective PARP inhibitors were developed and tested on multiple cancer types, including BRCA1 and BRCA2 mutant ovarian and breast cancers. Eventually, first-in-human studies of the PARP1/2 inhibitor olaparib as a single agent in advanced cancers were conducted. In an early phase 1 trial evaluating olaparib, any patient with refractory cancer was enrolled (50% had ovarian or breast cancer), but antitumor activity was observed only in patients with known BRCA mutations (42). Studies with expansion cohorts focused on patients with BRCA mutations, where progression-free benefit was demonstrated for only a subset of patients, indicating the need for an additional predictive biomarker (43,44). Additional benefit in patients with BRCA mutations was demonstrated in a randomized phase 2 study of olaparib for recurrent serous ovarian cancer (45,46). Although there is evidence that PARP inhibitors also benefit patients without germ line BRCA mutations in multiple cancer types, the recent regulatory approval of olaparib by the Food and Drug Administration was only for therapy for refractory ovarian cancer in patients with germ line BRCA1/2 mutations. Future selective PARP therapies likely will be expanded beyond the BRCA population and will target other tumor types and even cardiovascular or inflammatory disease (35).
Current Clinical Trials
As of March 2015, there were 116 active trials evaluating PARP inhibitors on ClinicalTrials.gov, and PARP was being used as a cancer therapy in 112 of these. Twenty of these trials specifically target breast or breast and ovarian cancer. An additional 27 trials were enrolling patients with any solid malignancy, likely resulting in a strong ovarian and breast cancer representation. Surprisingly, although 38 of these trials focus on BRCA mutation enrichment and 3 focus on other biomarkers, 71 of them do not select for any biomarker as a means of identifying patients who might respond to therapy (35).
How Can Patients Who Will Respond to PARP Inhibitors Be Identified?
Although promising preliminary results have been shown for PARP inhibitors, the heterogeneous clinical response, even within patient populations selected for BRCA mutations, indicates a need to predict and monitor the response to therapy. Without this ability, patients are exposed to months of costly and possibly ineffective treatment before a decision is made about therapeutic futility. One recent review (47) on the future clinical applications of PARP inhibitors stated, “At present, it remains unclear how to best identify patients who will respond to PARP inhibitors. Although tumor phenotypes can provide rough predictions…it seems that optimal clinical development might be advanced by improved understanding of both the mechanism of action of PARP inhibitors and mechanisms of resistance.” Another review (35) asked, “What molecular markers can be identified that may predict sensitivity to PARP1 inhibitors?” A third review (48) stated that a critical issue surrounding PARP investigations is “identification of patients who stand to benefit from such drugs.”
Imaging PARP1 Activity with PET
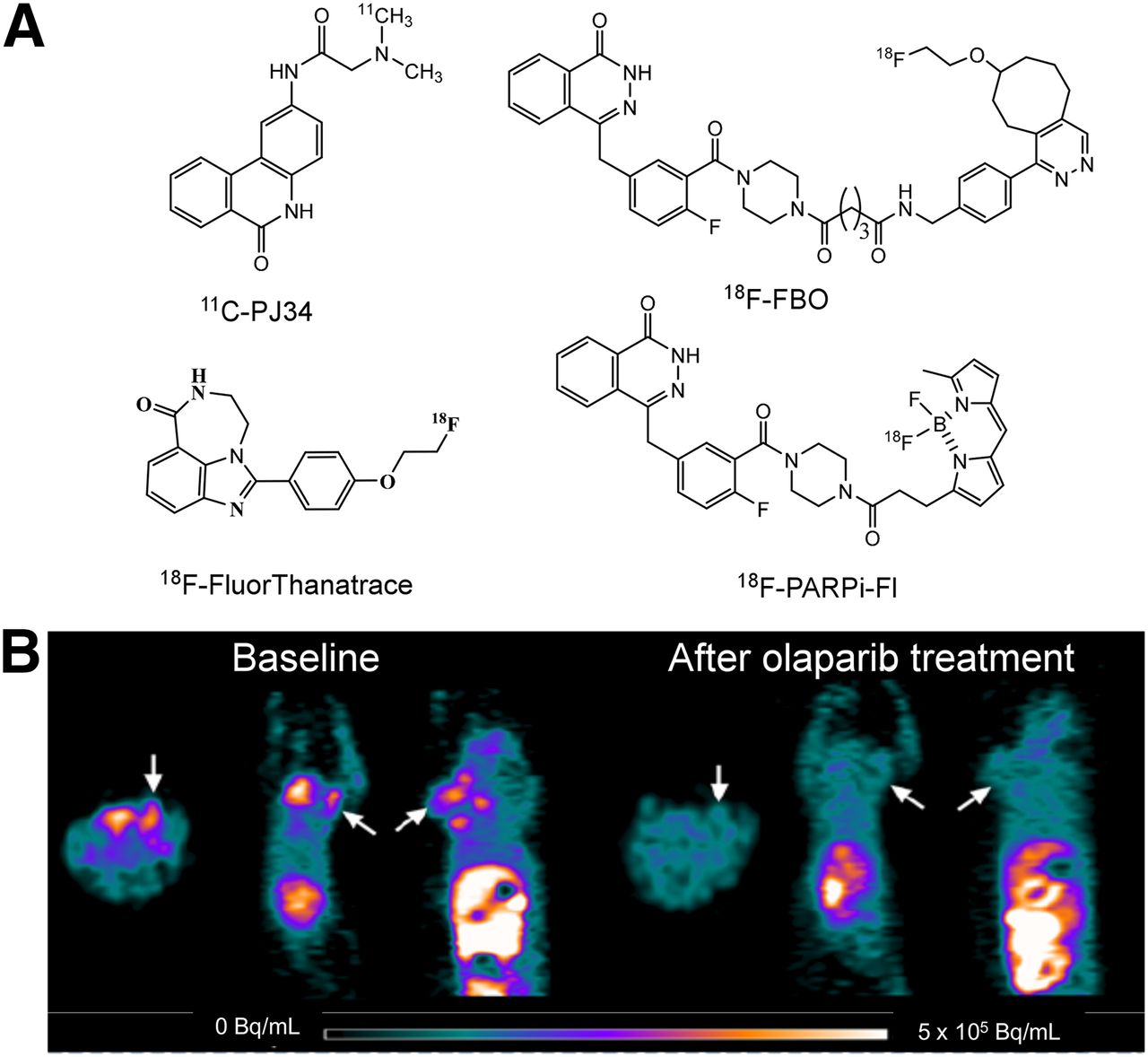
One method of identifying patients who likely will have a favorable response to PARP inhibitors is to measure PARP activity directly with PET. The availability of multiple PARP1 inhibitors as therapeutic agents led to their use as lead compounds for PET radiotracer development. Four radiotracers have been reported to date. PARP1 inhibitor PJ34 has been labeled with 11C, and promising results in an animal model of type 1 diabetes mellitus have been reported (49). However, no study evaluating this radiotracer in breast tumor models has been reported. Three 18F-labeled radiotracers have also been developed: 18F-FBO, which is an analog of olaparib (50); the dual-modality optical/PET probe 18F-PARPi-Fl, which is also based on olaparib (51); and 18F-fluorthanatrace, which is an analog of AG14699 (Fig. 5) (52). Small-animal PET studies of 18F-FBO revealed a strong correlation between radiotracer uptake and PARP1 expression in a panel of ovarian and pancreatic tumors. The low specific activity of 18F-PARPi-Fl, which is radiolabeled with 18F for 19F exchange, likely will limit the clinical utility of this probe. 18F-fluorthanatrace showed high uptake in a breast tumor with a high level of expression of PARP1; this uptake could be blocked with olaparib, suggesting that this uptake was specific to PARP1 expression (52). A phase 0 clinical trial of 18F-fluorthanatrace is currently being conducted at Washington University, and new trials on other tumor types and in various therapeutic settings are planned.
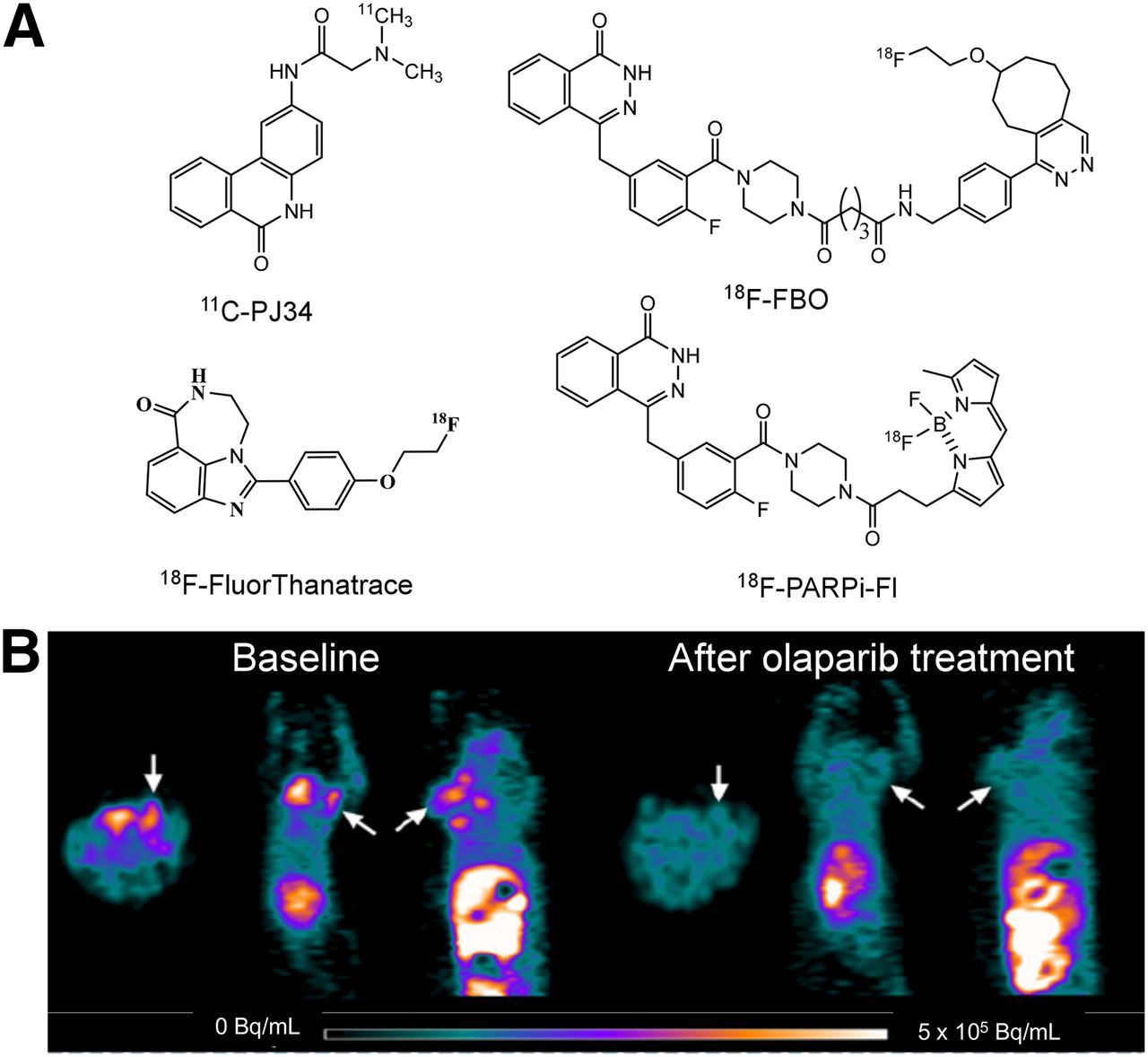
(A) Structures of PARP1 PET radiotracers reported to date. (B) Small-animal PET images of 18F-fluorthanatrace in murine model of breast cancer.
CONCLUSION
We have highlighted examples in which new molecular imaging agents or combinations of molecular imaging agents show potential to inform drug development and early testing of new breast cancer therapeutic strategies. For example, the imaging agents discussed here can potentially be used as predictive biomarkers: PARP imaging will directly measure the level of expression of the therapeutic target, whereas 18F-ISO-1 imaging will measure the proliferative status—which is important for cell cycle–dependent therapeutics. They can also be used to identify an early pharmacodynamic response; for example, the ability to differentiate between quiescent and proliferating cells could be an important means of monitoring the mechanistic efficacy of new CDK4/6 inhibitors that are expected to drive proliferating tumor cells into prolonged quiescence.
The role of cancer molecular imaging biomarkers in targeted drug development will continue to expand and enable a new era of personalized cancer therapy. The development and validation of novel molecular imaging probes designed to evaluate new targets or treatment strategies is essential to the practice of precision medicine for breast cancer.
DISCLOSURE
This work was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through grant UL1TR000003. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was also supported by a Komen leadership grant, AC140060, and a Department of Energy training grant, DE-SE0012476. No potential conflict of interest relevant to this article was reported.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication September 1, 2015.
- Accepted for publication November 2, 2015.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}