


Abstract
Nanobodies are single-domain antigen-binding fragments derived from heavy-chain antibodies that are devoid of light chains and occur naturally in Camelidae. We have shown before that their small size and high affinity and specificity for their target antigen make Nanobodies ideal probes for in vivo tumor imaging. In the present study, we have evaluated the use of Nanobodies as a generic method for imaging the in vivo biodistribution of specific immune cell types, using myeloid cells as an example. Methods: The cellular specificity of Nanobodies raised against murine bone marrow–derived dendritic cells was verified using flow cytometry on a range of myeloid and nonmyeloid cell types. The Nanobodies were then labeled with 99mTc and their biodistribution was analyzed using SPECT. The biodistribution was also assessed by measuring radioactivity in various organs and tissues. To verify whether the observed biodistribution was due to specific targeting through the antigen-binding loops, rather than retention in organs because of effects of the framework regions, we genetically grafted the antigen-binding loops of the Nanobodies onto the framework region of a Nanobody scaffold that by itself showed low background retention in the periphery. The cellular specificity and biodistribution of these grafted Nanobodies were determined as before. Results: Nb-DC2.1, which recognizes a wide range of myeloid cells, targets most strongly to the liver, spleen, and lungs. Nb-DC1.8, which recognizes immature bone marrow–derived dendritic cells in vitro, gives a much smaller signal in the liver and spleen than does Nb-DC2.1 but mainly targets to the lungs and gives a pronounced signal in the skin. Grafting of the antigen-binding loops of Nb-DC1.8 or Nb-DC2.1 to the scaffold of Nb-BCII10 alters the observed biodistribution of the Nanobodies to resemble that of the Nanobody from which the antigen-binding loops have been derived. Conclusion: The observed in vivo biodistribution of the Nanobodies reflects the main in vivo locations of the cells recognized by the Nanobodies and is determined by the antigen-binding loops of the Nanobodies. Thus, Nanobodies represent elegant targeting probes for imaging the in vivo biodistribution of specific immune cell types.
A multitude of radiopharmaceuticals are in use or under investigation as imaging tools that allow the monitoring of ongoing immune or inflammatory disease and response to therapy—including, for example, radiolabeled autologous leukocytes, 18F-FDG, and radiolabeled antigranulocyte antibodies (1). Currently, there is a great interest in imaging agents that accumulate and are retained in inflammatory foci by specific interaction with infiltrated and activated inflammatory cells and that could be developed into safe, off-the-shelf available radiopharmaceuticals (2). The potential of these agents can be expanded to other diseases, such as type I diabetes and atherosclerosis, in which activated inflammatory cells have a crucial role in the pathogenesis (3).
Macrophages and myeloid dendritic cells (DCs) are developmentally related myeloid lineage cells of the innate immune system that share the ability to act as antigen-presenting cells—engulfing, processing, and presenting antigens to T lymphocytes—and as such are functionally positioned at the junction of innate and adaptive immune defenses. Moreover, as professional phagocytes, involved in clearance and destruction of infiltrating infectious agents, and through the release of bactericidal and cytocidal mediators, myeloid cells—such as macrophages, neutrophils, or DCs—also constitute important innate effector cells. The ability to image the in vivo biodistribution of these myeloid cells would thus constitute an in vivo sensor for the status of ongoing immune and inflammatory responses (4,5).
The presence in serum of Camelidae of heavy-chain antibodies that have naturally evolved to be devoid of light chains (6), in conjunction with the possibility to immunize these animals, allows a straightforward cloning and selection of single-domain antigen-binding fragments that matured in vivo, termed Nanobodies (Ablynx) (7). Their high robustness and stability at elevated temperature allow the fast and easy labeling of their carboxy-terminal hexahistidine tail with 99mTc by tricarbonyl chemistry at elevated temperature and straightforward application as probes in pinhole SPECT/micro-CT. Using 99mTc-labeled Nanobodies targeting carcinoembryonic antigen (8) and epidermal growth factor receptor (9), we have shown before that these small (15 kD), high-affinity antigen-binding proteins are excellent tools for molecular tumor imaging. Indeed, they rapidly and specifically bind tumor antigens, whereas unbound Nanobodies are rapidly cleared from the blood by renal elimination. Hence, high tumor-to-background ratios can be obtained as early as 1 h after tracer injection. In these tumor imaging experiments, we have mainly been working in conditions in which the target cells of the Nanobodies are in a single, predefined location. Yet these results offer perspectives for using Nanobodies as generic tools for imaging the in vivo biodistribution of specific cell types. In the present study, we have evaluated the cellular specificity of Nanobodies raised against bone marrow–derived DCs and assessed their capacity to image the in vivo biodistribution of their target cells.
MATERIALS AND METHODS
Mice
Female C57BL/6J mice (6–8 wk old) were purchased from Harlan. Animal care and treatment conformed to institutional guidelines of the Belgian Council for Laboratory Animal Science, inspired by the recommendations of the Federation of European Laboratory Animal Science Associations.
Generation of DC-Specific Nanobodies
The generation of the anti-DC Nanobody library is part of a study that has been submitted elsewhere. In brief, a humoral immune response was induced in Lama glama by immunization with either immature or mature bone marrow–derived DCs without adjuvant. After cloning variable domains of the heavy-chain antibodies from the peripheral blood and lymph node lymphocytes, a library of phage-displayed Nanobodies was constructed and whole-cell panning was performed on bone marrow–derived DCs in suspension after a prior negative selection on a T-cell line (Jurkat) and a macrophage cell line (RAW264.7). Enriched clones were selected by flow cytometry. The gene segments encoding DC-binding Nanobodies were recloned into an expression vector harboring a hexahistidine tag–coding sequence. The Nanobodies used in this report are Nb-DC1.8 and Nb-DC2.1, next to the negative control Nb-BCII10, which is a binder against subunit 10 of the β-lactamase BcII enzyme of Bacillus cereus (10). The 3 Nanobodies are different in their amino-acid sequence, as shown in Figure 1.

Deduced amino-acid sequences of different Nanobodies, consisting of CDR sequences alternated with framework (FR) sequences. CDR sequences of each Nanobody are marked in color; amino acid differences between frameworks are marked in yellow. Determination of numbering corresponds to International ImMunoGeneTics information system amino-acid numbering (http://imgt.cines.fr/).
Grafting of Antigen-Binding Loops on Universal Antibody Scaffold
The complementarity-determining region (CDR)–H loops from loop donor Nanobodies DC1.8 (llama) and DC2.1 (llama) were transferred to the scaffold of Nb-BcII10 (recipient Nanobody) by polymerase chain reaction–based mutagenesis. The sequence of each CDR-H loop from the loop donor Nanobody was encompassed by 2 primers, 1 back and 1 forward, containing at the 5′ and the 3′ ends the sequences corresponding to the framework residues of the recipient Nanobody. The chimeras were constructed as described previously, with some minor modifications (11,12). The chimeric Nanobody constructs of Nb-DC1.8 and Nb-DC2.1 on Nb-BcII10 were digested with PstI and BstEII. Both fragments were cloned in the expression vector pHEN6 (10). The expression yield of both chimeras was comparable to the level of the humanized recipient Nanobody (2 mg/L of culture).
Expression and Purification of Nanobodies
Escherichia coli WK6 cells were transformed with the plasmid constructs. His6-tagged Nanobodies were expressed in the periplasm and purified as described previously (13). The purity of the proteins was evaluated by Coomassie-stained sodium dodecylsulfate polyacrylamide gels. The protein concentration was determined spectrophotometrically at 280 nm using the computed extinction coefficient of each Nanobody, as calculated from their amino-acid sequence (14).
Stability Measurements
Temperature-induced unfolding was followed by circular dichroism with a Jasco J715 spectropolarimeter in the far-ultraviolet region (205–250 nm), using a protein concentration of 0.2 mg/mL and a cell path length of 0.1 cm. A total volume of 300 μL of each sample was heated in 50 mM sodium phosphate buffer (pH 7.0). The temperature was increased from 35°C to 95°C at a rate of 1°C/min, and the signal intensity at 205 nm was recorded as a function of temperature. Data were acquired with a reading frequency of 1/20 s−1, an integration time of 1 s, and a bandwidth of 2 nm. Data were analyzed according to the method of Dumoulin et al. (15).
Alexa Fluor 488 Labeling of Nanobodies
The Nanobodies BCII10, DC1.8, DC2.1, DC1.8 graft, and DC2.1 graft were labeled with Alexa Fluor 488 dye using the Alexa Fluor 488 protein-labeling kit (Molecular Probes; Invitrogen) according to the manufacturer's recommendations. Labeled Nanobodies were purified by size-exclusion chromatography on AKTA explorer (Amersham Sciences) using a Superdex-75 column (Amersham) in phosphate-buffered saline. The protein concentration and the degree of labeling of the Alexa Fluor 488–coupled Nanobodies were determined by Spectrophotometer Nanodrop ND-1000 (Isogen Life Sciences).
Flow Cytometry
For flow cytometric analysis, the bone marrow–derived DC population was stained with antibodies (BD Biosciences) for 20 min at 4°C using standard protocols. Cells (106/100 μL) were incubated with 1 μg of allophycocyanin-conjugated CD11c (HL3) antibody and either 1 μg of fluorescein isothiocyanate–conjugated MHC-II (clone M5/114.15.2), fluorescein isothiocyanate–conjugated CD86 (Gl-1), or fluorescein isothiocyanate–conjugated CD40 antibody (clone 3/23). Cell recognition of different Nanobodies (at 1 μg) was monitored using 1 μg of Alexa 488–labeled Nanobody. Multiple parameters were acquired on a FACSCanto II (Becton Dickinson), followed by analysis with FlowJo (TreeStar).
In Vitro Competition Study
Of each bone marrow–derived DC population, 106 cells were used in 100 μL and preincubated with 40 μg of unlabeled Nanobody (DC1.8 graft or DC2.1 graft) for 20 min at 4°C using standard protocols. The cells were then incubated with 1 μg of Alexa 488–labeled Nb-DC1.8, Nb-DC2.1, Nb-DC1.8 graft, or Nb-DC2.1 graft. Multiple parameters were acquired on a FACSCanto II, followed by analysis with FlowJo.
99mTc Labeling of Nanobodies
Both Nanobodies were labeled with 99mTc at their hexahistidine tail as described before (16). For the labeling, [99mTc(H2O)3(CO)3]+ was synthesized by adding 1 mL of 99mTcO4− (0.74–3.7 GBq) to an Isolink kit (Mallinckrodt Medical BV) containing 4.5 mg of sodium boranocarbonate, 2.85 mg of sodium tetraborate·10H2O, 8.5 mg of sodium tartrate·2H2O, and 7.15 mg of sodium carbonate, pH 10.5. The vial was incubated at 100°C in a boiling bath for 20 min. The freshly prepared [99mTc(H2O)3(CO)3]+ was allowed to cool at room temperature for 5 min and neutralized with 125 μL of 1 M HCl to pH 7–8. [99mTc (H2O)3(CO)3]+ was added to 50 μL of a 1 mg/mL solution of Nb-BCII10, Nb-DC1.8, Nb-DC2.1, Nb-DC1.8 graft, or Nb-DC2.1 graft together with 50 μL of carbonate buffer, pH 8. The mixture was incubated for 90 min at 52°C in a water bath. The labeling efficiency was determined by instant thin-layer chromatography in acetone as the mobile phase and analyzed using a radiometric chromatogram scanner (VCS-201; Veenstra). The 99mTc-Nanobody solution was purified on a NAP-5 column (GE Healthcare) preequilibrated with phosphate-buffered saline. The collected fraction was then passed through a Millex-GV4 0.22-μm filter (Millipore).
Pinhole SPECT/micro-CT Procedure
C57BL/6J (original Nanobody, n = 9; grafted Nanobody, n = 3) mice were intravenously injected with 45–155 MBq of 99mTc-Nanobodies. At 120 min after injection, mice were anesthetized with a mixture of 18.75 mg of ketamine hydrochloride per kilogram (Ketamine 1000; CEVA) and 0.5 mg of medetomidin hydrochloride per kilogram (Domitor; Pfizer). Micro-CT (Skyscan 1178; Skyscan) was followed by pinhole SPECT (e.cam180; Siemens Medical Solutions) on separate imaging systems. With both modalities, the animals were imaged in the same animal holder, which included 2 plastic discs, each containing three 57Co (3.7 MBq) sources (Canberra-Packard). The six 57Co sources were detected on both micro-CT and pinhole SPECT and used for alignment of CT and SPECT images. A micro-CT scan was obtained using a dual-source CT scanner with 60 kV and 615 mA at a resolution of 83 mm. The total-body scan time was 2 min. Images were reconstructed using filtered backprojection (Nrecon; Skyscan). A total-body pinhole SPECT scan was obtained using a dual-head γ-camera, mounted with 2 multipinhole collimators (3 pinholes of 1.5 mm in each collimator, 200-mm focal length, and 80-mm radius of rotation). Images were acquired over 360° in 64 projections of 20 s each into 128 × 128 matrices, resulting in a total imaging time of 22 min. The micro-SPECT images were reconstructed using an iterative reconstruction algorithm modified for the 3-pinhole geometry (17) and automatically reoriented for fusion with CT using the six 57Co landmarks.
Image Analysis
Images were viewed and quantified using AMIDE's a Medical Image Data Examiner software (18). Ellipsoid regions of interest (ROIs) based on the CT images were drawn on the muscle, liver, lungs, and spleen; around the kidneys; and around the total body. Uptake was calculated as the counts in the tissue divided by the injected activity and normalized for the ROI size. A pinhole SPECT image of a syringe with 37 MBq of 99mTc was acquired as a reference for conversion of injected activity into measured counts in the image.
Ex Vivo Analysis
The C57BL/6J mice received via the lateral tail vein 22–96 MBq of 99mTc-BCII10, 99mTc-DC1.8 (original/grafted Nanobody), or 99mTc-DC2.1 (original/grafted Nanobody) in 100–150 μL of phosphate-buffered saline. After SPECT/CT had been performed at the different time points, mice were sacrificed with a lethal dose of pentobarbital (Nembutal; CEVA). Kidneys, liver, lungs, muscle, skin, spleen, heart, and blood were removed and weighed, and the radioactivity was measured using an automated γ-counter (Cobra II Inspector 5003; Canberra-Packard). Tissue and organ uptake was calculated as percentage of injected activity per gram of tissue, corrected for decay.
Statistical Analysis
Statistical analysis of imaging experiments and dissection analysis were performed using the unpaired 2-tailed t test. Results were considered statistically significant when the P value was less than 0.05.
RESULTS
In Vitro Characterization of Nb-DC1.8 and Nb-DC2.1
Cellular specificity of the anti–DC Nanobodies DC1.8 and DC2.1 was determined using flow cytometry analysis of Alexa 488–labeled Nanobodies. Nb-DC1.8 binds to immature bone marrow–derived DCs but not to lipopolysaccharide-matured bone marrow–derived DCs, whereas Nb-DC2.1 binds to both immature and mature bone marrow–derived DCs (Supplemental Fig. 1; supplemental materials are available online only at http://jnm.snmjournals.org). Besides DCs, Nb-DC2.1 also binds to a range of other myeloid cell types, including peritoneal macrophages, peripheral blood neutrophils, and total CD11b-positive splenic myeloid cells. Neither of these 2 Nanobodies binds to nonmyeloid cell types such as splenic CD4 T cells (Supplemental Fig. 3). To confirm the specificity of binding, blocking experiments with unlabeled Nanobody were performed. Binding of each of the Nanobodies to its target cells could be inhibited by prior incubation of the cells with an excess of unlabeled form of the same Nanobody but not by preincubation with the other anti-DC Nanobody or with the BCII10 control Nanobody (Supplemental Fig. 1).
Pinhole SPECT/micro-CT Analysis of 99mTc-Labeled Nb-DC1.8 and Nb-DC2.1
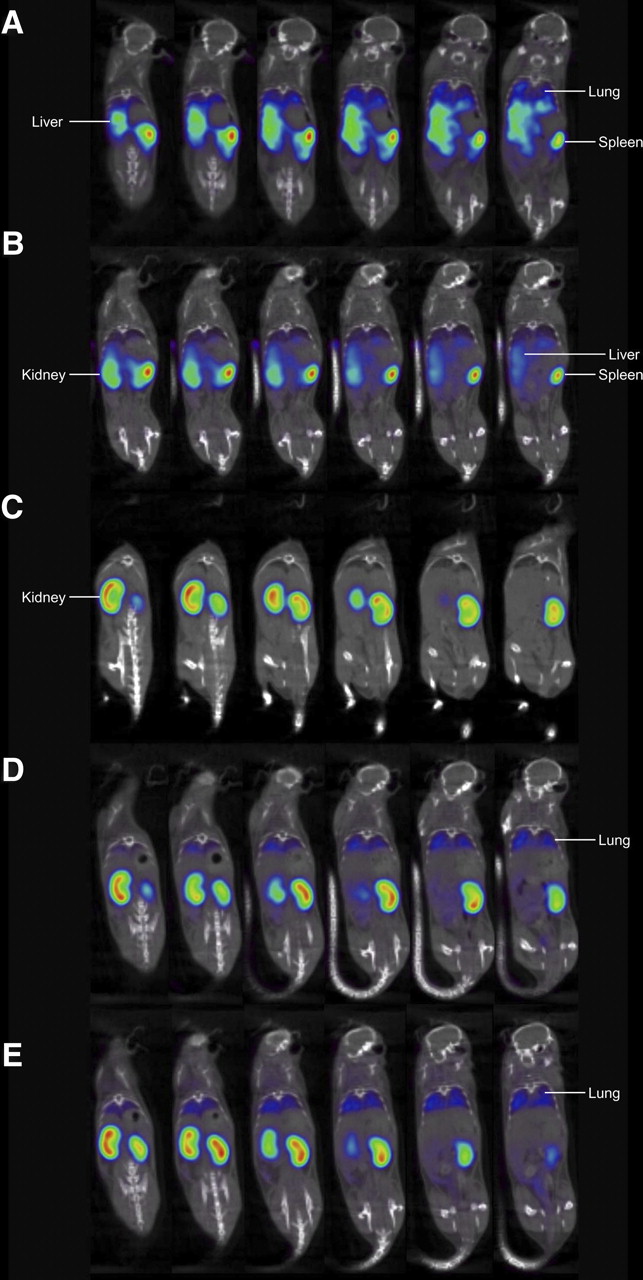
Nanobodies are particularly amenable to fast and efficient labeling of the hexahistidine tail with 99mTc at elevated temperatures by tricarbonyl chemistry without unfolding the protein or interfering with antigen binding (9,19). In fact, the hexahistidine tail at the Nanobodies carboxy terminus is situated on the opposite side of the antigen-binding side, as shown in crystal structures of Nanobodies in complex with antigen (12). Purified 99mTc-labeled Nanobodies were injected intravenously in 3 healthy C57BL/6J mice, and total-body scans were obtained using pinhole SPECT and micro-CT. The duration of 1 complete image acquisition was less than or equal to 30 min. Images acquired 2 h after injection for the control Nanobody BCII10 showed clearance of the Nanobody by the kidneys and low background retention in other organs such as the liver (Fig. 2; Table 1). In contrast, Nb-DC2.1 showed a profound reduction in the signal for the kidneys and intense uptake in mainly liver, spleen, and lungs. For Nb-DC1.8, a marked kidney and lung accumulation and low spleen uptake was recorded. Both Nb-DC2.1 and Nb-DC1.8, as compared with the control Nanobody BCII10, showed an increased signal in bone marrow.

Serial coronal pinhole SPECT/micro-CT images at 2 h after tracer injection. (A) Nb-DC 2.1 showing uptake in liver, spleen, and lungs. (B) Nb-DC 2.1 graft showing same pattern as Nb-DC 2.1, with slightly higher kidney elimination. (C) Nb-BCII10 showing only kidney elimination and no targeting. (D) Nb-DC 1.8 showing lung targeting and kidney elimination. (E) Nb-DC 1.8 graft showing lung targeting and kidney elimination identical to Nb-DC 1.8. Images are shown using National Institutes of Health color scale and are scaled to maximum in whole image.
Biodistribution of Nanobodies Based on Pinhole SPECT/Micro-CT at 2 Hours After Injection
In addition to calculating the in vivo biodistribution based on quantification of the pinhole SPECT/micro-CT images, mice were also sacrificed and the biodistribution was assessed by measuring radioactivity in various organs and tissues. The resulting data (Table 2) confirm the biodistribution obtained from the pinhole SPECT/micro-CT images. For Nb-DC1.8, measurements of radioactivity in skin sections reveal a weak signal—yet remarkably higher than for Nb-BCII10 and Nb-DC2.1. In contrast, the signal in blood is much higher for Nb-DC2.1 than for Nb-BCII10 and Nb-DC1.8. Notably, the ratio of kidney signals for Nb-DC1.8 and Nb-DC2.1 is lower using data obtained from postmortem dissection than from micro-SPECT images analysis. This low value is explained by the fact that it is difficult to delineate the kidneys on the Nb-DC 2.1 SPECT images, on which kidney activity is low; the liver and spleen, however, show intense uptake on these images and we had to ensure that the kidney ROI did not overlap with these high activities near the kidney. As a result, the kidney signal values for Nb-DC2.1 based on the images are likely underestimated. The value based on the dissection is more reliable in this case.
Biodistribution of Nanobodies Based on Dissection at 2.5 Hours After Injection
Grafting of Antigen-Binding Loops from Anti-DC Nanobodies to Universal Nanobody Scaffold
We next wanted to ascertain that the observed biodistribution of the anti-DC Nanobodies is due to specific recognition of the target antigen on specific myeloid cell populations present in the organs rather than retention in certain organs due to, for example, interactions with the framework sequences of the Nanobodies. To this aim, we genetically grafted the 3 antigen-binding loops or CDR of the DC1.8 and DC2.1 Nanobodies onto the framework of the control BCII10 Nanobody. The deduced amino-acid sequences of the original and the grafted Nanobodies are shown in Figure 1. We next confirmed the specificity of the binding of the grafted Nanobodies in flow cytometry. Similar to the original Nanobodies, binding of the Alexa 488–labeled Nb-DC1.8 graft to immature but not to lipopolysaccharide-matured DCs was detected, whereas the Alexa 488–labeled Nb-DC2.1 graft yielded a signal on both immature and mature DCs (Supplemental Fig. 2; data not shown for mature DCs). Moreover, in a competition study in which immature DCs were stained with the Alexa 488–labeled Nb-DC1.8 and Nb-DC2.1 after preincubation with an excess nonlabeled form of the grafted Nanobodies, only preincubation with the grafted Nanobody of which the CDR loops are corresponding interferes with the signal of the directly labeled DC Nanobodies (Supplemental Fig. 2). Similar results were obtained when using the original Nanobodies for inhibition of binding of Alexa 488–labeled grafted Nanobodies (data not shown).
Because the 99mTc-labeling procedure involves an incubation of the Nanobodies at 52°C, we wanted to ascertain that the grafting of the antigen-binding loops did not impair the thermal stability of the Nanobodies. In fact, in line with the higher thermal stability of the BCII10 scaffold than the DC1.8 and DC2.1 Nanobodies, the thermal stability of the grafted Nanobodies even tended to be increased, as compared with that of their native counterparts (Table 3).
Melting Temperature of Nanobodies
Pinhole SPECT/micro-CT Analysis of 99mTc-Labeled Nb-DC1.8 Graft and Nb-DC2.1 Graft
After having confirmed in flow cytometry that grafting of the antigen-binding loops transfers the cellular specificity of the Nb-DC1.8 and Nb-DC2.1 to the framework of the BCII10 control Nanobody and that the thermal stability of the grafted Nanobodies was sufficient to proceed with labeling, we evaluated the biodistribution of the DC1.8 graft and DC2.1 graft Nanobodies using pinhole SPECT/micro-CT analysis. As before, purified, 99mTc-labeled Nanobodies were injected intravenously in 3 healthy C57BL/6J mice, images were acquired at 2 h after injection, and the duration of 1 complete image acquisition was 30 min or less. Biodistributions of the grafted Nanobodies based on pinhole SPECT/micro-CT and dissection of organs at 2 h after injection are also shown in Figure 2 and Tables 4 and 5. Overall, genetically grafting the CDRs of the DC1.8 and DC2.1 Nanobodies onto the framework of the control BCII10 Nanobody altered the observed biodistribution of the Nanobodies to resemble that of the Nanobody from which the antigen-binding loops had been derived. Indeed, similar to the original DC1.8 and DC2.1 Nanobodies, the liver targeting of the Nb-DC2.1 graft but not that of the DC1.8 graft Nanobody was significantly higher than that of the BCII10 control Nanobody. The spleen targeting, however, was more pronounced for the Nb-DC1.8 graft, as compared with the Nb-DC2.1 graft. Ex vivo measurements of radioactivity revealed an increased blood signal for the Nb-DC2.1 graft, as compared with the control Nanobody, and a significantly higher skin signal for the Nb-DC1.8 graft than for the BCII10 Nanobody. These measurements are again similar to what was observed for the original DC1.8 and DC2.1 Nanobodies. Remarkably, also for the DC2.1 graft Nanobody, as compared with the BCII10 Nanobody, a significantly increased skin signal was recorded. However, whereas the skin signal for Nb-DC1.8 did not differ significantly from that of the Nb-DC1.8 graft (P = 0.27), the skin signal was still significantly lower for the Nb-DC2.1 graft than for the Nb-DC1.8 (P < 0.01) and Nb-DC1.8 grafts (P = 0.03).
Biodistribution of CDR-Grafted Nanobodies Based on Pinhole SPECT/Micro-CT at 2 Hours After Injection
Biodistribution of CDR-Grafted Nanobodies Based on Dissection at 2.5 Hours After Injection
DISCUSSION
A limited number of antibody-based radiopharmaceuticals are currently in use or under investigation as imaging tools to monitor ongoing immune or inflammatory disease, including, for example, 99mTc-labeled antigranulocyte antibodies or antilectinlike oxidized low-density-lipoprotein receptor 1 antibodies (1,20). Nanobodies offer the high binding affinity and specificity of antibodies, combined with the small size, stability, tissue penetration, and favorable pharmacokinetics of small molecules (16). Moreover, the compact, prolate shape of the Nanobodies allows them to interact with epitopes that are not antigenic for conventional antibodies (21), and the minimal size of Nanobodies allows them to target hidden or cryptic antigens that are inaccessible to larger antibodies (22). Finally, the recombinant, renewable nature of Nanobodies offers clear advantages at the level of generation, production, and molecular biologic manipulations such as sequence modification and transfer of the antigen specificity and affinity from one Nanobody to another (12).
In the current study, we wanted to assess the ability of Nanobodies to act as targeting probes for imaging the in vivo biodistribution of specific cell types, using a couple of Nanobodies raised against mouse DCs as proof-of-principle tools. The observed in vivo biodistribution for the 2 selected Nanobodies with different cellular specificities nicely reflects the main in vivo locations of the cells that have been determined in vitro to be recognized by the Nanobodies.
Nb-DC2.1, which recognizes a wide range of myeloid cells, targets most strongly to the liver, spleen, and lungs—tissues with pronounced macrophage populations. The increased signal in blood for this Nanobody, as compared with the other Nanobodies, fits with the binding of Nb-DC2.1 to peripheral blood neutrophils.
Nb-DC1.8, which recognizes immature bone marrow–derived DCs in vitro, gives a much lower signal in the liver and spleen than does Nb-DC2.1 but mainly targets to the lungs. The much higher signal in the skin for Nb-DC1.8 than for the other Nanobodies may reflect recognition of immature DC populations in the skin. Possibly, these skin DC populations express less of the antigen recognized by Nb-DC2.1. Alternatively, the strong retention of Nb-DC2.1 in the liver and spleen may reduce the amount of Nanobody remaining for targeting to the skin. Yet, we do not favor the hypothesis that the strong liver and spleen retention of Nb-DC2.1 a priori impairs its ability to target its antigen in other organs or tissues, because Nb-DC2.1 targets the bone marrow to an extent similar to that of Nb-DC1.8.
The specificity of Nanobodies is mainly determined by their antigen-binding loops, which are attached on the framework regions of the Nanobody. However, there are still some differences in the sequence of the framework regions for the different Nanobodies. Therefore, we cannot a priori exclude the possibility that at least part of the observed in vivo biodistribution may be caused by retention in certain organs due to interactions with the framework sequences of the Nanobodies. Indeed, in the case of tumor imaging, for example, the target antigen is not present in steady-state conditions and therefore the images obtained in steady-state conditions can be used to ascertain the absence of aspecific signals. This is, however, not the case for the anti-DC Nanobodies because the myeloid cell markers recognized by these Nanobodies are present in mice in steady-state conditions. To ascertain that the observed biodistribution of the anti-DC Nanobodies is due to specific recognition of the target antigen on specific myeloid cell populations present in the organs rather than retention in certain organs due to, for example, interactions with the framework sequences of the Nanobodies, we took advantage of experience in our group with genetic grafting of antigen-binding loops. In particular, we have shown before that the framework regions of Nb-BCII10 are able to act as a stable scaffold for the exchange of antigen specificities by CDR grafting (11). Genetically grafting the CDRs of the DC1.8 and DC2.1 Nanobodies onto the framework of the control BCII10 Nanobody altered the observed biodistribution of the Nanobodies to resemble that of the Nanobody from which the antigen-binding loops have been derived.
CONCLUSION
The observed in vivo biodistribution for 2 selected Nanobodies with different cellular specificities nicely reflects the main in vivo locations of the cells that have been determined in vitro to be recognized by the Nanobodies. Moreover, genetic grafting experiments of the antigen-binding loops of the Nanobodies onto the framework of the control BCII10 Nanobody have indicated that the observed biodistribution of these Nanobodies is mainly determined by their antigen-binding loops rather than by, for example, hydrophobic interactions with the framework sequences of the Nanobodies. These experiments substantiate the suitability of Nanobodies as generic targeting probes for imaging the in vivo biodistribution of specific cell types.
Acknowledgments
We acknowledge Cindy Peleman for her technical assistance with the small-animal pinhole SPECT/CT. The research at ICMI is funded by the Interuniversity Attraction Poles Program, Belgian State, Belgian Science Policy. Kurt De Groeve is a PhD fellow of the Horizontale Onderzoeks Actie (HOA) of the Vrije Universiteit Brussel. Tony Lahoutte is a Senior Clinical Investigator of the Research Foundation, Flanders (Belgium) (FWO).
Footnotes
-
COPYRIGHT © 2010 by the Society of Nuclear Medicine, Inc.
References
- Received for publication September 2, 2009.
- Accepted for publication November 30, 2009.





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Simultaneous selection of nanobodies for accessible epitopes on immune cells in the tumor microenvironment
- The Immunoimaging Toolbox
- Anti-CTLA-4 therapy requires an Fc domain for efficacy
- Delivering Type I Interferon to Dendritic Cells Empowers Tumor Eradication and Immune Combination Treatments
- PET Imaging of Macrophage Mannose Receptor-Expressing Macrophages in Tumor Stroma Using 18F-Radiolabeled Camelid Single-Domain Antibody Fragments
- Molecular Imaging with Macrophage CRIg-Targeting Nanobodies for Early and Preclinical Diagnosis in a Mouse Model of Rheumatoid Arthritis
- Targeting of Human Antigen-Presenting Cell Subsets
- SPECT Imaging of Joint Inflammation with Nanobodies Targeting the Macrophage Mannose Receptor in a Mouse Model for Rheumatoid Arthritis
- Generation and characterization of non-competitive furin-inhibiting nanobodies
- Nanobody-Based Targeting of the Macrophage Mannose Receptor for Effective In Vivo Imaging of Tumor-Associated Macrophages
- In Vitro Analysis and In Vivo Tumor Targeting of a Humanized, Grafted Nanobody in Mice Using Pinhole SPECT/Micro-CT