


Abstract
18F-FDG accumulates in glycolytically active tissues and is known to concentrate in tissues that are rich in activated macrophages. In this study, we tested the hypotheses that human granulocyte-macrophage colony-stimulating factor (GM-CSF), a clinically used cytokine, increases macrophage glycolysis and deoxyglucose uptake in vitro and acutely enhances 18F-FDG uptake within inflamed tissues such as atherosclerotic plaques in vivo. Methods: In vitro experiments were conducted on human macrophages whereby inflammatory activation and uptake of radiolabeled 2-deoxyglucose was assessed before and after GM-CSF exposure. In vivo studies were performed on mice and New Zealand White rabbits to assess the effect of GM-CSF on 18F-FDG uptake in normal versus inflamed arteries, using PET. Results: Incubation of human macrophages with GM-CSF resulted in increased glycolysis and increased 2-deoxyglucose uptake (P < 0.05). This effect was attenuated by neutralizing antibodies against tumor necrosis factor–α or after silencing or inhibition of 6-phosphofructo-2-kinase. In vivo, in mice and in rabbits, intravenous GM-CSF administration resulted in a 70% and 73% increase (P < 0.01 for both), respectively, in arterial 18F-FDG uptake in atherosclerotic animals but not in nonatherosclerotic controls. Histopathologic analysis demonstrated a significant correlation between in vivo 18F-FDG uptake and macrophage staining (R = 0.75, P < 0.01). Conclusion: GM-CSF substantially augments glycolytic flux in vitro (via a mechanism dependent on ubiquitous type 6-phosphofructo-2-kinase and tumor necrosis factor–α) and increases 18F-FDG uptake within inflamed atheroma in vivo. These findings demonstrate that GM-CSF can be used to enhance detection of inflammation. Further studies should explore the role of GM-CSF stimulation to enhance the detection of inflammatory foci in other disease states.
PET, in combination with 18F-FDG, has been shown to be useful for the identification of inflamed tissues. Over the past few years, 18F-FDG PET/CT imaging has been put into increasing clinical use to evaluate syndromes such as fever of unknown origin, cardiac sarcoidosis (1,2), prosthetic valve endocarditis (3), and infection of implanted devices (4). More recently, the use of 18F-FDG PET/CT as a research tool to characterize atherosclerotic cardiovascular disease has been increasing. However, more widespread use of 18F-FDG PET imaging of inflammatory foci has been limited, in part because of the relatively modest signal (5). Approaches to enhance 18F-FDG localization to inflamed tissues could improve the clinical utility of 18F-FDG PET imaging of inflammatory diseases.
Perhaps the most common chronic inflammatory disorder, atherosclerotic cardiovascular disease, remains the leading cause of mortality in the United States (6). Inflammation plays a pivotal role in atherogenesis, plaque progression, and thrombotic complications (7). In particular, the pathogenesis of atherosclerosis involves a myriad of immune mediators, with a well-accepted role for macrophages (8). A substantial body of cellular physiology literature has established that activated macrophages show upregulated glycolysis and hence avidly accumulate 18F-FDG (5,9). Key enzymatic mediators of macrophage activity include the glucose transporters, the upper part of glycolysis, and specifically the expression of the ubiquitous form of 6-phosphofructo-2-kinase (PFKFB3), which has been noted to be upregulated in stimulated macrophages (compared with the constitutively expressed liver type-6-phosphofructo-2-kinase [PFKFB1] isoenzyme, which is associated with lower rates of glycolysis) (9). Advances in 18F-FDG PET imaging have led to its use for the quantification of vascular wall inflammatory activity. 18F-FDG uptake has been shown to strongly correlate with arterial wall macrophage infiltration (10), systemic proinflammatory biomarkers (11), inflammatory cell gene expression (12), and increased risk for subsequent atherothrombotic events (13,14).
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a Food and Drug Administration–approved medication used to stimulate the production of white blood cells and thus prevent neutropenia after chemotherapy. It affects a wide range of immune cells, including macrophages, neutrophils, and dendritic cells (15–18). The drug is well tolerated, even among individuals with atherosclerosis or those with recent atherothrombotic events (19,20). Accordingly, we sought to evaluate whether GM-CSF could be used as an adjunctive agent to enhance the detection of inflamed foci. Specifically, we performed both in vitro and in vivo experiments to better understand the effect of GM-CSF on macrophage glycolytic flux and on 18F-FDG uptake, which is dependent on glucose flux. In these studies, we tested the hypotheses that GM-CSF augments glycolytic flux in macrophages in vitro and enhances 18F-FDG accumulation within inflammatory foci in vivo, thereby enhancing imaging sensitivity.
MATERIALS AND METHODS
Cellular Experiments
Chemicals
Reagents and antibodies were from Sigma-Aldrich, Roche, Invitrogen, R&D Systems, Santa Cruz Biotech, or Merck-Millipore. [U-14C]-2-deoxyglucose (9,250 kBq [250 μCi]/mmol) was from New England Nuclear/Perkin Elmer. Serum and media were from BioWhittaker. Murine and human GM-CSFs were from PeproTech or Gentaur.
Preparation of Human Monocytes/Macrophages
PBMCs were isolated from the blood of healthy donors by centrifugation on Ficoll-Hypaque Plus (GE Biotech) following the manufacturer’s protocol, and the CD14-enriched fraction was collected after binding to MACS-hCD14-magnetic beads (Miltenyi Biotec). Cells were differentiated with human CSF-1 (20 ng/mL, PeproTech) for 5 d in RPMI1640 supplemented with antibiotics and 10% fetal calf serum. After this period, the cells were kept for 48 h in medium lacking CSF-1 and treated with the indicated stimuli. The purity of all cultures was verified by CD14+ staining; on average, more than 95% of the cells were highly positive for this surface marker.
Tumor Necrosis Factor–α (TNF-α) Neutralization
The cells were maintained in culture and, 1 h before GM-CSF challenge, were treated with a 20 ng/mL concentration of anti-TNF-α neutralizing antibody (R&D Systems) as previously described (9), using a mouse IgG as control.
PFKFB3 Silencing in Macrophages
The cells were transfected overnight with lipofectamine and a mixture of 3 different Silencer Select predesigned silencer RNAs from Ambion/Invivogen, following the instructions of the supplier. Controls with scrambled (negative) RNAs were used to ensure the specificity of the silencing.
Measurement of Radiolabeled Deoxyglucose Accumulation
To evaluate 2-deoxyglucose uptake, the cells (6-cm dishes) were washed with warm Dulbecco modified Eagle medium lacking glucose and incubated with this medium containing 0.3 mM glucose and 92.5 kBq (2.5 μCi) of [U-14C]-2-deoxyglucose for 60–90 s, following a previous protocol (21). The reaction was stopped by rapid aspiration of the medium and addition of 1 mL of ice-cold Dulbecco modified Eagle medium containing 10 mM glucose and 0.1 mM phloretin. The cell layer was washed twice with ice-cold phosphate-buffered saline, and the cells were resuspended in 1 mL of 0.1 M NaOH in 0.1% sodium dodecyl sulfate. A linear incorporation of radioactivity was observed for at least 2 min by scintillation counting.
Preparation of Macrophage Extracts
The cell cultures were washed twice with ice-cold phosphate-buffered saline and homogenized in 0.2 mL of buffer containing 10 mM Tris-HCl, pH 7.5; 1 mM MgCl2; 1 mM ethylene glycol tetraacetic acid; 10% glycerol; 0.5% CHAPS buffer (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate); 1 mM β-mercaptoethanol; and 0.1 mM phenylmethylsulfonyl fluoride and a protease inhibitor cocktail (Sigma). The extracts were stirred in a vortex mixer for 30 min at 4°C and centrifuged for 15 min at 13,000g. The supernatants were stored at −20°C. Protein levels were determined using the Bio-Rad detergent-compatible protein reagent. All steps were performed at 4°C.
Western Blot Analysis
Samples of cell extracts containing equal amounts of protein (30 μg per lane) were boiled in 250 mM Tris-HCl, pH 6.8; 2% sodium dodecyl sulfate; 10% glycerol; and 2% β-mercaptoethanol and size-separated in 10%–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis. The gels were blotted onto a polyvinylidene fluoride membrane (GE Healthcare) and processed as recommended by the supplier of the antibodies against the following human antigens: signal transducer and activator of transcription 5 (STAT5), P-STAT5, hexokinase-1, hexokinase-2, PFK-1, PFKFB1, PFKFB3, glyceraldehyde 3-phosphate dehydrogenase, and β-actin. The blots were developed by ECL protocol (GE Healthcare), and different exposition times were performed for each blot with a charge-coupled device camera in a luminescent image analyzer (Molecular Imager; BioRad) to ensure the linearity of the band intensities.
Measurement of TNF-α, Lactate, and Fructose-2,6-Bisphosphate (Fru-2,6-P2)
TNF-α was measured in the cell culture medium using an ELISA kit (PeproTech). Lactate accumulation was also measured in the culture medium as previously described (9,22). The intracellular concentration of Fru-2,6-P2 was determined in cell extracts treated with 0.1 mL of NaOH (50 mM at 80°C) and heated at 80°C for 10 min. The metabolite was measured after the activation of the pyrophosphate-dependent 6-phosphofructo-1-kinase activity following a previous protocol (9,22).
Real-Time Quantitative Polymerase Chain Reaction
3-mo-old Apoe−/− mice were fed for 2 wk with a high-fat diet and intraperitoneally administered 37.5 μg/kg of GM-CSF on day 12. On day 14, they were sacrificed and the whole aorta was cleaned to remove the adventitial fat and connective tissue in the magnifying glass, always working over a sheet of ice. The aortas were transferred to RNAlater RNA stabilization reagent (QIAGEN) and then to dry ice. Aorta total RNA was isolated by homogenization in QIAzol (QIAGEN) by a Tissue Lyser LT (QIAGEN) and eluted in a volume of 15 μL using MinElute columns (QIAGEN). RNA integrity was assessed by RNA 6000 Nano Kit (Agilent). A 250-ng quantity of RNA was retrotranscribed using a high-capacity complementary DNA reverse transcription kit (Life Technologies). Real-time polymerase chain reaction was conducted with SYBR Green (Life Technologies) in a fast real-time polymerase chain reaction system (Life Technologies). The real-time polymerase chain reaction assay was performed with 5 ng of complementary DNA per well, and the thermocycling conditions were 95°C for 10 min and 40 cycles of 95°C for 15 s followed by 60°C for 1 min. Calculations were made from measurement of triplicates of each sample. The relative amount of messenger RNA (mRNA) was calculated with the comparative 2-ΔΔCt method. Gene expression was normalized to 36b4. Primer sequences are available on request.
Statistical Analysis
The data shown are the mean ± SD of 3–5 experiments. Statistical significance was estimated with the Student t test for unpaired observations or ANOVA followed by the Bonferroni test when appropriate. Differences with values of P less than 0.05 were considered statistically significant.
Animal Model Studies
Mouse Model
Animal care and experimental procedures were performed according to directive 2010/63/EU of the European Parliament, and the studies were approved by the Institutional Committee on Bioethics (authorization 28079-37A to the Instituto de Investigaciones Biomédicas).
PFKFB3 In Vivo Silencing
A mixture of at least 3 different Silencer Select predesigned silencer RNAs for PFKFB3 was obtained from different sources (Ambion/InvivoGen, OriGene, or Sigma-Aldrich). The transfection mixture was prepared using Invivofectamine 2.0 (InvivoGen) and was administered intraperitoneally at 5 mg/kg per dose, following the instructions of the supplier. Administration of the corresponding scrambled (negative) RNAs was used to ensure the specificity of the silencing.
Atherogenesis in ApoE-Deficient Mice and 18F-FDG PET Image Analysis
Thirty male ApoE-deficient mice 3–4 mo old were fed a high-fat/high-cholesterol diet for 3 wk, and after anesthesia with isoflurane, 18F-FDG (37 MBq/kg; 0.2 mL) was administered intraperitoneally and the 18F emission was analyzed in a small-animal CT/SPECT/PET system (Inveon; Siemens). The images were analyzed and quantified as previously described (9). Briefly, the first axial slice, representing the descending aorta (the first PET/CT slice clear of the aortic arch), and 5 consecutive slices at intervals of 3 mm were averaged to obtain the SUVmax. Measured background SUVs from the paraspinal muscles were used to obtain a corrected TBR. When PFKFB3 was silenced, the silencer RNAs were administered at days 3, 7, 10, and 12 after high-fat/high-cholesterol administration. A mixture of scrambled RNAs was used as control and administered at the same periods. GM-CSF (37.5 μg/kg) was intravenously administered on day 12. The animals were processed on day 14 for images and biochemical analyses.
Rabbit Model
Nine male New Zealand White rabbits (Charles River Breeding Laboratories) were included in the study. Seven of the rabbits were initiated on a 0.3% cholesterol, 4.7% peanut oil hyperlipidemic diet for 6 mo to precipitate the development of atherosclerosis. One week after beginning the high-cholesterol diet, the animals were briefly anesthetized using ketamine and xylazine, and aortoiliofemoral denudation was performed by balloon catheter injury using a modified Baumgartner technique (23). Additionally, 2 control rabbits of similar size and identical origin were maintained on standard rabbit chow for 6 mo. No catheterization or other invasive procedure was performed on the control animals.
Rabbit PET/CT Imaging Protocol
After 6 mo of the prescribed diet, 8 atherosclerotic animals and 2 healthy controls underwent 18F-FDG PET imaging. The animals were injected with a 37 MBq/kg dose of 18F-FDG, and PET images were obtained on a microPET P4 (Concorde MicroSystems) or similar system 3 h after 18F-FDG administration to allow for maximum tracer uptake. The choice of time interval after 18F-FDG injection was based on our previous work in animals (10). Images were obtained over 20 min and reconstructed using a filtered backprojection algorithm. The microPET P4 scanner is an animal PET tomograph with 32 planes over a 7.8-cm axial extent, 19-cm transaxial field of view, and 22-cm animal port. Within 6 d of PET imaging, multidetector CT imaging was performed for anatomic coregistration.
GM-CSF Administration to Rabbits
After baseline imaging, 6 animals (4 atherosclerotic animals and 2 healthy controls) were injected with 100 μg of sargramostim (yeast-derived recombinant human GM-CSF; Berlex) daily for a total of 3 d. Four atherosclerotic animals received saline injections at identical time points. One hour after the final injection on day 3, a 37 MBq/kg dose of 18F-FDG was administered, and PET imaging was performed 3 h afterward. The biodistribution methods and results are provided in the supplemental data, available at http://jnm.snmjournals.org (Supplemental Fig. 1).
18F-FDG PET Image Analysis in Rabbits
The PET images were analyzed with masking of treatment allocation and temporal sequence. First, localization of aortic 18F-FDG uptake was aided by coregistering the PET images with the multidetector CT images. The temporally masked baseline and follow-up PET image pairs were registered to their common multidetector CT images using a workstation that allows multimodal standard image fusion (REVEAL-MVS; Mirada Solutions) (24). Thereafter, 18F-FDG uptake could be compared for several stacked aortic segments over time. Aortic 18F-FDG uptake was quantified by drawing a circular region of interest around the aorta in the axial view, in 4-mm increments (yielding a stack of 4-mm-thick aortic slices composing the imaged aorta). For each region of interest, the maximal SUV was recorded. The SUV is the decay-corrected tissue concentration of 18F-FDG (in kBq/mL) divided by the injected dose per body weight. Background 18F-FDG uptake was measured in the paraspinal muscles as an average of ten 20-mm2 region-of-interest samples. SUVmax was then background-corrected to derive a target-to-background ratio (TBR) for each aortic slice.
Histologic Analysis in Rabbits
After final imaging assessments, 3 atherosclerotic animals were sacrificed using an overdose of sodium pentobarbital. The aortas were excised, placed in 10% buffered formalin, and decalcified according to the standard protocol. The aortic samples were sectioned transversely at 5-mm intervals and stained with rabbit macrophage-specific monoclonal antibody RAM11 (Dako Corp.). RAM11 staining was calculated as a percentage of the stained area over the total cross-sectional area of the transversely sectioned aortic wall. Multiple segments from each rabbit abdominal aorta were obtained and compared against the corresponding PET axial slices. The averaged RAM11 staining for each 5-mm aortic segment was also obtained and compared with the mean TBR for the same segments. The association between anatomic histologic segments and corresponding PET images was based on distances from the renal arteries measured ex vivo compared with the coregistered multidetector CT images. Control animals were not sacrificed for histologic analysis because previous work has demonstrated that there is no inflammation (percentage rabbit antimacrophage antibodies [%RAM11 staining]) within the aortic wall of control rabbits (24).
Statistical Analysis
Data were analyzed using SPSS, version 22 (IBM). Continuous parameters are reported as mean ± SEM. Statistical analysis comparing the change in the TBR across atherosclerotic segments (before and after treatment with GM-CSF or saline) was performed using the Wilcoxon signed-rank test. The Spearman method was used to assess the correlation between atherosclerotic tissue uptake of 18F-FDG after GM-CSF (measured by PET, as TBR) and the subsequent histopathologic assessment of inflammation in those same sections (%RAM11 staining). A P value of less than 0.05 was considered statistically significant.
RESULTS
GM-CSF Augments Glycolytic Flux via Upregulation of PFKFB3
In human macrophages, incubation with GM-CSF resulted in rapid phosphorylation of STAT5 in Y694 (Fig. 1A) and metabolic reprograming of macrophages (including modest increases in hexokinase-1 and hexokinase-2; Fig. 1B). Moreover, there was a substantial increase in PFKFB3, which was negligibly expressed before GM-CSF (Figs. 1B and 1C). The PFKFB3 isoform has a much higher net kinase activity than the constitutively expressed PFKFB1 isoenzyme. Thus, upregulation of PFKFB3 would be expected to produce substantially higher glycolytic flux. The specificity of this association was demonstrated using silencing RNA for PFKFB3, which resulted in attenuated induction of PFKFB3 (Fig. 1D), but not by the scrambled RNA sequence. Additionally, the selective PFKFB3 inhibitor 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) did not alter the induction of this gene by GM-CSF (Fig. 1D).

GM-CSF augments 2-deoxyglucose uptake and glycolytic flux in human macrophages via PFKFB3. (A and B) Human monocytes were differentiated to macrophages. Cells were exposed to human GM-CSF (10 ng/mL) for the specified time duration, and phosphorylation of STAT5 in Y694 (A) and levels of glycolytic enzymes (B) were determined. Addition of GM-CSF induced significant expression of hexokinase-1, hexokinase-2, and mainly PFKFB3. (C) Cells exposed to native GM-CSF demonstrated increased PFKFB3 expression compared with incubation with heat-inactivated GM-CSF (10 min at 80°C). (D) GM-CSF–mediated increase in PFKFB3 expression (right) was decreased with addition of silencer to PFKFB3 but not by selective PFKFB3 inhibiter 3PO, demonstrating specificity of silencer. Cells were treated with mixture of siPFKFB3 for silencing PFKFB3 18 h before challenge with GM-CSF. Results show mean ± SD. *P < 0.05 vs. same condition in control (absence of GM-CSF or 0 h). **P < 0.01 vs. same condition in control (absence of GM-CSF or 0 h). ##P < 0.01 for scRNA vs. siPFKFB3 with or without 3PO. a.u. = arbitrary units; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; HK = hexokinase; scRNA = scrambled RNA.
In line with the increased expression of glycolytic enzymes, GM-CSF resulted in a substantial increase in glycolytic flux (measured as Fru-2-6-P2 concentration, a potent activator of glycolysis; Fig. 2A). This augmented glycolytic flux was attenuated after inhibition of PFKFB3 by 3PO. In concert with the rise in Fru-2-6-P2 concentration, GM-CSF also increased lactate accumulation in the medium (Fig. 2B).

PFKFB3 mediates increase in 2-deoxyglucose uptake in human macrophages treated with GM-CSF. (A) Intracellular levels of Fru-2,6-P2 were determined at the indicated times with and without GM-CSF and in absence or presence of PFKFB3 inhibitor 3PO (5 μM). GM-CSF augments Fru-2,6-P2 levels, which are attenuated in presence of 3PO, demonstrating critical role of expression of PFKFB3 after GM-CSF treatment. (B) Lactate accumulation in culture medium was determined at 18 h with and without native or heat-inactivated GM-CSF and in absence or presence of 3PO (5 μM). Lactate levels (by-product of glycolysis) increase with GM-CSF exposure and are dampened by 3PO. (C) TNF-α levels were determined in culture medium after challenge with native or heat-inactivated GM-CSF. (D) Intracellular levels of Fru-2,6-P2 were determined at the indicated times with GM-CSF and in presence of neutralizing anti-TNF-α antibody. GM-CSF augments Fru-2,6-P2 levels, which are attenuated in presence of neutralizing anti-TNF-α antibody. (E) 2-deoxyglucose uptake was determined at the indicated times with and without GM-CSF and in absence or presence of neutralizing anti-TNF-α antibody (20 ng/mL) or PFKFB3 inhibitor 3PO (5 μM). 2-deoxyglucose significantly increases with addition of GM-CSF. This effect was diminished with neutralizing anti-TNF-α antibody and with 3PO. (F) To analyze contribution of PFKFB3 to enhancement of glycolytic flux elicited by GM-CSF, macrophages were transfected for 18 h with specific siRNA to silence PFKFB3 or with corresponding inactive RNA control (scRNA) and in absence or presence of 3PO, followed by activation for 18 h with GM-CSF. 2-deoxyglucose uptake was significantly decreased after silencing PFKFB3 compared with corresponding control (scRNA) or inhibition with 3PO. Data are mean ± SD. *P < 0.05 vs. same condition in control (absence of GM-CSF or 0 h). **P < 0.01 vs. same condition in control (absence of GM-CSF or 0 h). #P < 0.05 for GM-CSF vs. heated GM-CSF, with 3PO plus GM-CSF. ##P < 0.01 for GM-CSF vs. heated GM-CSF, with 3PO plus GM-CSF. IgG vs. anti-TNF-α antibody or scRNA vs. siPFKFB3 with or without 3PO. scRNA = scrambled RNA; siRNA = silencer RNA.
Additionally, GM-CSF resulted in a selective (not achieved by heat-denatured GM-CSF) and modest increase in proinflammatory activation of the macrophages (as evidenced by macrophage TNF-α production; Fig. 2C). Furthermore, antagonism of TNF-α (using a neutralizing anti-TNF-α antibody) attenuated the glycolytic flux (measured as Fru-2-6-P2 concentration) after GM-CSF (Fig. 2D).
GM-CSF Augments Deoxyglucose Accumulation Within Cultured Macrophages
Incubation of macrophages with GM-CSF upregulated the incorporation of [U-14C]-2-deoxyglucose in a manner that paralleled its impact on glycolytic flux (Figs. 2E and 2F). A time-dependent increase in 2-deoxyglucose uptake was observed after GM-CSF challenge (Fig. 2E). The rate of 2-deoxyglucose uptake was significantly greater than in control cells, was increased as early as 2–4 h after GM-CSF exposure, and persisted, at a more pronounced rate of increase, at later time points (P < 0.01). Also, in parallel with the impact of GM-CSF on glycolysis, blocking of TNF-α or inhibition (3PO) or silencing (siPFKFB3) of PFKFB3 resulted in reduced 2-deoxyglucose uptake (Figs. 2E and 2F).
GM-CSF Increases Atherosclerotic 18F-FDG Uptake In Vivo Without Altering Lesion Area
The in vivo impact of GM-CSF on chronic atherosclerotic inflammation was investigated in 2 animal models (mouse and rabbit). In Apoe−/− mice fed for 2 wk on a high-fat diet, administration of GM-CSF (on day 12 of the diet) significantly enhanced the aortic 18F-FDG uptake (TBR measured at 48 h after administration was increased by 70% relative to animals treated with vehicle; P < 0.01), whereas aortic 18F-FDG uptake did not increase in animals given both GM-CSF and silencer mRNA for PFKFB3 (Fig. 3A). When the lesion area was evaluated after oil red staining, the single-dose administration of GM-CSF did not influence the lipid accumulation (Fig. 3B). Moreover, the increased TBR was the result of a significant increase in target (arterial) activity, whereas the background activity did not significantly change with GM-CSF (Fig. 3C).
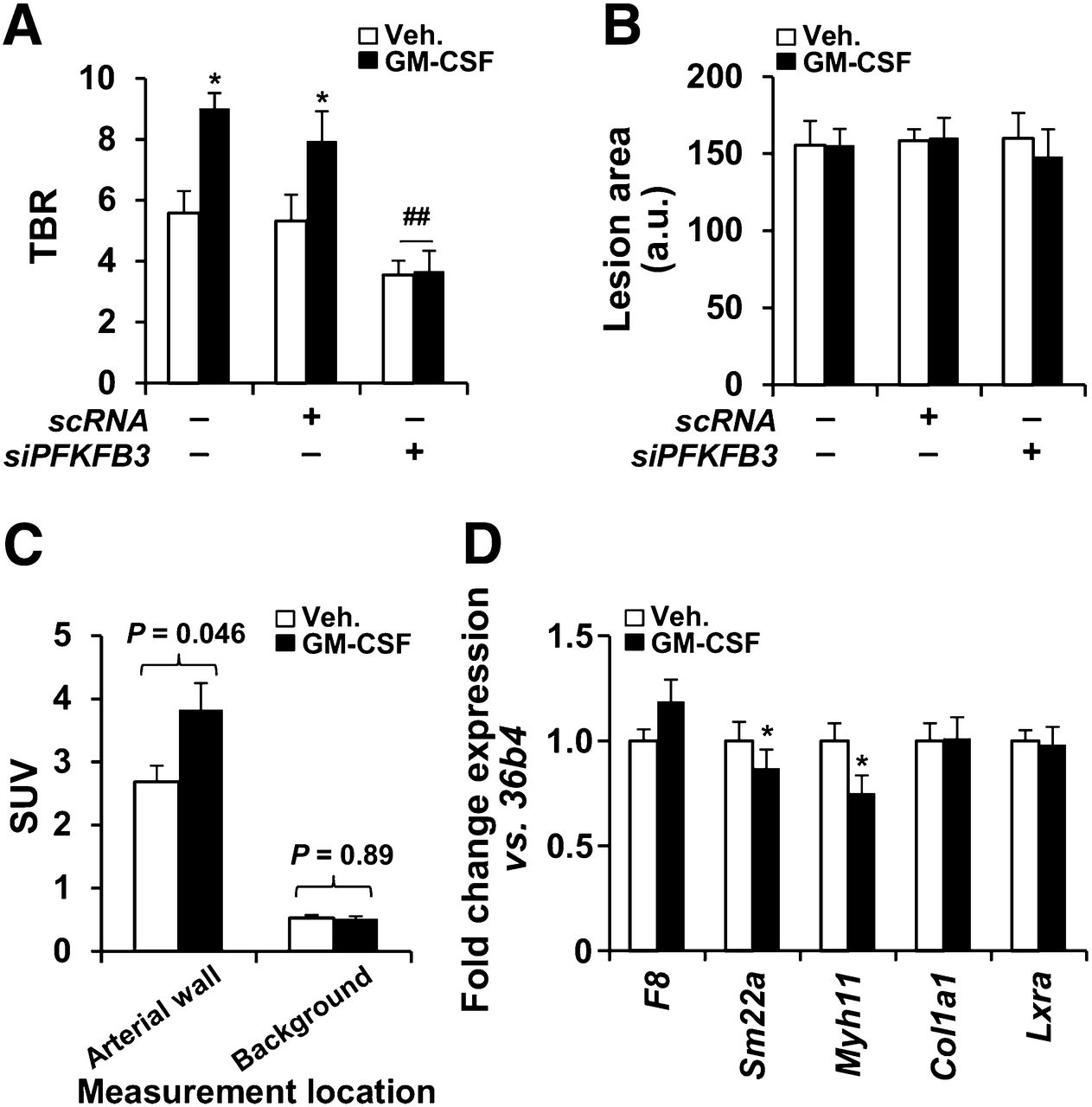
In vivo augmentation of 18F-FDG uptake after GM-CSF administration in mice. (A) 18F-FDG uptake in atherosclerotic mice (n = 30). Animals received siPFKFB3 (n = 10) or scRNA (n = 8) at days 3, 7, 10, and 12 after high-fat/high-cholesterol administration. GM-CSF (37.5 μg/kg; n = 15) or saline (n = 15) was intravenously administered on day 12, and 18F-FDG and PET analysis was performed on day 14. (B) Aortas were stained with oil red and images evaluated with Image J. (C) Average target and background SUVs from A were compared (n = 15 animals for each group). Target SUVs were higher with than without GMCSF (3.38 ± 0.46 vs. 2.70 ± 0.26; P = 0.046). Background SUVs were unchanged with vs. without GMCSF (0.54 ± 0.10 vs. 0.55 ± 0.10; P = 0.53). (D) In parallel experiment, aortas from untreated (n = 4) or GM-CSF–treated animals (n = 4) as described in A were isolated and RNA extracted for analysis of the indicated genes representative of endothelial cells (F8), smooth muscle cells (Sm22a, Myh11, Col1a1), and macrophages (Lxra). Data are mean ± SD. *P < 0.05 vs. same condition in absence of GM-CSF. ##P < 0.01 vs. same condition with scRNA. a.u. = arbitrary units; scRNA = scrambled RNA.
Further, to assess the impact of GM-CSF on potential infiltration of circulating monocytes into the aortic wall, the whole aorta from treated animals was analyzed using real-time quantitative polymerase chain reaction. As Figure 3D shows, the macrophage-specific gene, Lxrα, did not increase after GM-CSF administration when compared with specific endothelial and smooth muscle cell markers (i.e., F8, Sm22a, Myh11, and Col1a1). Together, these results suggest a minimal contribution of GM-CSF, if any, to enhance the recruitment of monocytes to the lesion area, in agreement with previous data (9).
In atherosclerotic rabbits, GM-CSF administration was associated with an approximately 73% increase in 18F-FDG uptake (5.87 ± 0.14 vs. 10.09 ± 0.40 before vs. after GM-CSF, arterial TBR ± SEM, P < 0.01; Figs. 4A and 4B). Atherosclerotic animals injected with saline showed no statistically significant change in TBR (4.17 ± 0.32 vs. 4.08 ± 0.20, P < 0.01) over the same time period. Further, in healthy rabbits without atherosclerotic inflammation, GM-CSF administration was associated with a decrease in the arterial signal (2.26 ± 0.07 vs. 1.47 ± 0.17, P < 0.01). In contrast, blood 18F-FDG activity did not differ between rabbits given GMCSF and those given saline (supplemental data). Collectively, these data demonstrate that administration of GM-CSF augments 18F-FDG uptake within the arterial wall in atherosclerotic but not in normal rabbits.
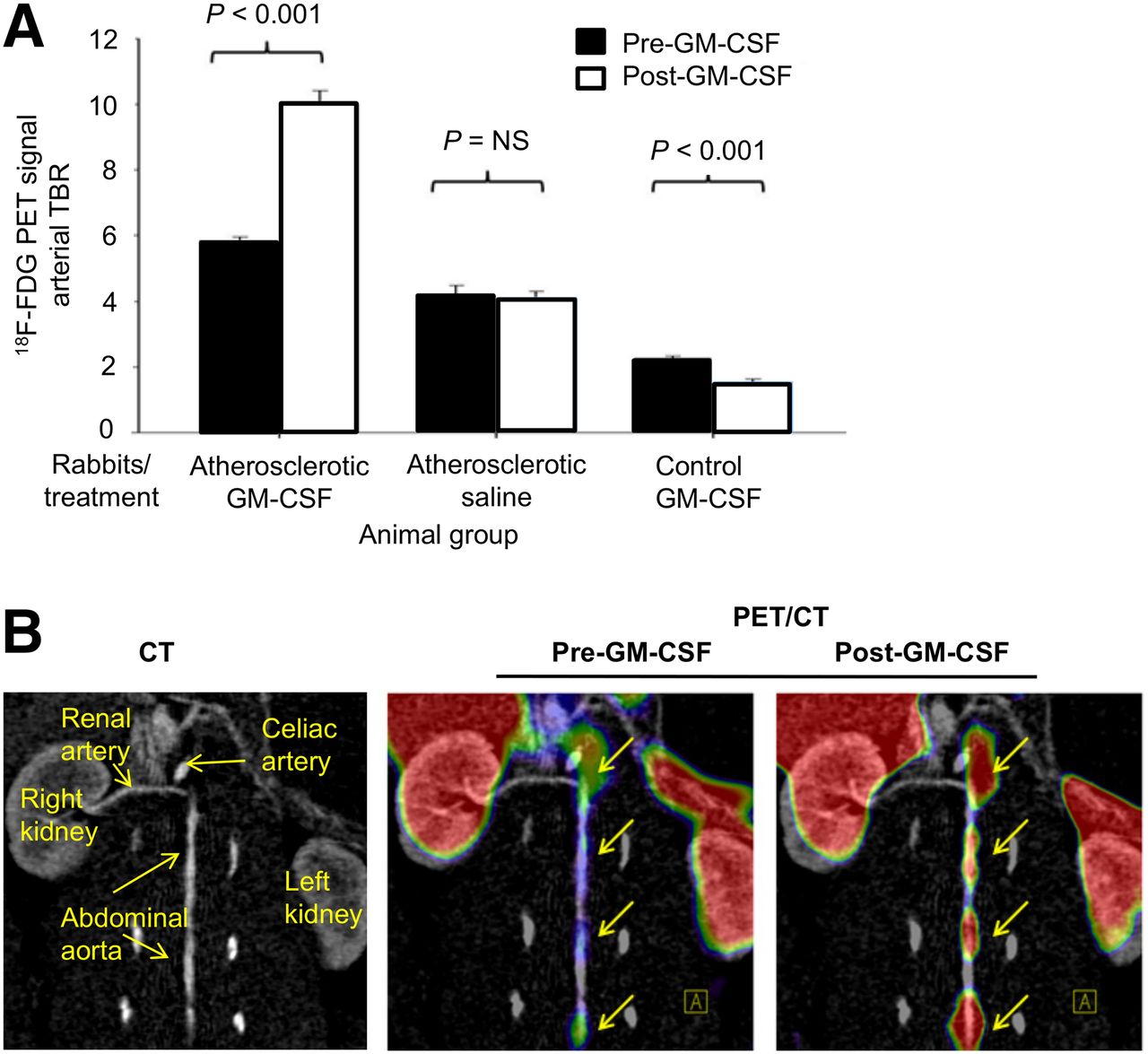
In vivo augmentation of 18F-FDG uptake after GM-CSF administration in rabbits. (A) 18F-FDG uptake in rabbits (n = 9) before and after challenge with cytokine GM-CSF or saline shows significant increase in 18F-FDG uptake in atherosclerotic rabbits (P < 0.001). 18F-FDG uptake is expressed as TBR compared with baseline imaging. (B) Representative image of 18F-FDG signal enhancement demonstrating increased 18F-FDG uptake in descending aorta before and after GM-CSF.
Atherosclerotic 18F-FDG Uptake After GM-CSF Correlates with Macrophage Density
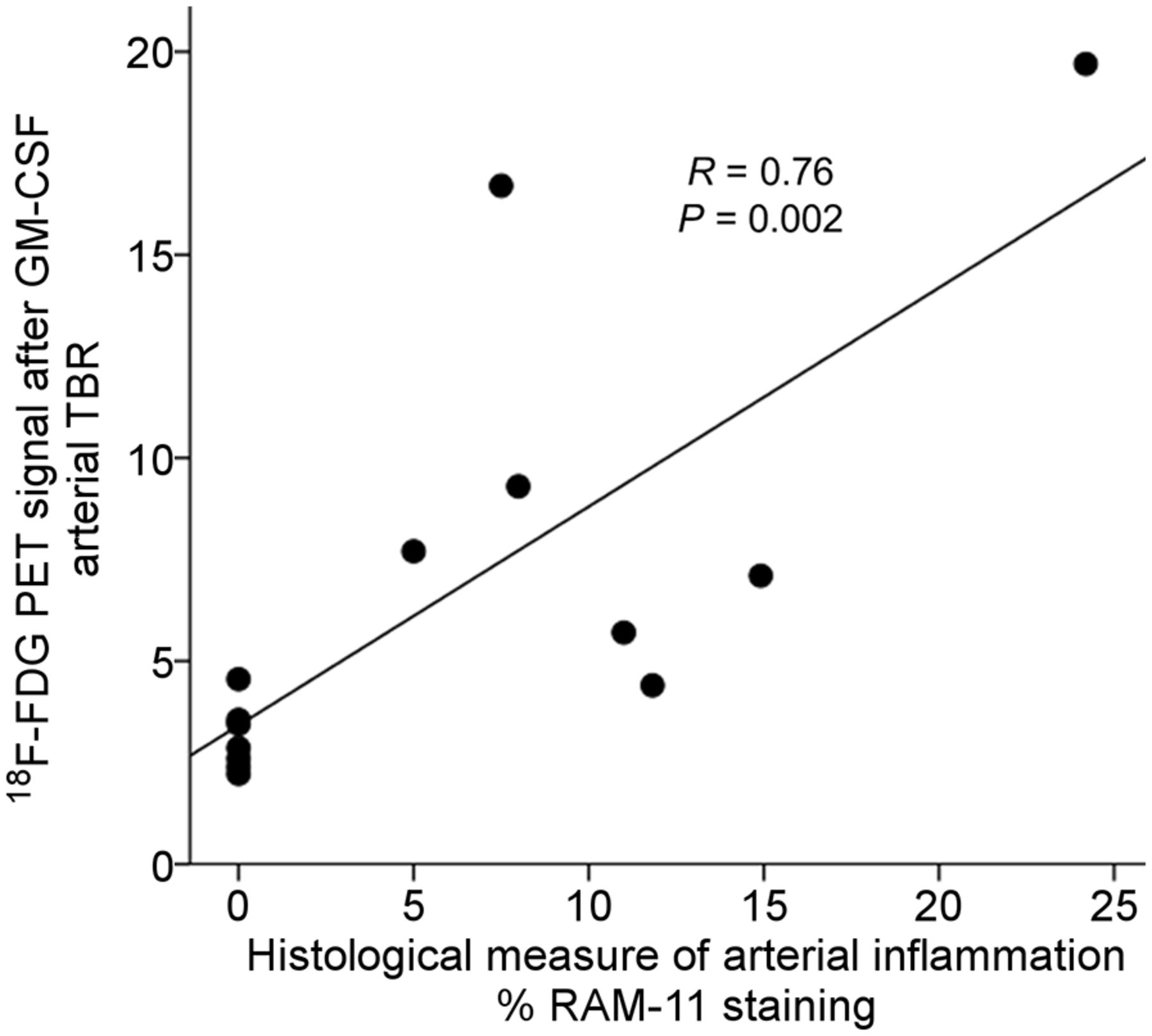
Next, we tested the hypothesis that arterial 18F-FDG uptake after GM-CSF correlates with histologic macrophage density. To do so, we compared 18F-FDG uptake in atherosclerotic rabbits with macrophage-specific %RAM11 staining in histologic segments of aortas from sacrificed animals. We observed that 18F-FDG uptake significantly correlated with %RAM11 staining (R = 0.76, P = 0.002; Fig. 5).

Relationship between arterial 18F-FDG uptake (by PET/CT) and histologic macrophage accumulation. Rabbits that had received GM-CSF (4 atherosclerotic and 2 control) were imaged using 18F-FDG PET/CT. Arterial 18F-FDG uptake was recorded as TBR. Subsequently, the animals were sacrificed and macrophage concentration within aortic wall was determined histologically (%RAM11 staining). There was significant relationship between macrophage staining and TBR (R = 0.76, P = 0.002).
DISCUSSION
Here, we have shown that GM-CSF augments the glycolytic flux in vitro (via a mechanism that depends on PFKFB3) and acutely increases 18F-FDG uptake within inflamed tissues in vivo (in 2 animal atherosclerotic models). These data suggest that GM-CSF, when administered as an adjunctive imaging agent, improves the sensitivity for detecting inflammatory foci. Further investigations to evaluate the clinical impact of GM-CSF in 18F-FDG PET imaging are warranted.
GM-CSF is an endogenously circulating cytokine that is present at basal levels in serum. Recombinant human GM-CSF, a widely available and clinically used immunomodulatory agent, was first approved by the Food and Drug Administration in 1991 for the acceleration of myeloid recovery after autologous bone marrow transplantation. Since that time, the use of GM-CSF has expanded. GM-CSF acts to mobilize peripheral-blood progenitor cells, resulting in shorter durations of neutropenia in patients receiving induction chemotherapy for hematologic malignancies (25). Further, it is widely appreciated that GM-CSF increases the hematopoietic 18F-FDG signal and that the heightened signal may persist for weeks after the last dose (16,26–31). The findings of this study provide further understanding of the mechanisms underlying this observation.
Here, we show that GM-CSF acutely increases the expression of PFKFB3, resulting in increased glycolytic flux compared with the constitutively expressed PFKFB1, since PFKFB3 has a much higher net kinase activity, therefore increasing the intracellular levels of Fru-2,6-P2 and accelerating the upper part of glycolysis. Further, we found that the upregulation of PFKFB3 was necessary for the increased glycolytic flux seen with GM-CSF in both in vitro (human macrophages) and in vivo models (mice): blocking PFKFB3 (with silencer RNA specific for PFKB3 or inhibiting its activity using 3PO) resulted in levels of glycolysis that were only slightly above baseline levels. Additionally, we observed that antagonism of TNF-α (using an antibody against TNF-α) resulted in an attenuation of glycolytic flux after GM-CSF. This finding is consistent with the prior observation that PFKFB3 activation results in augmented TNF-α production and that the TNF-α in turn leads to increased PFKB3 (via a mechanism that relies on HIF-1α (9).
Moreover, we found that GM-CSF provided a roughly 3-fold increase in glycolytic flux and a similar increase in macrophage 2-deoxyglucose uptake in vitro. Similarly, in the Apoe−/− mice and in the rabbit models of atherosclerosis, we observed an approximately 70%–73% increase in 18F-FDG uptake. Such increased 18F-FDG uptake might provide improved detection of infectious foci.
The studies examining the effects of chronic GM-CSF administration on atherosclerosis have provided inconsistent findings (32). In one study, hyperlipidemic LDLR−/− mice with a GM-CSF deficiency exhibited a 20%–50% decrease in aortic lesion size, depending on the location of the lesions and the sex of the animals. In another, Apoe−/− mice treated with GM-CSF chronically manifested an increased atherosclerotic-lesion area. However, in another study, Apoe−/− mice with a genetic deletion of GM-CSF demonstrated an increased atherosclerotic-lesion size (without changes in plasma cholesterol). It follows then, that both proatherogenic and antiatherogenic properties have been attributed to GM-CSF. Accordingly, the consequences of long-term GM-CSF administration have yet to be determined. However, clinical experience with short-term GM-CSF administration suggests no increased risk of cardiovascular complications (33).
Clinical Implications
The concept of transiently “stimulating” macrophages with GM-CSF to improve the signal-to-noise ratio of inflammatory foci is analogous to the use of exercise stress testing to enhance the detection of myocardial ischemia. With electrocardiographic exercise treadmill testing, the detection of occlusive coronary lesions (those that cause myocardial ischemia) is limited under resting conditions. However, when exercise is used to temporarily induce myocardial ischemia (leading to diagnostic changes on the electrocardiogram), a substantial boost in test sensitivity is yielded. At the same time, the induction of transient ischemia during exercise has been shown to be remarkably safe. Analogously, we propose further examination of the use of GM-CSF to transiently and safely provoke an inflammatory signal. This transient provocation may substantially improve the detection of inflammatory lesions, such as atherosclerotic or infectious foci.
Importantly, GM-CSF has a relatively safe track record, even when used for extended periods. The in vivo data presented within this study and from others provide some explanation of why GM-CSF has a relatively modest proinflammatory profile compared with some other cytokines. We previously reported that lipopolysaccharide induces a 50- and 15-fold greater increase in TNF-α mRNA and protein production, respectively, compared with GM-CSF (9). In that experiment as well, GM-CSF resulted in a 6-fold increase in Arg-1 mRNA, a prototypical marker of M2 polarization. In fact, the increase in Arg-1 mRNA exceeded the increase in TNF-α mRNA, supporting the notion that GM-CSF provides a balanced increase in M1 as well as M2 polarization, yielding an intermediate polarization profile.
Moreover, the safety of GM-CSF has already been established in patients who have a preponderance of atherosclerotic risk factors, as well as in critically ill patients (33). In fact, GM-CSF may have benefits in chronic atherosclerosis (potentially related to augmentation of progenitor cell release) and is being examined as a therapy for chronic peripheral arterial disease (ClinicalTrials.gov identifier NCT01408901) (34,35).
18F-FDG PET imaging has already become a critical tool in the localization of infections in the setting of fever of unknown origin (36,37). Our findings offer justification to further explore the use of GM-CSF as an adjunctive agent for 18F-FDG PET imaging of inflammation, in both acute and chronic inflammatory diseases (such as atherosclerosis and infections). In such cases, GM-CSF would be expected to transiently increase the 18F-FDG signal within inflammatory cells and thus enhance the sensitivity, and possibly the specificity, of PET/CT imaging for detecting an inflammatory lesion.
Use of GM-CSF as an Adjunctive Imaging Agent in Other Settings
GM-CSF might also aid in the characterization of malignancies. 18F-FDG PET/CT imaging has proven to be an invaluable tool in the workup of oncologic processes (38). The biologic basis of 18F-FDG accumulation in malignant tumors results in part from the relatively high glycolytic rates found within malignant cells. However, tumor-associated macrophages also constitute an important fraction of oncologic tissues and provide an additional locus of intratumor 18F-FDG accumulation (10). Occasionally, one cannot differentiate whether the 18F-FDG signal originates within tumor or immune cells (especially after some chemotherapeutic regimens, which can activate immune cells while decreasing tumor activity). In these cases, the use of GM-CSF may improve diagnostic accuracy; an increase in lesional 18F-FDG uptake on serial 18F-FDG PET/CT imaging (repeated after GM-CSF) may suggest that the activity was localized to immune cells.
Furthermore, GM-CSF may prove useful as an adjunctive agent for other approaches targeting inflammation. GM-CSF might enhance macrophage activation sufficiently to increase uptake of other tracers targeting inflammation, such as gallium DOTATATE PET and superparamagnetic iron oxide nanoparticles. This is particularly facilitated by the fact that GM-CSF activates macrophages via mechanisms that promote both M2 (phagocytic) and mild M1 actions. With M2 activation comes an increase in uptake of nanoparticles such as superparamagnetic iron oxide (39). GM-CSF–enhanced imaging of superparamagnetic iron oxide and other nanoparticles should be further explored.
Limitations
This study is not without limitations. The optimal duration and dosing of GM-CSF was not examined in this study (only a 3-d GM-CSF regimen was examined). Thus, it is not clear from these data whether a single injection of GM-CSF would suffice to provide the desired boost in 18F-FDG uptake. A multiday regimen would come at increased complexity, cost, and potential risks. Further, since only one imaging interval was studied, the optimal time point for 18F-FDG PET/CT imaging after GM-CSF administration has not been determined. Future human studies are needed to assess the impact of a simplified GM-CSF regimen on 18F-FDG PET/CT imaging.
CONCLUSION
GM-CSF augments macrophage glycolytic flux in vitro via a mechanism dependent on PFKFB3 and increases 18F-FDG uptake in vivo in animal atherosclerotic models. GM-CSF is an approved and clinically used medication with an established safety profile. Future larger studies should explore the potential of using GM-CSF as an adjunctive imaging tool to improve detection of inflammation.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. James H.F. Rudd is in part supported by the NIHR Cambridge Biomedical Research Centre, the British Heart Foundation, and HEFCE. Zahi A. Fayad is supported by NIH/NHLBI R01 HL071021, R01 HL078667, NIH/NBIB R01 EB009638, and the NIH/NHLBI Program of Excellence in Nanotechnology (PEN) Award, contract HHSN268201000045C. Parmanand Singh was supported by a grant from the NHLBI (5T32 HL076136) and is funded by the Marfan Foundation. Lisardo Boscá was supported by SAF2014-52492R, RTC2015-3741 from MINECO, and RD12/0042/0019. Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas is funded by the Instituto de Salud Carlos III and S2010/BMD-2378 from Comunidad de Madrid. Ahmed Tawakol is supported by NIH/NHLBI R01 HL122177. No other potential conflict of interest relevant to this article was reported.
Footnotes
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication September 24, 2015.
- Accepted for publication March 7, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.