


Abstract
Cyclooxygenase (COX), a prostanoid-synthesizing enzyme, is considered to be involved in the neuroinflammatory process of neurodegenerative diseases. However, the role of COX in the progression of neurodegeneration is not well understood. We hypothesized that in vivo imaging of COX by PET will contribute to elucidation of the function of COX during the neurodegenerative process in Alzheimer’s disease (AD). 11C-labeled ketoprofen methyl ester (racemic (RS)-11C-KTP-Me) developed recently by our group is a useful PET probe for in vivo imaging of COX-1 during neuroinflammation. The (S)-enantiomer of ketoprofen is known to be pharmacologically more active than the (R)-enantiomer. We thus synthesized 11C-labeled (S)-ketoprofen methyl ester ((S)-11C-KTP-Me) as an improved PET probe specific for COX-1 and applied it for investigation of the changes in COX-1 during the progression of AD in a mouse model. Methods: The specificity of (S)-11C-KTP-Me for COXs was examined in PET studies with rats that had intrastriatal injection of lipopolysaccharide. To determine the details of changes in COX-1 during progression of amyloid-β (Aβ) plaque formation in amyloid precursor protein transgenic (APP-Tg) mice, we performed immunohistochemical studies and ex vivo autoradiography with (S)-11C-KTP-Me. Results: PET studies using hemispheric lipopolysaccharide injection into rats revealed that the sensitivity of (S)-11C-KTP-Me in neuroinflammation was much higher than that of (RS)-11C-KTP-Me and (R)-11C-KTP-Me; these results closely corresponded to the inhibitory activities of each enantiomer against COX-1 estimated by an in vitro assay. In APP-Tg mice, (S)-11C-KTP-Me administration resulted in progressive and significant increases in accumulation of radioactivity in the brain from 16 to 24 mo old in accordance with the histopathologic appearance of abundant Aβ plaques and activated microglia, whereas few changes in radioactivity accumulation and few Aβ plaques were seen in age-matched wild-type control mice. High-radioactivity accumulation by (S)-11C-KTP-Me was markedly observed in the frontal cortex and hippocampus in which COX-1–expressing activated microglia tightly surrounded and enclosed large and more intensely stained Aβ plaques, indicating neuroinflammation that originated with Aβ. Conclusion: (S)-11C-KTP-Me is a potent PET probe that is highly selective for COX-1. Studies using APP-Tg mice demonstrated that (S)-11C-KTP-Me could detect activated microglia that are associated with amyloid plaque progression, suggesting the involvement of COX-1 in the neuroinflammatory process in AD.
Neuroinflammation has been hypothesized to be critical in the pathologic cascades of Alzheimer’s disease (AD), in which the deposition of amyloid-β (Aβ) leads to aggregates of hyperphosphorylated tau protein, synaptic disruption, neuronal cell death, and ultimately, dementia (1–3). Prolonged inhibition of cyclooxygenase (COX) by nonsteroidal antiinflammatory drugs (NSAIDs) reduces the risk and provides therapeutic value in both AD patients and animal disease models (4). Both COX-1 and COX-2 are changed in the brain of AD patients (5,6). However, the relative contribution of each COX to AD pathogenesis and progression is not well understood.
Recently, we developed a COX imaging probe, 11C-labeled ketoprofen methyl ester (11C-KTP-Me), as an esterified proradiotracer for brain penetration by increasing lipophilicity of its active form, ketoprofen, which is selective for COX-1. 11C-KTP-Me is promptly and almost completely hydrolyzed to acid form, 11C-KTP, in rat brain within 5 min after the intravenous injection and can be used with PET to visualize time-dependent changes in COX-1 in activated microglia during neuroinflammatory processes (7,8). This finding provided evidence that COX-1 plays a critical role in microglial activation during acute neuroinflammation in vivo. Ketoprofen is generally available as a racemate. Indeed, its (S)-enantiomer is the active compound that shows antiinflammatory effects (9). Therefore, in the present study, we established a solid chiral resolution method with semipreparative high-performance liquid chromatography (HPLC) to obtain 11C-labeled (S)-ketoprofen methyl ester ((S)-11C-KTP-Me) and (R)-ketoprofen methyl ester ((R)-11C-KTP-Me) from the racemic mixture, with the objective of evaluating the specificity and sensitivity for COX-1 to assess the function of COX-1 in the pathologic process of AD progression in vivo. The characteristics of each enantiomer, (S)- or (R)-11C-KTP-Me, as an imaging agent for neuroinflammation were investigated using a rat model created by hemispheric intrastriatal injection of lipopolysaccharide and also using an AD model animal, amyloid precursor protein transgenic (APP-Tg) mice, to elucidate the role of COX-1 in the pathogenesis and progression of AD.
MATERIALS AND METHODS
Radiochemistry
Detailed procedures are provided in the supplemental data (available at http://jnm.snmjournals.org). According to a previous report, 11C-labeled ketoprofen methyl ester as a racemate ((RS)-11C-KTP-Me) was synthesized by the reaction of 11C-CH3I and (3-benzophenyl)acetic acid methyl ester in the presence of bases (7). The radioactivity of the final samples including (RS)-11C-KTP-Me was 2.5–4.0 GBq, and the specific activity was 30–50 GBq/μmol. The chemical purity as determined by UV-HPLC analysis at 255 nm was greater than 99%, and the radiochemical purity as determined by radio-HPLC analysis was greater than 99%. The reaction mixture containing (RS)-11C-KTP-Me was injected into a semipreparative HPLC system with a chiral column to separate (R)- and (S)-11C-KTP-Me (Supplemental Fig. 1). The total synthesis time, including the 11C-labeling reaction, chiral resolution by HPLC, and radiopharmaceutical formulation, was less than 30 min. The radioactivity values of the samples of (R)- and (S)-11C-KTP-Me were 1.4 and 1.0–1.6 GBq, respectively, and the specific activities of (R)- and (S)-11C-KTP-Me were 20 and 20–50 GBq/μmol, respectively. The 11C-CH3I–based decay-corrected radiochemical yields of (R)- and (S)-11C-KTP-Me were calculated to be 20% and 14%–23%, respectively. The chemical purity as determined by UV-HPLC analysis at 255 nm was greater than 99%, and the radiochemical purity as determined by radio-HPLC analysis was greater than 99%. The enantiomeric excess as determined by chiral HPLC analysis was greater than 99% each for (R)- and (S)-11C-KTP-Me.
In Vitro Assay for COX Inhibitory Activity
The inhibitory activity of (RS)-, (R)-, and (S)-ketoprofen for the recombinant COX-1 and -2 enzymes was measured using a colorimetric COX (ovine) inhibitor screening assay kit (Cayman Chemical Co.). All inhibitors added to the reaction system were dissolved in dimethyl sulfoxide and prepared just before use. In this assay, the COX activity was measured using N,N,N′,N′-tetramethyl-p-phenylenediamine as a cosubstrate with arachidonic acid (reduction of prostaglandin G2 to prostaglandin H2). N,N,N′,N′-tetramethyl-p-phenylenediamine oxidation was monitored spectrophotometrically with a 96-well plate reader at 590 nm. The 50% inhibitory concentration (IC50) was calculated from a concentration–inhibition response curve.
Animals and Surgery
All experimental protocols were approved by the Animal Care and Use Committee of RIKEN Kobe Institute (MAH21-21-4) and were performed in accordance with the Principles of Laboratory Animal Care (10). Three to 4 rats or 4–5 mice were housed per cage under a 12-h light–dark cycle (lights off at 20:00) at 23 ± 1°C and 60% ± 5% humidity and were allowed access to food and water ad libitum. We used male Wistar rats aged 10–13 wk from CLEA Japan, Inc., for preparation of the rat model of acute neuroinflammation. Under 50 mg of sodium pentobarbital anesthesia per kilogram, lipopolysaccharide (strain 026:B6; Sigma-Aldrich. Co. Ltd.) diluted in saline was injected into the striatum (0.5 μg/μL at a rate of 0.2 μL/min for 5 min) of the rat using a 26-gauge needle controlled by an automated syringe pump (Muromachi Kikai Co., Ltd.). The stereotactic coordinates from bregma were as follows: anteroposterior, + 0.2 mm; lateral, + 3.2 mm; and ventral, −5.5 mm from the dura. At the end of the injection, the needle was left in place for 5 min before being slowly removed.
Mice that are models of AD were purchased from Taconic Farms, Inc. The APPSWE (Tg2576) mouse model (11) carries a transgene coding for the 695-amino acid isoform of human APP with the Swedish mutation (K670N, M671 L) (12). Non-Tg littermates were used as wild-type controls.
PET Imaging Studies
The rats were anesthetized with a mixture of 1.5% isoflurane and nitrous oxide/oxygen (7:3) and placed on the bed of a small-animal PET scanner (microPET Focus-220; Siemens AG). A venous catheter for PET probe injection was inserted into the tail vein before PET scanning. (RS)-, (R)-, or (S)-11C-KTP-Me (∼70 MBq per animal) dissolved in 1 mL of saline was injected via the cannula inserted into the tail vein for 10 s, and emission data were then acquired for 45 min using the 3-dimensional list-mode method of data acquisition. The data were reconstructed with a standard 2-dimensional filtered backprojection (ramp filter, cutoff frequency at 0.5 cycles per pixel). Regions of interest were placed on the ipsilateral and contralateral sides of the striatum using image processing software (Pmod, version 3.2; PMOD Technologies Ltd.) according to the fused PET/MR T1-weighted image. Regional uptake of radioactivity in the brain was decay-corrected to the injection time and expressed as the SUV: SUV = tissue radioactivity concentration (MBq/mL)/injected radioactivity (MBq) × body weight (g).
Ex Vivo Autoradiography
Ex vivo autoradiographic assessments of regional brain radioactivity were conducted for Tg2576 and wild-type mice. Because a rapid and transient brain time–activity curve was observed in our preliminary PET study of mice using (S)-11C-KTP-Me, we decided to use the time point at 15 min to avoid the effect of cerebral blood flow. Fifteen minutes after (S)-11C-KTP-Me (∼1 MBq per g of body weight) injection, mice were sacrificed under deep anesthesia with 3.0% isoflurane, and the brain was sliced into coronal sections (1 mm thick) using a brain matrix (RBM-2000C; ASI Instruments, Inc.) on ice. The coronal slices were placed on an imaging plate (BAS-SR2040; Fuji Photo Film Co.) for 1 h. The exposed imaging plate was then scanned with a bio-imaging analyzer (FLA-7000IR; Fuji Photo Film Co.). To obtain quantitative data from ex vivo autoradiograms, regions of interest were manually placed on the brain regions according to the digital photograph obtained from a photo scanner. Brain regional radioactivity was represented by relative intensity of photostimulated luminescence (PSL/mm2) and calibrated to the uptake value (percentage injected dose per gram), which was defined as 100 × tissue activity concentration (MBq/g)/injected dose (MBq) using whole-brain radioactivity measured by a γ-well counter (Wizard 1480; PerkinElmer, Inc.).
Immunohistochemistry
The mice were deeply anesthetized and perfused transcardially with ice-cold 0.9% saline followed by 4% paraformaldehyde solution. The brains were removed and postfixed with formalin solution for 6 h, placed in a 30% sucrose solution, and embedded in optimal-cutting-temperature compound (Sakura Finetek Japan Co.). Serial coronal sections (20 μm thick) were obtained with a cryostat and thaw-mounted on glass slides. The antibodies used in this study were COX-1 (goat IgG [Santa Cruz Biotechnology, Inc.], 1:100), COX-1 (rabbit IgG [Cayman Chemical Co.], 1:500), COX-2 (goat IgG [Santa Cruz Biotechnology, Inc.], 1:100), Iba-1 (rabbit IgG [Wako], 1:1,000), CD11b (rat IgG [AbD Serotec], 1:500), and Aβ1–16 (mouse IgG [Covance], 1:1,000). After overnight incubation of the brain sections with the mixed antibodies at room temperature, the primary antibodies were visualized with a secondary antibody labeled with Alexa Fluor 488 (Invitrogen, 1:500), Cy3- (Jackson ImmunoResearch, 1:500), or Cy5 (Jackson ImmunoResearch, 1:500). Fluorescent images were captured with a 3-laser confocal microscope (Olympus Optical Co.).
Statistical Analysis
Statistical analysis was performed using the Student t test. P values of less than 0.05 were considered to be statistically significant.
RESULTS
Radiochemistry
Figure 1 illustrates the synthetic scheme of 11C-labeled (RS)-, (R)-, and (S)-KTP-Me. (R)- and (S)-11C-KTP-Me were successfully obtained with sufficient purity using chiral column separation.

Synthesis of (R)- and (S)-11C-KTP-Me via rapid C-11C-methylation and chiral resolution.
Inhibitory Activity of Ketoprofen Enantiomers Against COXs In Vitro
As shown in Table 1, (RS)-ketoprofen and (S)-ketoprofen showed inhibitory activity against COXs. The inhibitory activity of (RS)-ketoprofen and (S)-ketoprofen was highly selective for COX-1, and the COX-1/COX-2 ratios of (RS)-ketoprofen and (S)-ketoprofen were 33.5 and 17.7, respectively (Table 1). On the other hand, (R)-ketoprofen showed less inhibitory activity against both COX-1 and COX-2 (IC50 of 126.7 and 470.7 μM, respectively). In agreement with previous reports showing that the (S)-enantiomers of 2-arylpropionic acid derivatives in the profen family are efficient natural antiinflammatory agents (9), our results also clearly indicate that the (S)-enantiomer of ketoprofen was the active form and was specific for COX-1.
Inhibitory Effects of Ketoprofen Enantiomers on Ovine COX-1 and COX-2 Activities (IC50, μM)
In Vivo PET Imaging of Neuroinflammation in Rat Brain with (R)- and (S)-11C-KTP-Me
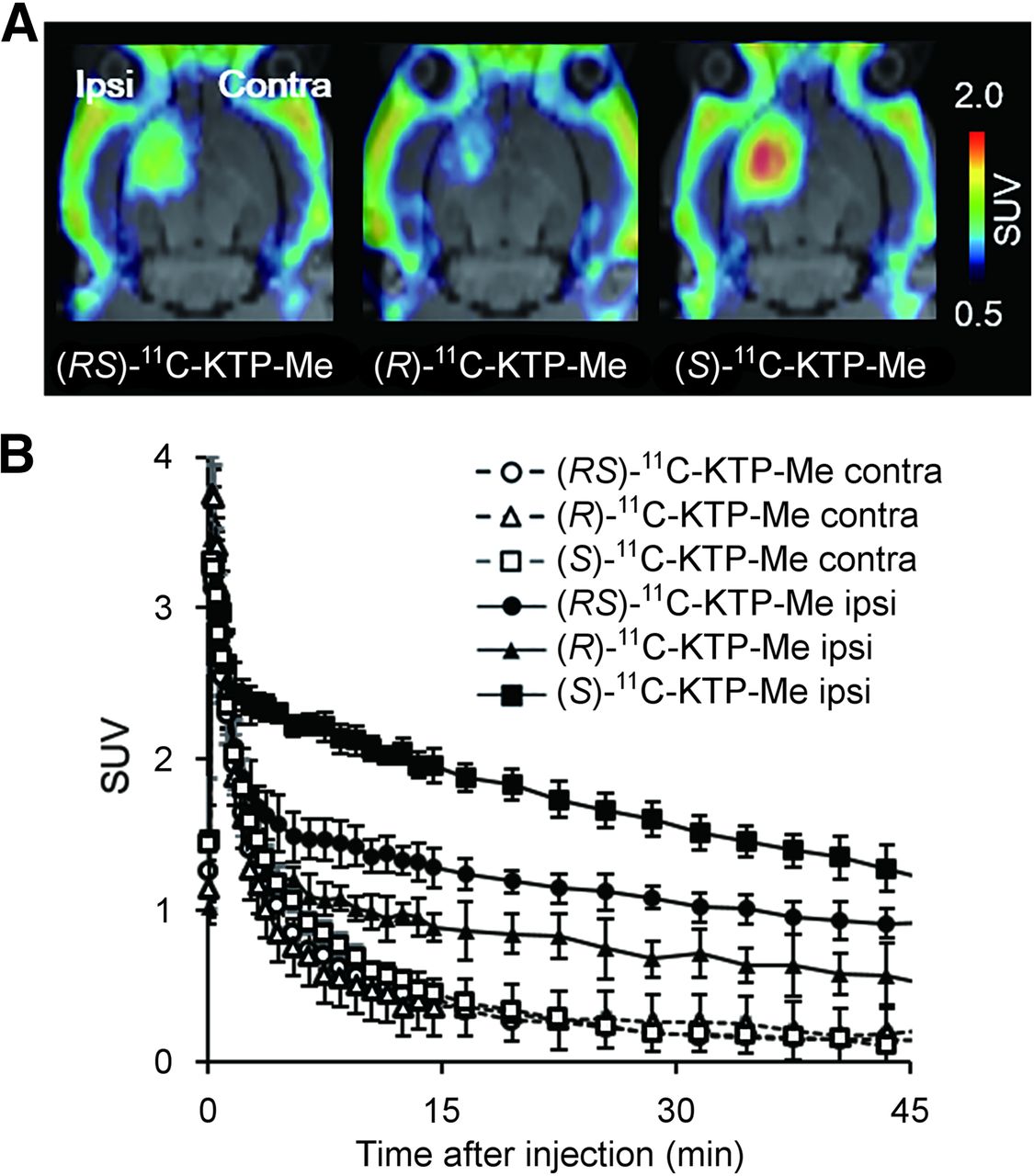
PET images generated by averaging dynamic data from 5 to 45 min after tracer injection revealed that (S)-11C-KTP-Me displayed high radioactivity accumulation in the ipsilateral striatum injected with lipopolysaccharide compared with the contralateral side (Fig. 2A). Quantification with region-of-interest base analysis showed that the uptake value of radioactivity in the inflamed region after (S)-11C-KTP-Me injection was almost 1.5-fold higher than that of (RS)-11C-KTP-Me (P < 0.001, t test, Table 2). On the other hand, (R)-11C-KTP-Me showed a low level of radioactivity accumulation and it was not specific because intravenous administration of unlabeled (R)-KTP-Me (10 mg/kg) did not produce any changes in brain radioactivity accumulation (Supplemental Fig. 2). The time–activity curves of (S)-11C-KTP-Me showed that the peak was reached within 1 min after injection on both the ipsilateral and the contralateral sides, and the maximum uptake value was comparable to that of (RS)-11C-KTP-Me and (R)-11C-KTP-Me (Fig. 2B). The time–activity curves in the contralateral striatum as the reference region revealed that all radiotracers were rapidly washed out in the normal condition by 15 min after injection. In the lipopolysaccharide-injected neuroinflammatory striatum, (RS)-11C-KTP-Me and (S)-11C-KTP-Me, but not (R)-11C-KTP-Me, showed a delay in clearance within 5 min and remained for 45 min after injection, resulting in high accumulation in the inflamed brain region. The retention rate of radioactivity after (S)-11C-KTP-Me injection was dramatically improved compared with that of (RS)-11C-KTP-Me (the ratio of radioactivity at 45 min to the maximum uptake value, 38.6% and 28.5%, respectively).

Representative PET images and time–radioactivity curves of each 11C-KTP-Me enantiomer in rat brain after lipopolysaccharide injection. (A) Transaxial rat brain views of SUV-summed PET images from 5 to 45 min after tracer injection were coregistered with individual MR images at baseline. (S)-11C-KTP-Me showed higher accumulation and superior specificity in rat brain hemisphere than (RS)- and (R)-11C-KTP-Me. (B) Quantitative time–radioactivity curves of each 11C-KTP-Me enantiomer in contralateral (Contra) and ipsilateral (Ipsi) striatum. Data are expressed as SUV and are mean ± SD ((R)-[11C]KTP-Me, n = 3; others, n = 4).
Regional Brain Accumulation of 11C-KTP-Me Enantiomers in Rat Brain After Intrastriatal Injection of Lipopolysaccharide
Imaging of COX-1 in APP-Tg Mouse Brain by (S)-11C-KTP-Me
To analyze the details of COX-1 changes during the process of AD progression, we performed an ex vivo autoradiography study using APP-Tg mice. Before that, the pharmacokinetic profiles of intravenously administered (S)-11C-KTP-Me in the brains of mice were investigated by in vivo PET to determine the timing of sacrifice for brain sampling. Because (S)-11C-KTP-Me showed rapid uptake and washout and became stable at 15 min after injection, we performed the ex vivo autoradiography study with APP-Tg mice at 15 min after (S)-11C-KTP-Me injection (Supplemental Fig. 3). In ex vivo autoradiography of APP-Tg mice by (S)-11C-KTP-Me, a remarkable increase in radioactivity accumulation was observed compared with that in wild-type mice (Fig. 3). Brain accumulation of radioactivity after (S)-11C-KTP-Me injection was elevated in an age-dependent manner and became significant from 16 mo old. This was particularly remarkable in the hippocampus and cerebral cortex. Quantitative region-of-interest base analysis also clearly showed age-dependent increases in radioactivity accumulation by (S)-11C-KTP-Me in all brain regions of APP-Tg mice (Fig. 4). At 24 mo old, the increased levels in the hippocampus and cerebral cortex of APP-Tg mice were approximately 2-fold higher than those of age-matched wild-type mice.

Age-dependent changes in (S)-11C-KTP-Me accumulation in brain of APP-Tg 2576 and wild-type mice. Representative ex vivo autoradiographs of coronal sections of mice at 15 min after PET tracer injection. Progressive and remarkable increases in (S)-11C-KTP-Me in frontal cortex, parietal occipital cortex, and hippocampus of APP-Tg mice from 16 mo old. Range bar represents intensity of photostimulated luminescence (PSL/mm2).
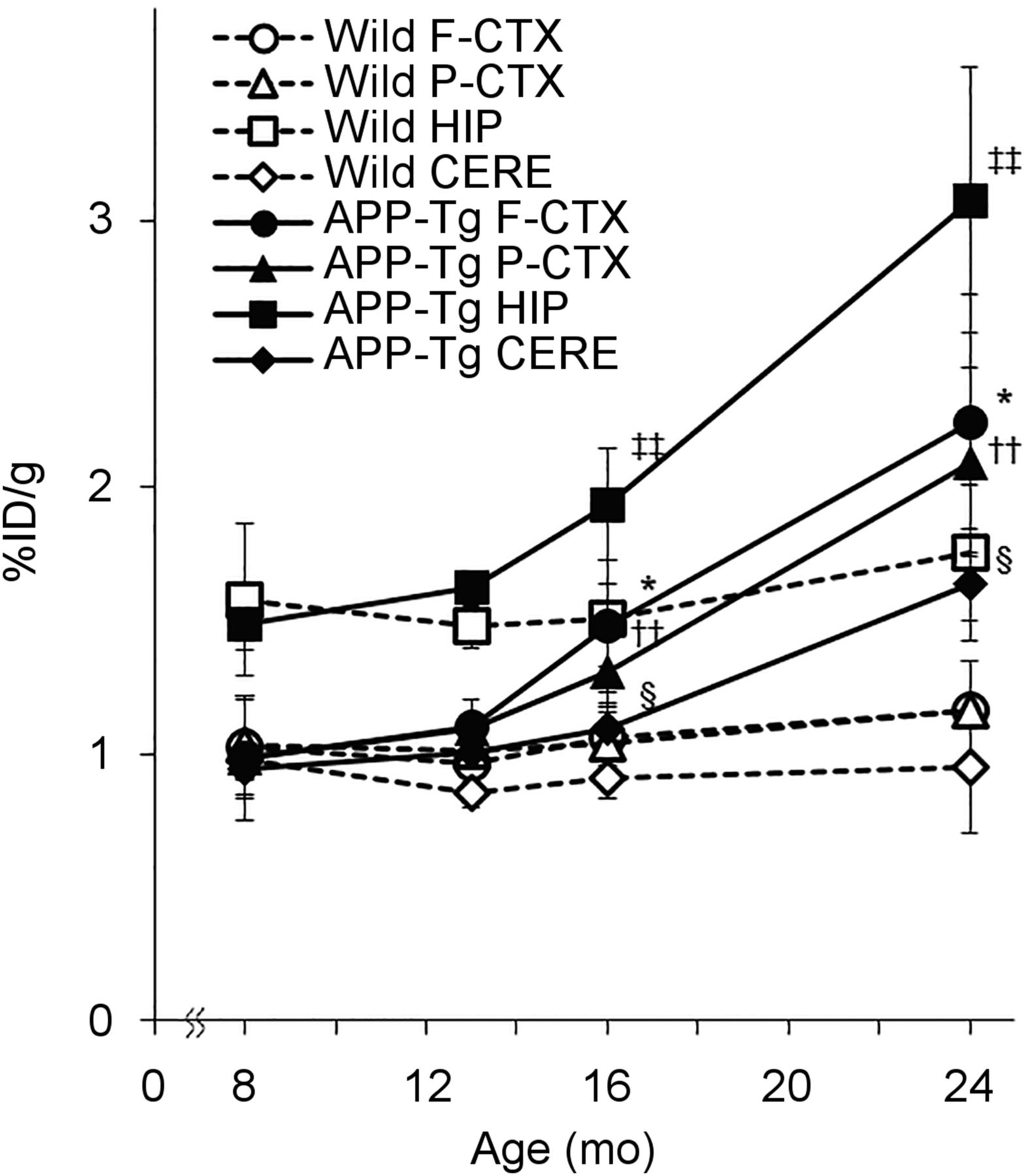
Age-dependent changes in regional brain uptake of (S)-11C-KTP-Me in APP-Tg 2576 and wild-type mice obtained from quantification of ex vivo autoradiography. Significant increases in (S)-11C-KTP-Me accumulation in APP-Tg mice are shown in all brain regions at 16 and 24 mo old compared with wild-type mice. Data are expressed as percentage injected dose per gram (%ID/g) and are mean ± SD (8 mo, n = 3; 13 and 24 mo, n = 4; 16 mo, n = 5). *P < 0.05, frontal cortex of APP-Tg mice vs. wild-type mice. ††P < 0.01, parietal cortex of APP-Tg mice vs. wild-type mice. ‡‡P < 0.01, hippocampus of APP-Tg mice vs. wild-type mice. §P < 0.05, cerebellum of APP-Tg mice vs. wild-type mice. CERE = cerebellum; F-CTX = frontal cortex; HIP = hippocampus; P-CTX = parietal cortex; STR = striatum.
Changes in COXs During Pathologic Process in APP-Tg Mice
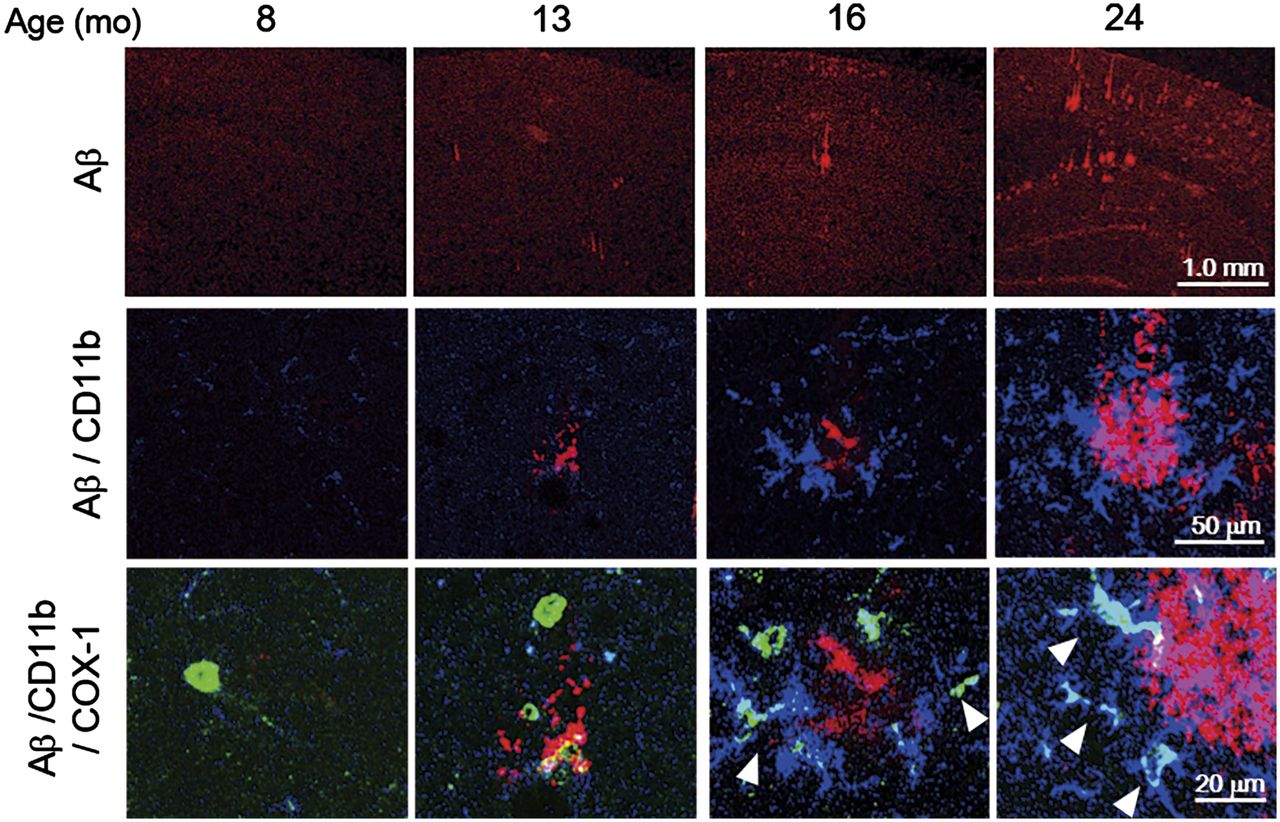
The immunohistochemical analysis of Aβ demonstrated that Aβ plaques were abundant in the hippocampus and cerebral cortex, in which significantly high accumulations of (S)-11C-KTP-Me were observed in the 24-mo-old APP-Tg mice (Supplemental Fig. 4A). To identify the cellular origin of (S)-11C-KTP-Me accumulation in APP-Tg mice, we performed triple immunostaining for COX-1 or COX-2 with microglial markers Iba-1 or CD11b and Aβ1–16. Aβ plaques were age-dependently increased in size and number, and COX-1 immunopositive microglia were also found in association with areas of abundant Aβ (Fig. 5). At 24 mo old, several Iba-1–positive activated microglia surrounding Aβ plaques in the hippocampus of APP-Tg mice also showed COX-1 coexpression, whereas COX-2 was not coexpressed with Iba-1–positive microglia (Supplemental Figs. 4B and 4C). As shown in 3-dimensional images of immunohistography of COX-1, CD11b, and Aβ plaques, COX-1 immunopositive microglia represented the active morphology and showed tight contact structures with Aβ plaques (Supplemental Fig. 5).

Age-dependent correlation between COX-1–expressing activated microglia and Aβ plaque formation in APP-Tg 2576 mouse brain. Representative photomicrographs of triple immunofluorescent labeling for Aβ1–16 (red), CD11b (blue), and COX-1 (green) in hippocampus of APP-Tg mice at 8, 13, 16, and 24 mo old. Arrows indicate cells coexpressing CD11b and COX-1. Population and fluorescence intensity of COX-1–expressing CD11b-positive activated microglia showed age-dependent increase from 16 mo old, which was well correlated with formation of Aβ oligomers.
DISCUSSION
Recently, much attention has been paid to the beneficial effects of NSAIDs due to the role of COX activity in the inflammatory cascade in AD (4,13). However, clinical epidemiologic analysis is still controversial, and the details of the mechanism are also not well understood. In the present study, we established a live imaging technique for monitoring neuroinflammation in vivo using PET with (S)-11C-KTP-Me for COX-1. PET images of (S)-11C-KTP-Me specifically detected and clearly visualized the changes in COX-1 in activated microglia during the formation of amyloid plaques in the brain of AD model mice. This finding indicates the possibility that neuroinflammation driven by COX-1 activity in activated microglia may be involved in the pathogenesis of AD and provides new evidence for the mechanism of the therapeutic effect of NSAIDs in AD patients.
According to the classic view of 2 COX isoenzymes, constitutive COX-1 has only homeostatic and housekeeping functions, and inducible COX-2 is mainly involved in inflammation (14,15). Therefore, COX-2 has been considered a pharmacologic target for selective therapies for neuroinflammation. However, several trials with COX-2–selective inhibitors such as celecoxib and rofecoxib have failed to ameliorate AD, whereas COX-1–selective or nonselective NSAIDs such as indomethacin and ibuprofen show Aβ-lowering properties in both AD transgenic mice and cell cultures (16–20).
Through research with imaging techniques including radiotracers and fluorescent probes, more attention has been paid to COX-2 than COX-1 as a molecular target for inflammation, and this has resulted in a failure to develop reliable probes for neuroinflammation (21,22). In the present study, (S)-11C-KTP-Me showed higher radioactivity accumulation in lipopolysaccharide-induced neuroinflammation than (RS)- and (R)-11C-KTP-Me, which was well correlated with the inhibitory activity against COX-1 by each enantiomer of ketoprofen. Though inhibitory activity of (S)-ketoprofen against COX-2 was also higher than that of (R)-ketoprofen, (S)-11C-KTP-Me is much more selective for COX-1 than COX-2, even in vivo, indicating that COX-1 in activated microglia may play a critical role in the process of neuroinflammation. Because (RS)-, (R)-, and (S)-11C-KTP-Me are proradiotracers, the hydrolysis rates are assumed to affect their dynamics in vivo. However, metabolic analysis of (RS)-11C-KTP-Me in our previous report demonstrated that the rate of hydrolysis did not show any differences between contralateral intact and neuroinflammatory brain region, indicating that the specificity of the radiotracer should be dependent on the inhibitory activity against COX-1.
The role of COX-1 in neuroinflammation is also supported by the facts that COX-1 inhibition using drug or gene deletion reduces neuronal damage and inflammatory responses after injection of lipopolysaccharide or Aβ in mouse brain (23,24). Moreover, the COX-1–selective inhibitor SC-560 reduces amyloid deposits and tau hyperphosphorylation and improves spatial learning and memory in triple transgenic AD mice (25). In our study of APP-Tg mice, (S)-11C-KTP-Me demonstrated age-dependent changes that closely corresponded to the increase in amyloid plaques as revealed by immunohistochemistry with an Aβ1–16 antibody. Importantly, expression of COX-1 was observed in activated microglia, which were tightly surrounded and enclosed by amyloid plaques. We previously reported that the expression level of COX-1 was constitutive and was not changed even in the lipopolysaccharide-induced inflamed area of the brain with activated microglia (8). Considering these results from our study, (S)-11C-KTP-Me could detect the increased numbers of activated microglia that is closely associated with development of Aβ plaques in AD progression. As in our results using AD model mice, previous studies with postmortem brain from AD patients showed an increase in COX-1–expressing microglia surrounding Aβ plaques (5,6). In addition, COX-2 is expressed in neurons in the early phase of the disease but not in microglia or astrocytes (26).
PET imaging of neuroinflammation may be a good surrogate marker involved in the amyloid plaque progression and tau aggregation (27,28). Neuroinflammatory responses induced by extracellular Aβ, which later become enhanced by aggregates of tau, have been suggested to be closely associated with synaptic disruption and neuronal cell death (1–3). (S)-11C-KTP-Me specifically detected microglial activation because COX-1 was expressed in microglia but not in astrocytes. Therefore, PET imaging using (S)-11C-KTP-Me could be useful for imaging of activated microglia and diagnosis of neuroinflammation in central nervous system diseases including AD.
Further studies on the role of COX-1 in AD progression are required to determine whether PET imaging of COX-1 using (S)-11C-KTP-Me could act as a valuable diagnostic tool for AD patients.
Microglial activation is classified into 2 phenotypes, proinflammatory (M1) and antiinflammatory or healing (M2), which are analogous to those of macrophages (29). Increasing evidence indicates that switching of the microglial phenotype from M2 to M1 occurs in the brain of AD and plays a significant role in disease progression (30). Targeting neuroinflammatory events in AD, primarily microglial activation, remains debatable as a promising therapeutic option because microglial phagocytosis may also contribute to the clearance of Aβ plaques from brain tissue. In the field of investigation of the specific role of COX-1 in activated microglia in the evolution of senile plaques in AD, (S)-11C-KTP-Me could be a valuable tool for understanding the role of activated microglia during AD progression.
CONCLUSION
We have established a live imaging technique for monitoring COX-1 in vivo using PET with (S)-11C-KTP-Me and have succeeded in visualizing the expression of COX-1 in activated microglia surrounding Aβ plaques in an AD model mice. Our results provide evidence that COX-1 is strongly involved in neuroinflammation during Aβ deposition and reinforce the potential of COX-1–selective NSAIDs for clinical use in AD. In addition, we propose that PET imaging of COX-1 with (S)-11C-KTP-Me could be a valuable diagnostic tool for monitoring activated microglia in neuroinflammation of central nervous system diseases including AD.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This study was supported by the Molecular Imaging Research Program from Japan’s Ministry of Education, Culture, Sports, Science, and Technology; Research Center Network for Realization of Regenerative Medicine from Japan Agency for Medical Research and Development; and a Grant-in-Aid for Young Scientists (B) of the Japan Society for the Promotion of Science. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Masahiro Kurahashi (Sumitomo Heavy Industry Accelerator Service Ltd.), Yasuhiro Wada, and Emi Hayashinaka (RIKEN) for their assistance in the PET study.
Footnotes
Published online Nov. 19, 2015.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 28, 2015.
- Accepted for publication October 16, 2015.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Synthesis and Preclinical Evaluation of 18F-Labeled Ketoprofen Methyl Esters for Cyclooxygenase-1 Imaging in Neuroinflammation
- A modest increase in 11C-PK11195-PET TSPO binding in depression is not associated with serum C-reactive protein or body mass index
- Evaluation of Two Potent and Selective PET Radioligands to Image COX-1 and COX-2 in Rhesus Monkeys