


Abstract
The P2X7 receptor (P2X7R) orchestrates neuroinflammation, and this is the basis for an increased interest in the development of antagonists inhibiting P2X7R function in the brain. This study provides the preclinical evaluation of 11C-JNJ-54173717, a PET tracer for P2X7R in both rats and nonhuman primates. Methods: 11C-JNJ-54173717 is a high-affinity radiotracer for the human P2X7R (hP2X7R). Biodistribution and radiometabolite studies were performed. Viral vectors encoding either enhanced green fluorescent protein-hP2X7R or 3flag-hP2X7R were engineered and validated in cell culture. hP2X7R was regionally overexpressed in the rat striatum after stereotactic injection of viral vectors. Dynamic small-animal PET studies were performed in vector-injected rats and in healthy monkeys using 11C-JNJ-54173717. Results: The affinity of JNJ-54173717 was 1.6 ± 0.1 nM in a rat cortex P2X7R membrane binding assay. In a functional assay at the recombinant human and rat P2X7R orthologs, the half maximal inhibitory concentration (IC50) of JNJ-54173717 was 4.2 ± 0.01 nM and 7.6 ± 0.01 nM, respectively. The rat biodistribution study showed that 11C-JNJ-54173717 crossed the blood–brain barrier and was cleared from plasma mainly via the hepatobiliary pathway. A polar radiometabolite was found in rat plasma. No radiometabolites were detected in rat brain. Dynamic small-animal PET showed binding of 11C-JNJ-54173717 in the striatum expressing hP2X7R, with rapid washout from the noninjected control striatum and other brain regions. Likewise, 11C-JNJ-54173717 PET signal was blocked by a chemically distinct P2X7R ligand, indicating specific binding to P2X7R in the monkey brain. Conclusion: JNJ-54173717 is a high-affinity P2X7R antagonist. An animal rat model stably expressing hP2X7R was developed and validated, identifying favorable characteristics for 11C-JNJ-54173717 as a PET radioligand for in vivo visualization of hP2X7R. 11C-JNJ-54173717 selectively visualized P2X7R in the monkey brain, and this radioligand will be further evaluated in a clinical setting to study P2X7R expression levels in neurodegenerative disorders.
Increasing evidence suggests an active role of neuroinflammation in the progression of neurodegenerative disorders such as Alzheimer's disease and Parkinson's disease (1–3). Neuroinflammation is a well-orchestrated, dynamic, multicellular process. Microglial cells make up the innate immune system of the central nervous system and are key cellular mediators of neuroinflammatory processes. Upon neuronal injury or infection, they become activated, thereby releasing neurotoxic substances, amplifying neuroinflammation and leading to neurodegeneration (M1 phenotype). On the other hand, they also provide tissue repair by releasing antiinflammatory cytokines and neurotrophic factors (M2 phenotype). However, the current repertoire of activated microglia phenotypes and phenotype development might be more variable and complex (4). PET provides a sensitive noninvasive imaging technique to study and quantify receptor and enzyme expression. A radiolabeled tracer for a protein expressed during neuroinflammation and more specifically for overactivated microglia would allow in vivo detection and quantification of neuroinflammation in an early stage of the disease and could facilitate the development and evaluation of specific antineuroinflammatory treatment (5).
Until now, translocator protein (TSPO) was the most studied biomarker to image neuroinflammation using PET. However, recent findings indicate that TSPO polymorphism results in differences in binding affinity of PET radioligands for TSPO (6). As a consequence, knowledge of the binding characteristics of a PET ligand for TSPO of patients is required to correctly interpret PET images, complicating image analysis. In contrast to TSPO, more is known about the pharmacology of the P2X7 receptor (P2X7R) and its role during inflammation. The mechanism of action involves inflammasome activation and IL-1β release. Moreover, caspase-1 activation, needed for IL-1β release, occurs within the NLRP3 inflammasome in response to an increased concentration of extracellular adenosine triphosphate and thus P2X7R activation. In turn, IL-1β is one of the proinflammatory cytokines that initiate the neuroinflammatory cascade (7,8). In addition, several animal models for neurodegenerative disorders such as Alzheimer's disease and Huntington’s disease (9–11) showed that proinflammatory cytokines correlated with upregulated P2X7R expression and increased adenosine triphosphate sensitivity, putting forward P2X7R as a valuable alternative marker for microglial activation and inflammation (8,12). In a recent study, 3H-A-804598 was used to determine the ex vivo brain P2X7R occupancy of different P2X7R antagonists, indicating that this class of molecules crosses the blood–brain barrier and supporting the feasibility of in vivo visualization of P2X7R with PET in the brain (13–15). An additional hurdle, however, is the P2X7R interspecies difference reported for several molecules, giving rise to different binding affinities for mouse, rat, and human P2X7R (hP2X7R) (16–18).
In this study, we developed a humanized animal model by stable expression of the hP2X7R in rat brain, which can be applied in the validation and evaluation of P2X7R PET tracers. To obtain efficient gene transfer, we applied viral vector technology, engineering a hP2X7R reporter system using HIV-based lentiviral vectors (LVs) for validation in cell culture settings, and recombinant adeno-associated viral (rAAV) vectors in the brain in vivo (19). JNJ-54173717 (20,21), which has nanomolar affinity for rat P2X7R and hP2X7R, was radiolabeled with 11C and evaluated in this rat model using small-animal PET. In contrast to several molecules described in the literature (16–18), this compound shows little interspecies differences. This PET tracer was further validated in vivo with biodistribution studies and plasma and brain radiometabolite analysis after intravenous injection in rats and in vitro by performing autoradiography binding studies on normal rat brain slices, slices of a rat brain overexpressing hP2X7R in striatum, and wild-type (WT) and P2X7R knockout mouse brain slices. Finally, a monkey small-animal PET study was performed with 11C-JNJ-54173717 in baseline and blocking conditions using 2 chemically distinct P2X7R selective ligands.
MATERIALS AND METHODS
The methods used for the in vitro pharmacology, radiosynthesis of the radioligand, biodistribution studies, viral vector construction and production, rat plasma and perfused rat brain radiometabolite analysis, cell culture and transduction, Western blot analysis, stereotactic injections, immunocytochemistry, cell uptake experiments, and in vitro autoradiography have been described in the supplemental materials (available at http://jnm.snmjournals.org). The research protocol was approved by the local Animal Ethics Committees and was performed according to guidelines of the European Ethics Committee.
Small-Animal PET Imaging
Imaging experiments were performed on a Focus 220 microPET scanner (Concorde Microsystems). Baseline scans were obtained in 3 rats at 10 wk after viral vector injection. For a pretreatment experiment, the authentic reference JNJ54173717 was dissolved in a vehicle containing 20% (2-hydroxypropyl)-β-cyclodextrine and 2 equivalents HCl. This formulation was injected subcutaneously 45 min before tracer injection at a dose of 10 mg/kg (n = 3; pretreatment protocol based on ex vivo receptor occupancy study results).
Rats were injected with approximately 74 MBq of 11C-JNJ-54173717 via a tail vein under isoflurane anesthesia (2.5% in oxygen at a flow rate of 1 L/min). List-mode dynamic 90-min small-animal PET scans were acquired. Acquisition data were Fourier-rebinned in 24 time frames (4 × 15, 4 × 60, 5 × 180, 8 × 300, and 3 × 600 s) and reconstructed using maximum a posteriori iterative reconstruction. The images were spatially aligned to a rat brain 18F-FDG template in Paxinos coordinates (22) using affine transformation, allowing the use of a predefined volume-of-interest map. Time–activity curves were generated for the striatum, cerebellum, and total brain for each individual scan using PMOD software (version 3.4; PMOD Technologies).
Kinetic modeling based on the simplified reference tissue model (SRTM) (23) was performed to quantify differences in radioactivity concentration between the left and right striatum. The left striatum was evaluated as a reference tissue.
Small-Animal PET Study in Rhesus Monkeys
Two juvenile male rhesus monkeys (Macaca mulatta, 5 and 6.8 kg) were used in this small-animal PET study. Sedation was performed by an intramuscular injection of a combination of 0.3 mL of xylazine (Rompun [Bayer], 2% solution) and 0.35 mL of ketamine (Nimatek [Eurovet], 100 mg/mL). About 60 min after the first injection, the monkey received an additional dose of 0.15 mL of xylazine and 0.175 mL of ketamine via intravenous injection. During the last part of the scan, dosing was done less frequently, based on the heartbeat frequency. PET imaging was performed using a Focus 220 tomograph (Siemens/Concorde Microsystems), which has a nominal transaxial resolution of 1.35 mm (full width at half maximum). The O2 and CO2 saturation in the blood and the heartbeat were constantly monitored. Temperature was regulated via a heating pad. The breathing frequency and the eye response were checked visually. The monkey was injected with ±185 MBq of 11C-JNJ-54173717 via the vena saphena. For the self-block experiment, the authentic reference JNJ54173717 was dissolved in a vehicle containing 20% (2-hydroxypropyl)-β-cyclodextrine and 2 equivalents HCl. In a second pretreatment experiment, JNJ-42253432 (a central nervous system–penetrant P2X7R antagonist and structurally unrelated compound (15)) was dissolved in a vehicle containing 20% (2-hydroxypropyl)-β-cyclodextrine and 0.75 equivalents HCl (no vehicle was injected in the baseline scan). These formulations were injected intravenously 10 min before tracer injection at a dose of 2.5 mg/kg. List-mode dynamic 90-min small-animal PET scans were acquired. Acquisition data were Fourier-rebinned in 24 time frames (4 × 15, 4 × 60, 5 × 180, 8 × 300, and 3 × 600 s) and reconstructed using maximum a posteriori iterative reconstruction. Data were analyzed using PMOD software, and volumes of interest were defined on the summed images and time–activity curves were drawn. During both baseline and pretreatment scans, blood samples were collected at 10, 30, and 60 min after tracer injection via the other vena saphena, this to determine radiometabolites of 11C-JNJ-54173717 in plasma.
RESULTS
In Vitro Pharmacology of JNJ-54173717
The compound displayed high potency in a functional assay. The half maximal inhibitory concentration (IC50) of JNJ-54173717 was 4.2 ± 0.01 nM and 7.6 ± 0.01 nM, at the human and rat P2X7R orthologs, respectively. Consistent with the functional inhibition, JNJ-54173717 displayed high-affinity binding at the native P2X7R channels in the rat cortex. The affinity of the compound was 1.6 ± 0.1 nM at the rat brain P2X7R channel (Fig. 1).
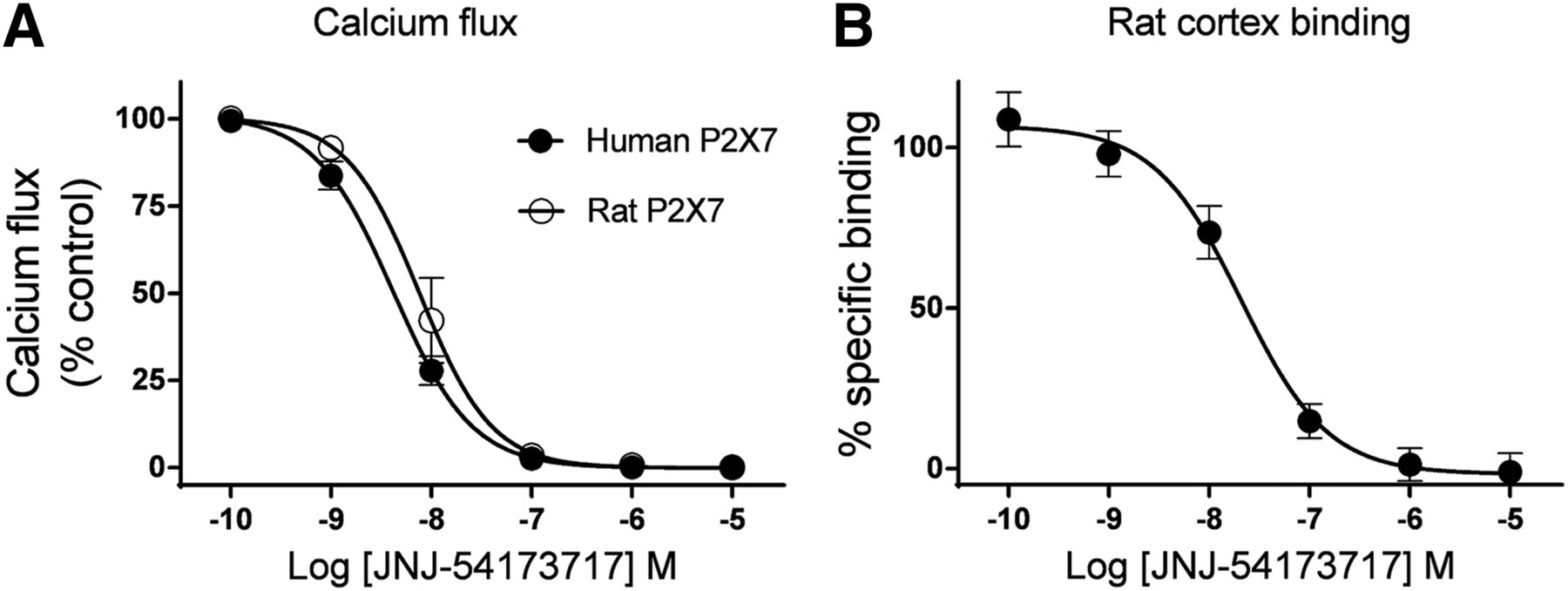
In vitro pharmacology of JNJ-54173717. (A) IC50 of JNJ-54173717 was 4.2 ± 0.01 nM and 7.6 ± 0.01 nM, at the human and rat P2X7 orthologs, respectively. (B) Affinity of compound was 1.6 ± 0.1 nM at the native P2X7 channels in rat cortex. Data are expressed as mean ± SEM from 3 independent experiments in duplicate (each symbol is a mean of 6 data points).
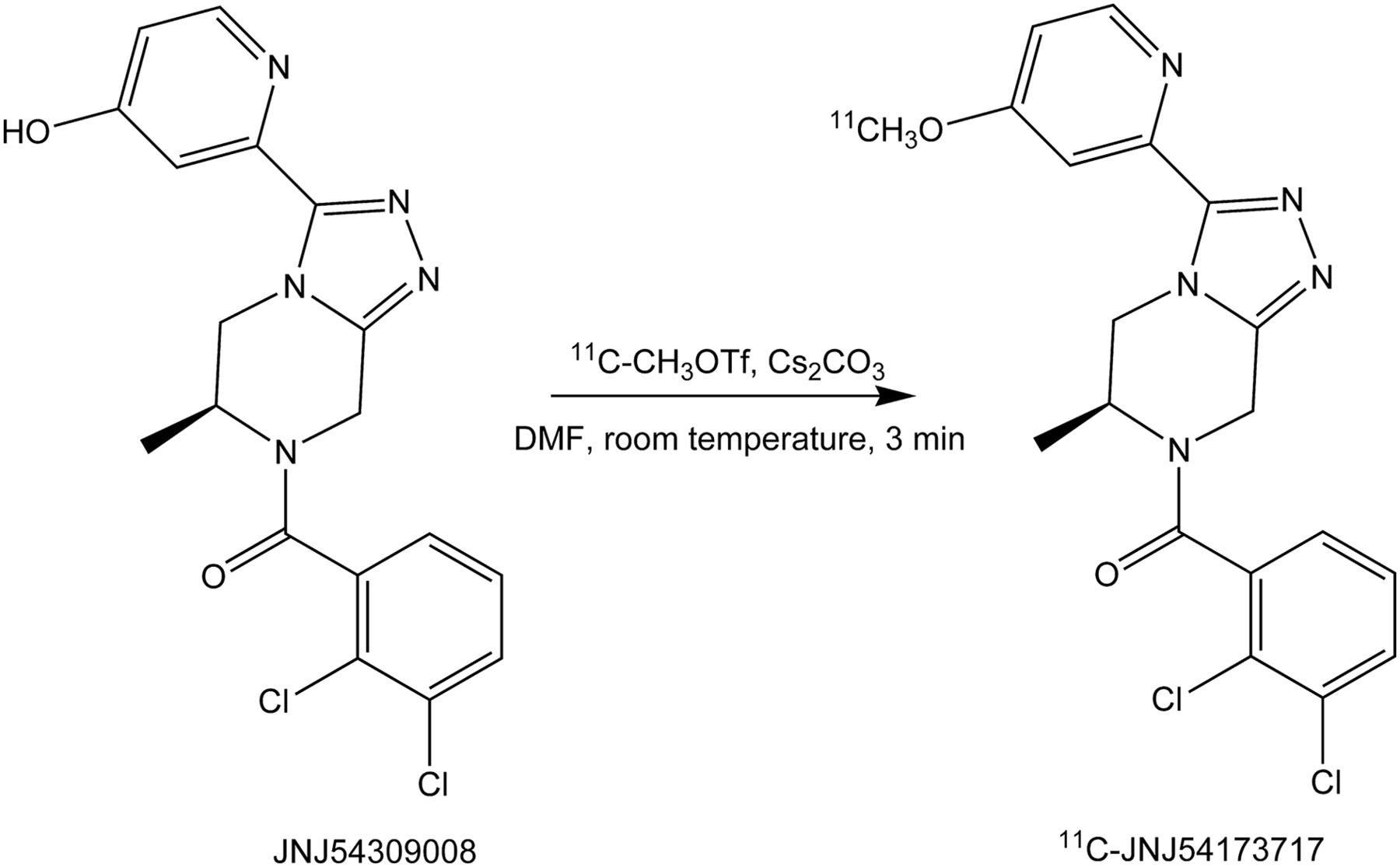
Radiosynthesis
11C-JNJ-54173717 was synthesized via methylation of the hydroxyl precursor with 11C-methyl triflate (11C-MeOTf) in the presence of cesium carbonate in dimethyl formamide at room temperature (Fig. 2). The crude radiolabeling mixture was purified using semipreparative reversed-phase high-pressure liquid chromatography (HPLC) affording 11C-JNJ-54173717 in good radiochemical yields (40%–60%, relative to starting radioactivity of 11C-MeOTf, non–decay-corrected, n = 12), with a radiochemical purity greater than 98% after HPLC purification and an average specific radioactivity of 233 ± 99 GBq/μmol at the end of synthesis (n = 12). The identity of 11C-JNJ-54173717 was confirmed by coelution with the nonradioactive analog after coinjection onto the analytic HPLC system (chromatogram not shown).
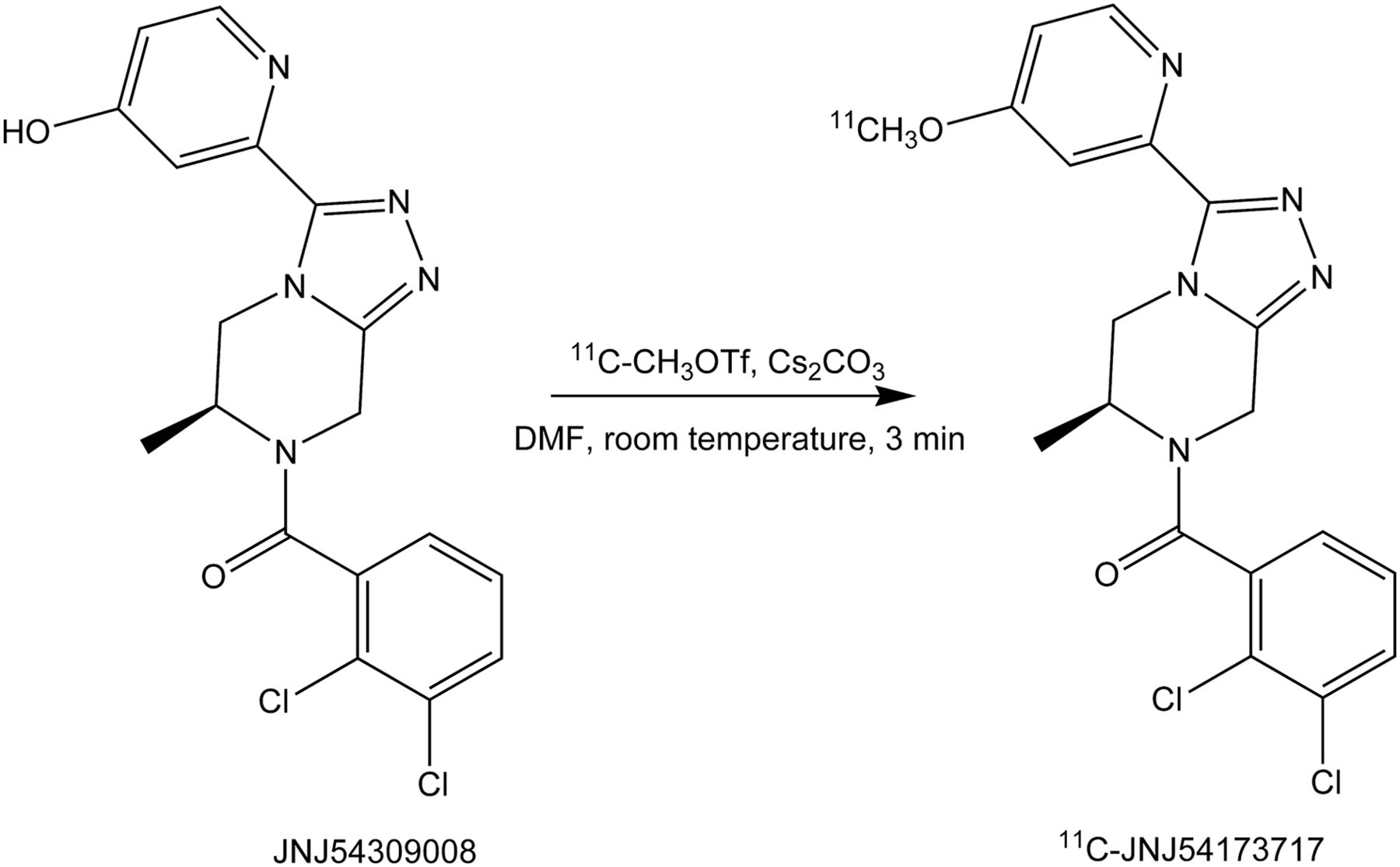
Radiosynthesis of 11C-JNJ-54173717. 11C-methyl triflate (11C-MeOTf) was bubbled with a flow of helium through a solution of precursor (0.2 mg) and Cs2CO3 (1–3 mg) in anhydrous dimethyl formamide (DMF; 0.2 mL). Reaction mixture was left at room temperature for 3 min and purified with reversed-phase high-pressure liquid chromatography.
Biodistribution and Radiometabolite Analysis in WT Rats
The biodistribution study of 11C-JNJ-54173717 was performed in healthy nonperfused female Wistar rats (body weight, 185–220 g) at 2, 30, and 60 min after tracer injection (n = 3/time point). Ten percent of the injected dose was detected in blood at 2 min after injection, which cleared to 5.3% at 60 min after tracer injection. The initial total brain uptake of 11C-JNJ-54173717 was 0.6% at 2 min after injection, and this cleared to 0.4% at 60 min after tracer injection. At 60 min after tracer injection, 44% of the injected dose was found in the liver and the intestines. Urinary excretion of the radiotracer was minimal, with 3% of the injected dose in urine and kidneys at 60 min after tracer injection (Supplemental Table 1).
In Supplemental Table 2, the SUVs for different brain regions and blood are shown. At 2 min after tracer injection, the radioactivity concentration in the cerebellum was highest of all studied brain regions. Clear washout was observed between 2 and 30 min after tracer injection for all brain regions with relative washout ratios (2-to-30-min) higher than 1.3. The radioactivity concentration at 30 and 60 min after tracer injection was comparable for all studied brain regions and also for total brain and blood at the 3 studied time points.
The relative amounts of intact tracer and radiometabolites were determined in rat plasma at 30 min after tracer injection (n = 2). The reconstructed radiochromatogram demonstrated 2 peaks, corresponding to a polar radiometabolite eluting at 2 min and the intact 11C-JNJ-54173717 eluting at 10 min (chromatograms not shown). At 30 min after radiotracer injection, 70% ± 6% of the recovered radioactivity in the plasma was in the form of intact tracer, and 30% ± 6% was in the form of polar radiometabolites. The fraction of more lipophilic radiometabolites eluting after the intact tracer was negligible (<1.5%).
The reconstructed radiochromatograms from perfused rat cerebellum and cerebrum HPLC analysis at 30 min after tracer injection showed only 1 radioactive peak corresponding to intact 11C-JNJ-54173717 eluting at 9 min (n = 2; chromatograms not shown). The fraction of both more polar and more lipophilic radiometabolites was negligible (<2%). The recovery of the HPLC (XBridge) column–-injected radioactivity was 85% ± 6%.
Evaluation of 11C-JNJ-54173717 in hP2X7R Cell Model: Validation in Cell Culture by Western Blot Analysis, Immunocytochemistry, and Cell Uptake Experiments
SHSY5Y (human dopaminergic neuroblastoma) cell lines stably expressing hP2X7R were generated via transduction with HIV-based LVs (LV_eGFP-hP2X7R and LV_3flag-hP2X7R) encoding human P2X7R N-terminally fused to eGFP or a 3flag tag driven from a ubiquitous human cytomegalovirus immediate-early promoter (eGFP is enhanced green fluorescent protein) (Supplemental Figs. 1A and 1D).
Expression of full-length P2X7R was confirmed by Western blot analysis for SHSY5Y cells transduced with LV_eGFP-hP2X7R and LV_3flag-hP2X7R, showing a band of 100 kDa corresponding to the eGFP-tagged hP2X7R protein and a band of 80 kDa corresponding to the 3flag tagged hP2X7R (Supplemental Figs. 1B and 1E, respectively). In parallel, we verified subcellular distribution of the tagged hP2X7R by immunocytochemistry (Supplemental Figs. 1C and 1F). Both protein fusions were primarily located at the plasma membrane, indicating that the tagging did not affect the subcellular distribution. No immunoreactivity was seen in nontransduced SHSY5Y cells. As a proof of principle, we verified 11C-JNJ-54173717 uptake for SHSY5Y cells overexpressing hP2X7R. The percentage of total radioactivity in the cell fraction per milligram of protein was calculated for LV_eGFP-hP2X7R, LV_eGFP, LV_3flag-hP2X7R, LV_3flag-eGFP, and nontransduced SHSY5Y cells. The ratio of total uptake of radioactivity in cells transduced with LV_eGFP-hP2X7R to cells transduced with LV_eGFP was 5.1 ± 0.6. The ratio of total activity bound by LV_3flag-hP2X7R cells to total activity bound to cells transduced with LV_3flag-eGFP was 3.6 ± 0.4.
Evaluation of 11C-JNJ-54173717 in hP2X7R Locoregional Transgenic Rat Model: Validation of rAAV_3flag-hP2X7R by In Vitro Autoradiography, Small-Animal PET, and Histology
In Vitro Autoradiography
Higher tracer binding was observed in WT mouse brain than in P2X7R knockout mouse brain (ratio of WT total binding to knockout total binding = 3.3), though the latter showed some specific binding (Supplemental Figs. 2A–2D; ratio of knockout total binding to knockout nonspecific binding = 2.0; ratio of WT total binding to WT nonspecific binding = 8.9; n = 4). The in vitro binding study on the transversal rat brain sections of vector-injected rats showed higher tracer binding in the rAAV_3flag-hP2X7R injected striatum (right striatum) than in the rAAV_3flag-eGFP–injected striatum (left striatum) (Fig. 3A; Supplemental Fig. 3). The right-to-left activity ratio was 3.1 ± 0.9 (n = 4). Tracer binding could be blocked using 10 μM A-740003 (Fig. 3B). Supplemental Figures 3G and 3H present the results of the in vitro autoradiography study on brain sections of WT rats (n = 3). Highest binding is observed to white matter, which can also be blocked with 10 μM A-740003. Displaceable binding is also observed to the colliculus, cortex, and striatum.
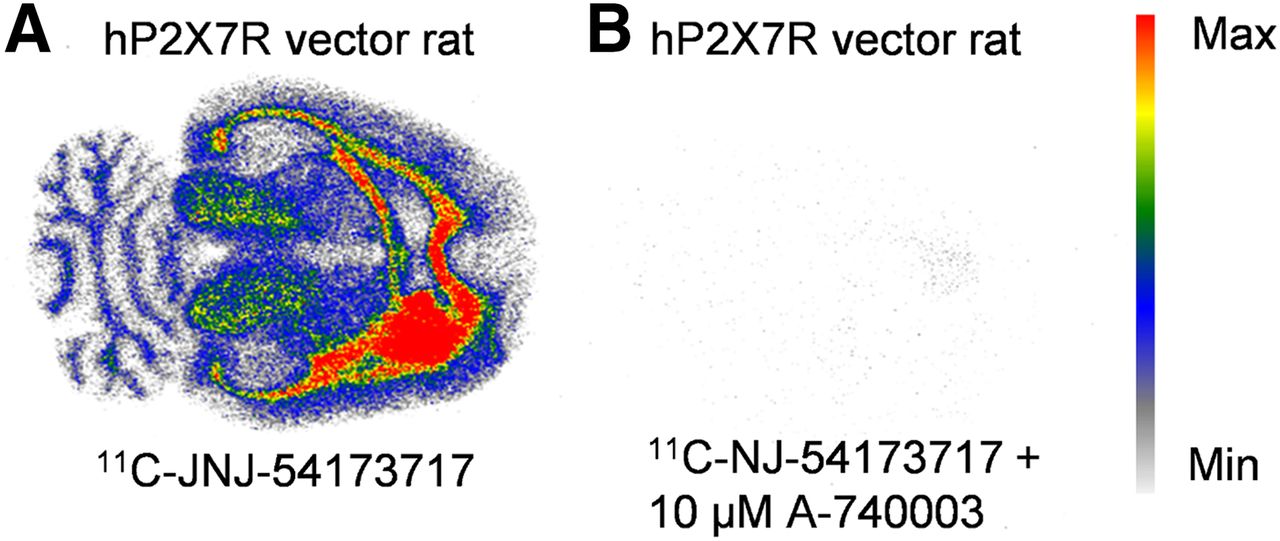
In vitro autoradiography binding studies on slices of rat brain injected with hP2X7R vector using 11C-JNJ-54173717 (n = 4). 11C-JNJ-54173717 shows specific binding to rAAV_3flag-hP2X7R–injected site (A), which can be blocked with 10 μM A-740003 (B). Max = maximum; min = minimum.
Small-Animal PET Imaging of hP2X7R Expression
A high-intensity signal was observed in the hP2X7R vector–injected right striatum compared with only background radioactivity in the eGFP control vector injected (Fig. 4A). This was also reflected in the baseline time–activity curves in which accumulation of radioactivity was observed in the right striatum with washout from the left striatum (Fig. 4C). The average binding potential (±SD) calculated with SRTM was 0.63 ± 0.16 (n = 3). Pretreatment by subcutaneous injection of JNJ-54173717 (10 mg/kg) at 45 min before tracer injection resulted in a significant decrease of the radioactivity concentration in the hP2X7R-expressing right striatum (n = 3) (Figs. 4B and 4D).
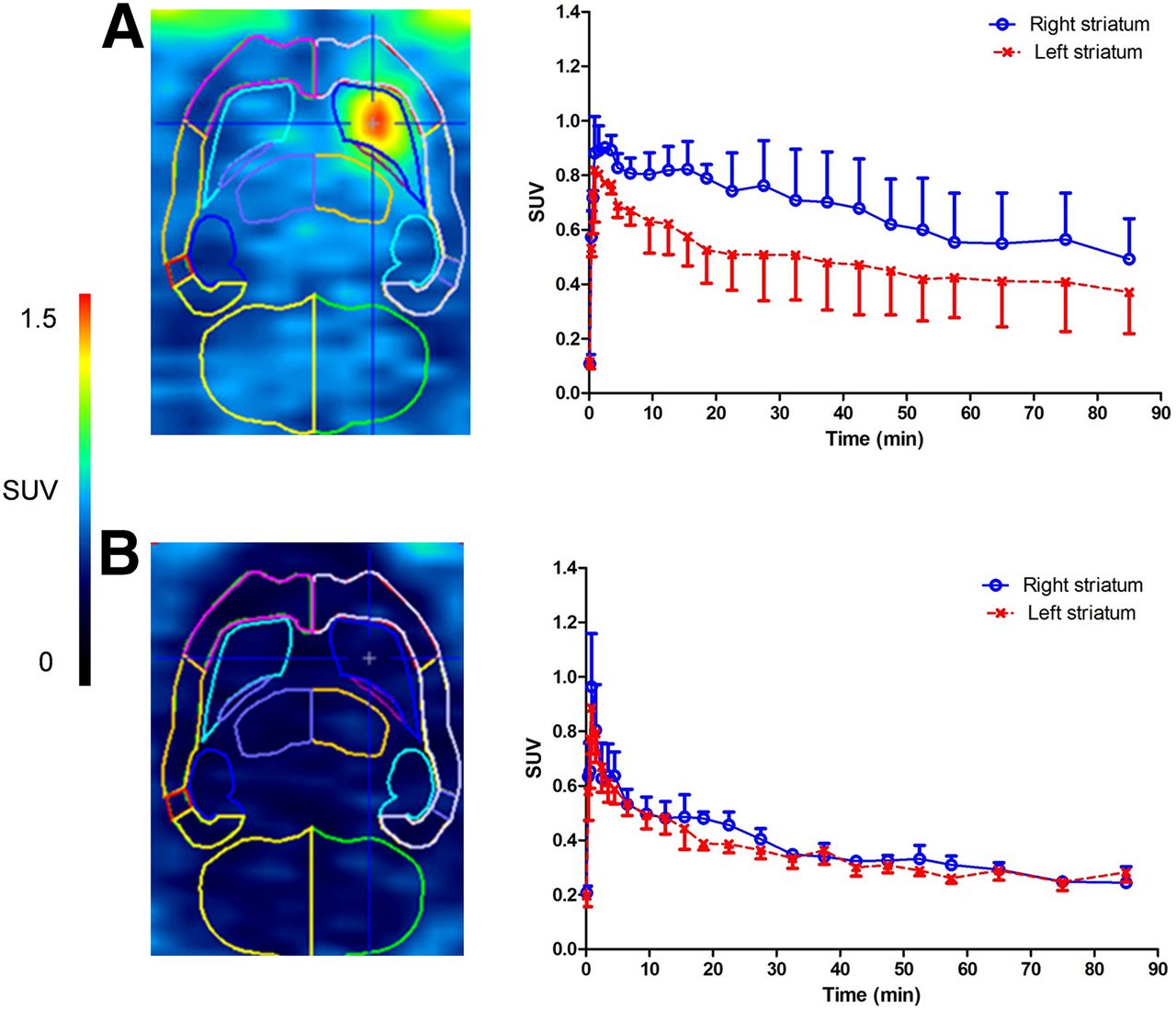
Small-animal PET data for 11C-JNJ-54173717 in hP2X7R rat model (right striatum, rAAV_3flag-hP2X7R, and left striatum, rAAV_3flag-GFP). Representative averaged small-animal PET images (same animal) of whole scan (90 min) were acquired at baseline with corresponding time–activity curves (A; n = 3) and after pretreatment with authentic reference material with corresponding time–activity curves (B; n = 3). Data were normalized for injected radioactivity and body weight of animal (SUV). Error bars correspond to SD.
Histology
To corroborate expression of the viral vectors, 3 animals were sacrificed after the small-animal PET scan to perform histology. The expression of the viral vectors was in accordance with the radioactivity accumulation observed in the small-animal PET data (n = 3; Supplemental Fig. 4).
Small-Animal PET Study in Rhesus Monkeys
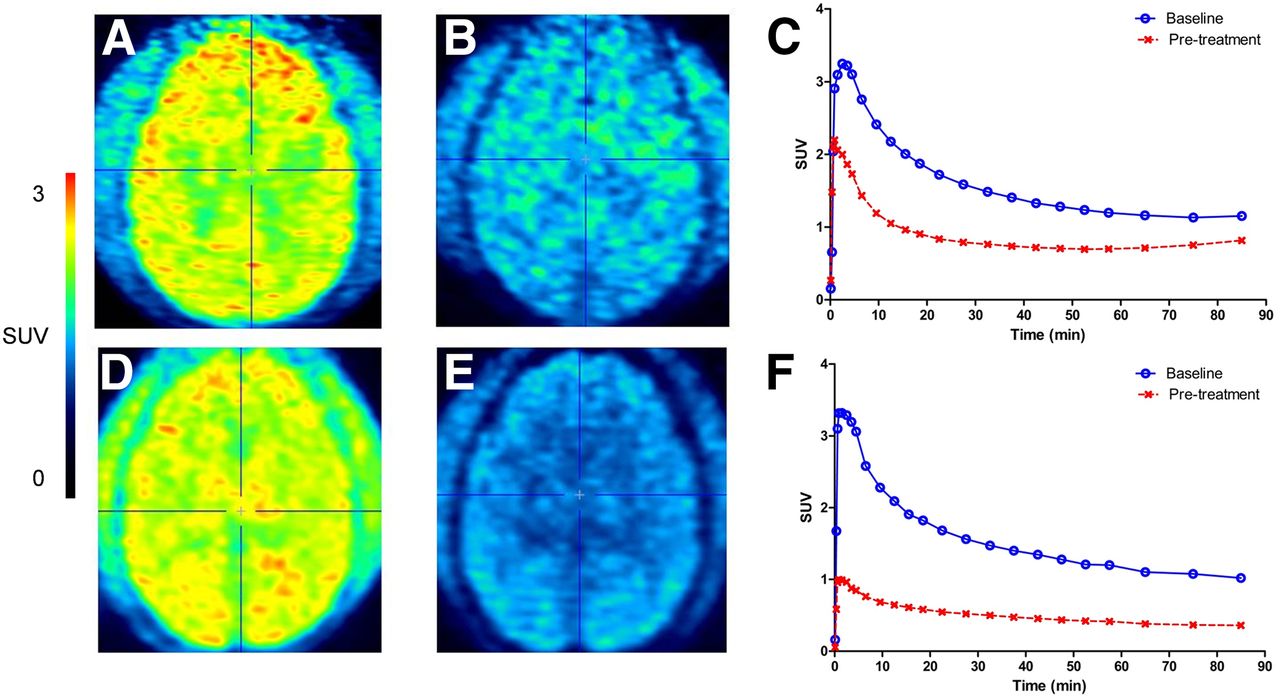
A relatively high initial brain uptake of 11C-JNJ-54173717 was observed in the monkey small-animal PET study (SUV, 3.3). The uptake of 11C-JNJ-54173717 was homogeneous throughout the monkey brain. Reduced uptake of 11C-JNJ-54173717 was observed after pretreatment (2.5 mg/kg intravenously, 10 min before tracer injection) with cold JNJ-54173717 and with JNJ-42253432 (Fig. 5) (15). The plasma SUVs of intact 11C-JNJ-54173717 for the first baseline scan were 0.55, 0.33, and 0.24, and for pretreatment (with JNJ-54173717) they were 0.52, 0.34, and 0.25 at 10, 30, and 60 min after tracer injection, respectively. The plasma SUVs of intact 11C-JNJ-54173717 for the second baseline scan in a different monkey were 0.69, 0.39, and 0.26, and for pretreatment (with JNJ-42253432) they were 0.54, 0.37, and 0.27 at 10, 30, and 60 min after tracer injection, respectively.

11C-JNJ-54173717 PET in rhesus monkey. Time-weighted averaged small-animal PET images (10–30 min) of baseline study (A) and after pretreatment with authentic reference material (2.5 mg/kg intravenously 10 min before tracer injection) (B). (C) Corresponding total brain time–activity curves at baseline and after pretreatment. Time-weighted averaged small-animal PET images (10–30 min) of second PET scan in different monkey at baseline (D) and after pretreatment with JNJ-42253432 (2.5 mg/kg intravenously 10 min before tracer injection) (E). (F) Corresponding total brain time–activity curves at baseline and after pretreatment.
DISCUSSION
The P2X7R plays an important role during neuroinflammation and therefore synthesis of small molecules that can act as antagonists, inhibiting P2X7R function in the brain, are of particular interest. The interspecies difference of P2X7R may, however, be a major hurdle in the development of P2X7-targeted drugs. Most P2X7R antagonists described in the literature are not active or weakly active against P2X7R isoforms present in mouse and rat, underscoring the need of an animal model expressing the hP2X7R. During the last decade, PET has become a valuable tool in the development of new drugs. Noninvasive imaging of P2X7R using PET would allow studying this receptor in both healthy and diseased conditions. Furthermore, it would be useful in the drug development process because it gives direct insight in the relation between P2X7R occupancy and the dose of the candidate antagonist, allowing the validation or invalidation of a new drug candidate at an early stage, thereby saving a lot of effort and money, making the drug development process more efficient.
In this study, we therefore aimed to develop a cell and animal model expressing the human isoform of the P2X7R and validate these humanized models with a newly developed high-affinity P2X7R PET tracer.
Unlike 11C-A-740003, a P2X7R-selective tracer developed by Janssen et al. (24), 11C-JNJ-54173717 was able to cross the blood–brain barrier as was shown in biodistribution studies in normal rats. Excretion was mainly hepatobiliary, as was expected in view of the lipophilicity of the tracer. To use a PET radioligand for accurate measurements of target density and target occupancy in drug dose occupancy studies, knowledge of radiometabolites is required, more particularly to which extent radiometabolites are present in plasma and their ability to pass the blood–brain barrier and contribute to specific or nonspecific binding. Therefore, the in vivo stability of 11C-JNJ-54173717 was examined in plasma and the perfused brain of normal rats. In plasma, 1 unidentified polar radiometabolite was detected, probably generated by cleavage of the amide bond. The radiotracer appeared to be, however, quite stable because still 70% of the radioactivity in plasma was in the form of intact tracer 30 min after radiotracer injection. In the brain, ideally, no radiometabolites should be present, because they may complicate PET quantification. A negligible fraction of radiometabolites (<2%) of 11C-JNJ-54173717 was found in the cerebrum and cerebellum 30 min after injection.
In vitro autoradiography studies with 11C-JNJ-54173717 performed on rat brain slices showed higher tracer binding to the hP2X7R viral vector–injected striatum than the contralateral eGFP viral vector–injected striatum, proving the specificity of tracer binding to hP2X7R. Interestingly, in vitro autoradiography binding studies on WT mouse brain slices showed specific binding, suggesting this tracer has also affinity for the mouse P2X7R. In WT rat brain, high displaceable binding to white matter was observed. This binding pattern was, however, not observed in the binding study with 3H-A-804598 performed by Able et al. (13) nor in the binding studies with 11C-JNJ-54173717 on the mouse brain sections.
Further evaluation and validation of our model and tracer was done using in vivo small-animal PET studies. Time–activity curves showed 11C-JNJ-54173717 binding in the hP2X7R vector–injected striatum and fast washout from the contralateral eGFP vector–injected striatum. Pretreatment experiments with the authentic reference material showed that tracer binding is specific. Binding potential values were calculated using the SRTM with the contralateral left striatum as a reference region. The obtained binding potential values were not that high. This can be explained by the fact that the transfected brain volume is much smaller than the striatum itself. Nevertheless, the use of mirror brain volume of interest is optimal for SRTM kinetic modeling, and the fits were robust. Because there is also endogenous expression of rat P2X7R throughout the rat brain (also in the contralateral striatum), this measures the increase in binding potential mediated by the expression of the hP2X7R. Confirmation of the expression of the injected viral vectors was done by histology on rat brain sections showing clear expression of the corresponding proteins.
In the monkey brain, pretreatment with the cold JNJ-54173717 and with the P2X7R antagonist JNJ-42253432 (15) reduced uptake of 11C-JNJ-54173717, suggesting P2X7R-selective binding of 11C-JNJ-54173717 in the rhesus monkey. Tracer binding in surrounding tissue was also lower after pretreatment, suggesting that P2X7R also blocks tracer retention in extracerebral tissues in-line with the ubiquitous expression of P2X7. The increase of tracer concentration at the end of the scan may indicate accumulation of radiometabolites in the brain. This was, however, not observed in the second study in a different monkey (baseline and pretreatment with JNJ-42253432) nor was it observed in rat studies. Almost no difference was observed in the metabolism of 11C-JNJ-54173717 between the baseline and pretreatment study, indicating absence of inhibition of 11C-JNJ-54173717 metabolism by authentic JNJ-54173717 or by JNJ-42253432.
CONCLUSION
We have developed a new PET brain reporter gene system based on hP2X7R expression via viral vector technology, which was validated with a new selective P2X7R PET tracer, 11C-JNJ-54173717. This humanized animal model can be used in the development and screening of new drugs targeting the hP2X7R. 11C-JNJ-54173717 selectively visualized P2X7R in the monkey brain, and this radioligand will be further evaluated in a clinical setting to study P2X7R expression levels in neurodegenerative disorders. We expect that also in humans, endogenous expression will be high enough to be quantified by 11C-JNJ-54173717 PET, and application of the pseudoreference tissue model (25) may be a valid approach for quantification.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. Dieter Ory is a fellow of the Research Foundation Flanders. Koen Van Laere is a Senior Research Fellow for the Research Foundation Flanders. Viral vectors were engineered and produced by the Leuven Viral Vector Core (Leuven, Belgium). No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Ann Van Santvoort, Julie Cornelis, and Ivan Sannen for their assistance in the animal work.
Footnotes
↵* Contributed equally to this work.
Published online May 19, 2016.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication November 19, 2015.
- Accepted for publication April 5, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- TSPO Versus P2X7 as a Target for Neuroinflammation: An In Vitro and In Vivo Study
- Preclinical Evaluation and Nonhuman Primate Receptor Occupancy Study of 18F-JNJ-64413739, a PET Radioligand for P2X7 Receptors
- P2X7 PET Radioligand 18F-PTTP for Differentiation of Lung Tumor from Inflammation
- 18F-JNJ-64413739, a Novel PET Ligand for the P2X7 Ion Channel: Radiation Dosimetry, Kinetic Modeling, Test-Retest Variability, and Occupancy of the P2X7 Antagonist JNJ-54175446