


Abstract
Small biomolecules are typically radiolabeled with 18F by binding it to a carbon atom, a process that usually is designed uniquely for each new molecule and requires several steps and hours to produce. We report a facile method wherein 18F is first attached to aluminum as Al18F, which is then bound to a chelate attached to a peptide, forming a stable Al18F-chelate-peptide complex in an efficient 1-pot process. Methods: For proof of principle, this method was applied to a peptide suitable for use in a bispecific antibody pretargeting method. A solution of AlCl3·6H2O in a pH 4.0 sodium-acetate buffer was mixed with an aqueous solution of 18F to form the Al18F complex. This was added to a solution of IMP 449 (NOTA-p-Bn-CS-d-Ala-d-Lys(HSG)-d-Tyr-d-Lys(HSG)-NH2) (NOTA-p-Bn-CS is made from S-2-(4-isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid; HSG is histamine-succinyl-glycine) and heated to 100°C for 15 min. In vitro and in vivo stability and targeting ability of the Al18F-IMP 449 were examined in nude mice bearing LS174T human colonic tumors pretargeted with an anti-CEACAM5 bispecific antibody (TF2). Results: The radiolabeled peptide was produced in 5%−20% yield with an estimated specific activity of 18,500–48,100 GBq (500–1,300 Ci)/mmol. The Al18F-IMP 449 was stable for 4 h in serum in vitro, and in animals, activity isolated in the urine 30 min after injection was bound to the peptide. Nonchelated Al18F had higher tissue uptake, particularly in the bones, than the chelated Al18F-IMP 449, which cleared rapidly from the body by urinary excretion. Tumor uptake was 30-fold higher with TF2-pretargeted Al18F-IMP 449 than with the peptide alone. Dynamic PET showed tumor localization within 30 min and rapid and thorough clearance from the body. Conclusion: The ability to bind highly stable Al18F to metal-binding ligands is a promising new labeling method that should be applicable to a diverse array of molecules for PET.
PET has become one of the most prominent functional imaging modalities in diagnostic medicine, with high sensitivity (fmol), high resolution (e.g., 4–10 mm), and tissue accretion that can be adequately quantitated (1). Although 18F-FDG is the most widely used functional imaging agent in oncology (2), there is a keen interest in developing other labeled compounds for functional imaging to complement and augment anatomic imaging methods (3), especially with hybrid PET/CT systems. Thus, a facile method of conjugating positron-emitting radionuclides to various molecules is needed.
Although there are several PET radionuclides, 18F (β+, 0.635 MeV [97%]; half-life, 110 min) has nearly ideal properties for PET, such as low positron energy, lack of side emissions, and a suitable half-life. Peptides and other small molecules are excellent candidates for use as 18F-labeled agents, because they have similar properties to FDG (i.e., they rapidly enter the extracellular space from the blood, where they are accessible to the cells and then clear just as quickly from the body). However, preparing 18F-conjugates can be an arduous and lengthy process, particularly given the short half-life (110 min) of 18F. For example, peptides are conventionally labeled with 18F in a 2- or 3-step process involving the labeling and purification of a small molecule (prosthetic group) and subsequent conjugation/purification of the conjugate (4). The first step typically involves the purification of 18F, and the second usually follows the path established for the production of 18F-FDG (5). The purified 18F is mixed with potassium carbonate, a crown-ether (Kryptofix 222; Merck), and boiled to dryness. The sample is then azeotropically dried twice with acetonitrile. Subsequently, the 18F is attached to a prosthetic group, which is present in great excess compared with the moles of 18F. The radiolabeled prosthetic group should be purified; the prosthetic group is then conjugated to the desired targeting molecule, and the product is purified again to obtain a radiolabeled targeting agent with high specific activity. Some examples include succinyl 18F fluorobenzoate (6–8), 4-18F-fluorobenzaldehyde (9–12), other acyl compounds (13–16), or click chemistry adducts (17). The total synthesis and formulation time for these methods ranges between 1 and 3 h, with most of the time dedicated to the high-performance liquid chromatography (HPLC) purification of the labeled peptides to obtain the specific activity required for in vivo targeting.
Since fluoride binds to most metals (18–20), we speculated that an 18F-metal complex could be bound to a chelate on a targeting agent in a similar manner to most radiometal-labeling procedures for antibodies and peptides—procedures that are typically accomplished within 15 min and with quantitative yields. Because we have reported previously the synthesis of several synthetic hapten-conjugated peptides that contain a variety of chelating groups for binding radiometals used for in vivo targeting of cancer with a bispecific antibody (BsmAb) pretargeting system (21–24), this system was selected as a testing platform for evaluating Al18F peptides. This procedure has been shown to be highly sensitive and specific for detecting cancers in humans (25), and animal testing has shown improved and more rapid tumor visualization than has been possible with directly radiolabeled antibodies, even providing more sensitive imaging than 18F-FDG in animal studies (26,27).
In this report, we describe our initial work leading to the discovery of a suitably stable first-generation ligand that is capable of binding the Al18F complex to a hapten-peptide. When used in a pretargeting setting, the pretargeted 18F-hapten-peptide was stable and provided excellent tumor localization within 1 h.
MATERIALS AND METHODS
Reagents
The p-SCN-Bn-NOTA was purchased from Macrocyclics, Inc.; protected amino acids, other peptide synthesis reagents, and resins from Creosalus, Chem Impex, Bachem, PepTech Corp., and EMD Biosciences; aluminum chloride hexahydrate from Sigma-Aldrich; and other solvents and reagents from Fisher Scientific or Sigma-Aldrich. The analytic and preparative reverse-phase HPLC (RP-HPLC) columns were purchased from Phenomenex or Waters Corp., and size exclusion HPLC (SE-HPLC) columns from BioRad. 18F and FDG were supplied by IBA Molecular.
The recombinant, humanized, tri-Fab BsmAb, TF2, was provided by IBC Pharmaceuticals, Inc. TF2 binds divalently to carcinoembryonic antigen (CEACAM5, CEA, CD66e) and monovalently to the synthetic hapten, HSG (histamine-succinyl-glycine) (28). The BsmAb was more than 95% immunoreactive against CEACAM5 and the divalent-HSG NOTA-peptide, IMP 449, using a SE-HPLC method described previously (28).
Preparation of Chelate-Hapten-Peptide
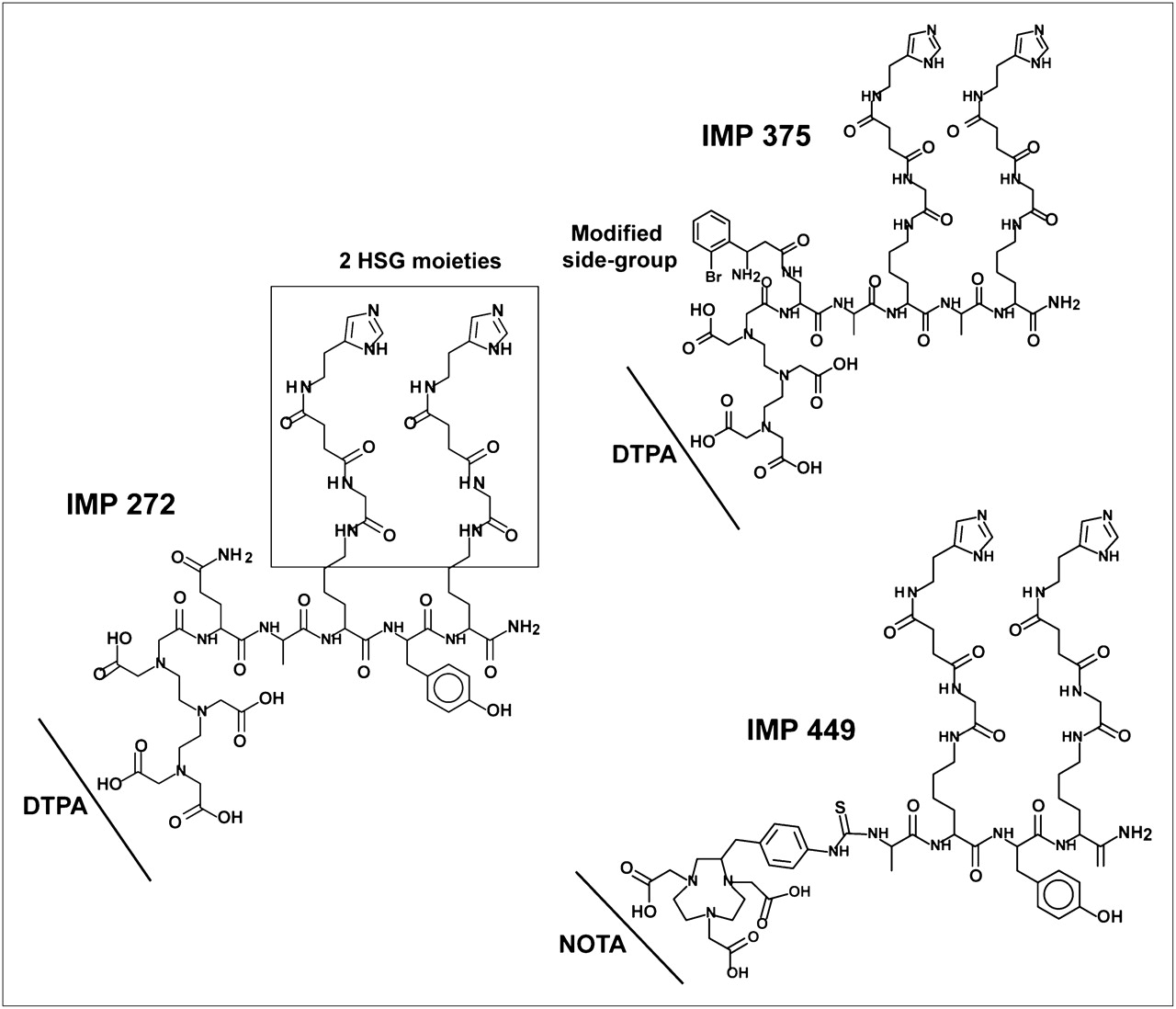
IMP 272 (DTPA-Gln-Ala-Lys(HSG)-d-Tyr-Lys(HSG)-NH2, MH+ 1,512), IMP 375 (DTPA-Dpr((R)-3-amino-3-(2-bromophenyl)-propionyl)-d-Ala-d-Lys(HSG)-d-Ala-d-Lys(HSG)-NH2), and IMP 449 (NOTA-p-Bn-NH-CS-d-Ala-d-Lys(HSG)-d-Tyr-d-Lys(HSG)-NH2, MH+ 1,459) were prepared as described previously (29,30).
Preparation of IMP 449 Labeling Kits
The peptide, IMP 449, was formulated with the addition of ascorbic acid. For this, 8 mg of lyophilized IMP 449 (5.48 μmol) were mixed with 0.1549 g of L-(+)-ascorbic acid and dissolved in 10.5 mL of deionized water. The liquid was dispensed in 1.0-mL aliquots into 2-mL lyophilization vials. The solutions were frozen, lyophilized, and sealed under vacuum.
Preparation of Al18F
AlCl3 hexahydrate was used to prepare a 2 mM Al3+ stock solution in a 0.1 M pH 4 sodium acetate buffer. Al18F was prepared using a 3-μL (6 nmol) aliquot of the aluminum stock solution, which was mixed with 50 μL of the 18F received from the commercial supplier (e.g., 370 MBq [10 mCi] in ∼0.5 mL of water). This Al18F was used for the preparation of IMP 272, but later, we purified the 18F to remove contaminating metals that might otherwise compete for binding to the chelates (31). Briefly, a Sep-Pak Light Accell Plus QMA cartridge (Waters) was washed with 10 mL of 0.4 M KHCO3 followed by 10 mL of deionized water. 18F, received in 2 mL of water (∼1.48 GBq [40 mCi]), was loaded onto the cartridge. The cartridge was washed with 5 mL of deionized water. 18F was then eluted from the cartridge in 0.2-mL fractions with 0.4 M KHCO3. Most of the activity was in the second 0.2-mL fraction. Three microliters (6 nmol) of the stock aluminum acetate solution were added to this fraction, which was then added to IMP 449.
18F Radiolabeling of IMP 272
Three microliters of a 2 mM IMP 272 stock solution in 0.1 M acetate buffer solution, pH 4.0, were added to the Al18F mixture (53 μL) prepared using the unpurified 18F. This was placed in a heating block at 110°C for 15 min and analyzed by RP-HPLC on a Gemini 5-μm C18110 A, 250 × 4.60 mm, column (Phenomenex) using 0.1% NH4OAc buffers (flow rate: 1 mL/min; buffer A: 0.1% NH4OAc in water; buffer B: 0.1% NH4OAc in 90% acetonitrile, 10% water; gradient: 100% buffer A to 100% buffer B over 30 min). Because the initial HPLC analysis showed poor incorporation, an additional 10 μL of the IMP 272 stock solution were added, and it was heated again and analyzed by RP-HPLC.
18F Radiolabeling of IMP 449
The Al18F solution prepared from the purified 18F was injected into the lyophilized IMP 449 vial and then heated at 100°C for 15 min. The reaction solution was RP-HPLC–purified using a monolithic C-18 column (100 × 4.5 mm; Phenomenex) under the following conditions: flow rate: 3 mL/min; buffer A: 0.1% trifluoroacetic acid (TFA) in deionized water; buffer B: 0.1% TFA in 90% acetonitrile; gradient: 100% buffer A to 75% buffer A/25% buffer B over 15 min using a linear gradient. The HPLC eluent was collected with a fraction collector in 30-s increments. Unbound 18F and Al18F are eluted in the void volume. As the gradient proceeds, the Al18F IMP 449 is eluted first, followed by the unlabeled IMP 449 about 1 min later. Any Al-IMP 449 (without 18F) coelutes with the Al18F IMP 449. The fraction containing the Al18F IMP 449 was diluted with water and loaded onto an Oasis 1-mL hydrophilic-lipophilic-balanced (HLB) column (part number 186001879; Waters). The HLB column was washed with three 1-mL aliquots of water to remove the acetonitrile and TFA. The labeled peptide was then eluted with 2 × 200 μL of 1:1 EtOH:H2O into a vial containing 15 mg of ascorbic acid neutralized to pH 6.6 with NaOH.
To estimate the specific activity of the labeled products, we assumed that the specific activity of 18F is approximately 370 TBq (10,000 Ci)/mmol (the theoretic specific activity of 18F is about 62,900 TBq/mmol, but because of 19F contamination it is generally considered to be the lower assumed value). The specific activity of the labeled peptide is controlled by the amount of aluminum that is added to the labeling reaction, since 18F will not bind to the peptide without the metal. As an example, in one study, 4.4 GBq, or 1.2 × 10−5 mmol of total fluoride, was added to 6 × 10−6 mmol of the aluminum solution, which was added to 5.2 × 10−4 mmol of peptide. The crude reaction mixture was purified to obtain 329 MBq in 2 main fractions (201 and 128 MBq). The 201-MBq fraction was then further purified on an HLB column to remove the acetonitrile and TFA, resulting in a final product yield of 174.6 MBq. It is assumed that each fraction contained the same amount of the Al-IMP 449, so since the 201-MBq fraction contained 60.6% of the labeled peptide, it is also assumed that it contained 60% of the aluminum added or 3.63 × 10−6 mmol of the aluminum peptide; therefore, the theoretic specific activity of the labeled peptide is 0.1746 GBq/3.63 × 10−6 mmol = 48,100 GBq (1,300 mCi)/mmol.
Al19F IMP 449
A 960-μL aliquot of a 0.02 M solution of AlCl3·6H2O in 0.5 M NaOAc, pH 4, was mixed with 192 μL of NaF in 0.5 M NaOAc, pH 4. The solution was added to 0.0280 g (MH+ 1,459; 1.919 × 10−5 mol) of IMP 449, heated in a 100°C heating block for 17 min, and then purified by RP-HPLC using a 30 × 150 mm Sunfire C-18 column (Waters) eluting with 0.01% ammonium acetate buffers. The HPLC buffers were as follows: buffer A, 0.1% NH4OAc in H2O, and buffer B, 0.1% NH4OAc 90% CH3CN 10% H2O. The HPLC gradient went from 100% A to 80:20 A:B over 80 min with a flow rate of 45 mL/min. The HPLC fractions were collected and lyophilized to obtain the aluminum-NOTA-IMP 449 complex, 0.0068 g, MH+ 1,482.6534 (C66H92N19O17S1Al1, theoretic 1,482.6527), and 2 aluminum fluoride IMP 449 complexes: RT 9.73 min, 0.0106 g, MH+ 1,502.6614 (C66H93N19O17S1Al1F1, theoretic 1,502.6589) and RT 9.90 min, 0.0068 g, MH+ 1,502.6588 (C66H93N19O17S1Al1F1, theoretic 1,502.6589).
Stability Testing
Al18F-IMP 449 (1,850 kBq [50 μCi], 50 μL) was added to 0.5 mL of freshly collected and sterile-filtered human serum. The sample was incubated at 37°C in a humidified 5% CO2 incubator. At approximately 1 and 4 h, the sample was analyzed by RP-HPLC, as well as by SE-HPLC after mixing with TF2 to determine the percentage immunoreactivity with HSG. HPLC systems were equipped with in-line ultraviolet and radiation detectors. A Bio-Sil SE 250 column (Bio-Rad Laboratories, Inc.) attached to a guard column was eluted with a buffer containing 0.2 M sodium phosphate, 0.02% sodium azide, and 10 mM ethylenediaminetetraacetic acid, pH 7.0. RP-HPLC studies were performed on an RCM 8 × 10 C18 NovaPak (4-μM) column (Waters), eluted using a gradient of 100% solvent A to 45% solvent B, 55% solvent A in 15 min, then at 100% B for 5 min before equilibration to initial conditions. The flow rate was 1.5 mL/min, solvent A was 0.075% TFA in H2O, and B was 0.075% TFA in 75% CH3CN and 25% H2O.
In vivo stability was examined by injecting 18.5 MBq of Al18F-IMP 449 intravenously to 3 non–tumor-bearing mice, and then 30 min later, the animals were anesthetized, bled, and then necropsied to remove urine from the urinary bladder. Samples were analyzed by RP- and SE-HPLC.
Biodistribution and Small-Animal PET
All studies were performed with the approval of the institutional animal care and use committee. The human colonic cancer cell line, LS174T (ATCC), was implanted subcutaneously in 6-wk-old NCr ν-m female nude mice (Taconic). When tumors were visible, 162 μg (∼1 nmol/0.1 mL) of TF2 were injected intravenously in pretargeted animals, and then 16–18 h later, about 0.1 nmol of Al18F-IMP 449 (3.11 MBq [84 μCi]/0.1 mL) was injected intravenously. Other nonpretargeted control animals received 18F alone (5.5 MBq [150 μCi]), Al18F complex alone (5.55 MBq [150 μCi]), the Al18F-IMP 449 peptide alone (3.11 MBq [84 μCi]), or 18F-FDG (5.55 MBq [150 μCi]). 18F and 18F-FDG were obtained on the day of use. Animals receiving 18F-FDG were kept fasting overnight, but water was given ad libitum.
At 1.5 h after the radiotracer injection, the animals were anesthetized, bled intracardially, and necropsied. Tissues were weighed and counted together with a standard dilution prepared from each of the respective products. Because of the short physical half-life of 18F, standards were interjected between each group of tissues from each animal. Uptake in the tissues is expressed as the counts per gram divided by the total injected activity to derive the percentage injected dose per gram.
Two types of imaging studies were performed. In one set, 3 nude mice bearing small LS174T subcutaneous tumors received either the pretargeted Al18F-IMP 449, Al18F-IMP 449 alone (not pretargeted) (both forms: 135 μCi [5 MBq]; 0.1 nmol), or 18F-FDG (5 MBq [135 μCi]). At 2 h after the intravenous radiotracer injection, the animals were anesthetized with a mixture of O2/N2O and isoflurane (2%) and kept warm during the scan. The mice were placed supine on the bed of an Inveon animal PET scanner (Siemens Preclinical Solutions). This scanner has an intrinsic spatial resolution of 1.5 mm. Emission scans were acquired over 15 min (18F-FDG) or 30 min (Al18F-IMP 449). Scans were reconstructed using Inveon Acquisition Workplace software (IAW, version 1.2) using an ordered-set expectation maximization 3-dimensional/maximum a posteriori (OSEM3D/MAP) algorithm with the following parameters: matrix, 256 × 256 × 159; pixel size, 0.43 × 0.43 × 0.8 mm; and MAP prior of 0.5 mm. Representative coronal cross-sections (0.8 mm thick) in a plane located approximately in the center of the tumor were displayed, with intensities adjusted until pixel saturation occurred in any region of the body (excluding the bladder) and without background adjustment.
In a separate dynamic imaging study, a single LS174T-bearing nude mouse that was given the TF2 BsmAb 16 h earlier was anesthetized with a mixture of O2/N2O and isoflurane (2%), placed supine on the camera bed, and then injected intravenously with 8.1 MBq (219 μCi) of Al18F IMP 449 (0.16 nmol). Data acquisition was initiated immediately and continued for 120 min. Data were graphed in 24 frames of 5 min each. The scans were reconstructed using OSEM3D/MAP with the same parameters as described above. Each of the 24-image time frames was examined. For presentation, time frames ending at 5, 15, 30, 60, 90, and 120 min (i.e., the 5-min image is for the period from time zero to 5 min) were displayed for each cross-section (coronal, sagittal, and transverse). For sections containing tumor, at each interval the image intensity was adjusted until pixel saturation first occurred in the tumor. Image intensity was increased as required over time to maintain pixel saturation within the tumor. Coronal and sagittal cross-sections without tumor taken at the same interval were adjusted to the same intensity as the transverse section containing tumor. Background activity was not adjusted.
RESULTS
Initial Test Compounds
Because diethylenetriaminepentaacetic acid (DTPA) has been used widely for binding a variety of metals, including aluminum (32), a DTPA-hapten-peptide, IMP 272, was prepared (Fig. 1). On the basis of the prior experience of Martin et al. (33), labeling was performed at approximately pH 4.0, since this was reported to be optimal for the formation of AlFn species in water and the same pH range has been used to bind Al3+ to ligands (34). Under the initial reaction conditions (6 nmol of aluminum solution added to 18F plus 6 nmol of IMP 272, pH 4.0; heated at 100°C for 15 min), RP-HPLC showed that only 7% of the activity was bound. However, when 26 nmol of peptide were added to this same reaction mixture and heating was continued for a further 15 min, the yield increased to 92%. Although the yields were good, the Al18F-IMP 272 was unstable in water (17% loss of 18F in 40 min at room temperature). Since chelates vary in their affinity for different metals, we tested other 18F-metal complexes (Ga, In, Zr, Lu, and Y) for their binding to IMP 272. These complexes also bound to the peptide, but not as well as Al18F, and they too were unstable in water, as is consistent with the findings of others (20,28). In an attempt to better stabilize the Al18F complex, several other DTPA-hapten-peptides were synthesized by altering the residues adjacent to the DTPA. One 18F-labeled peptide, IMP 375 (Fig. 1), was stable in water, with 98% radiolabeling yield, but this peptide, like the others, was not sufficiently stable in human serum in vitro.
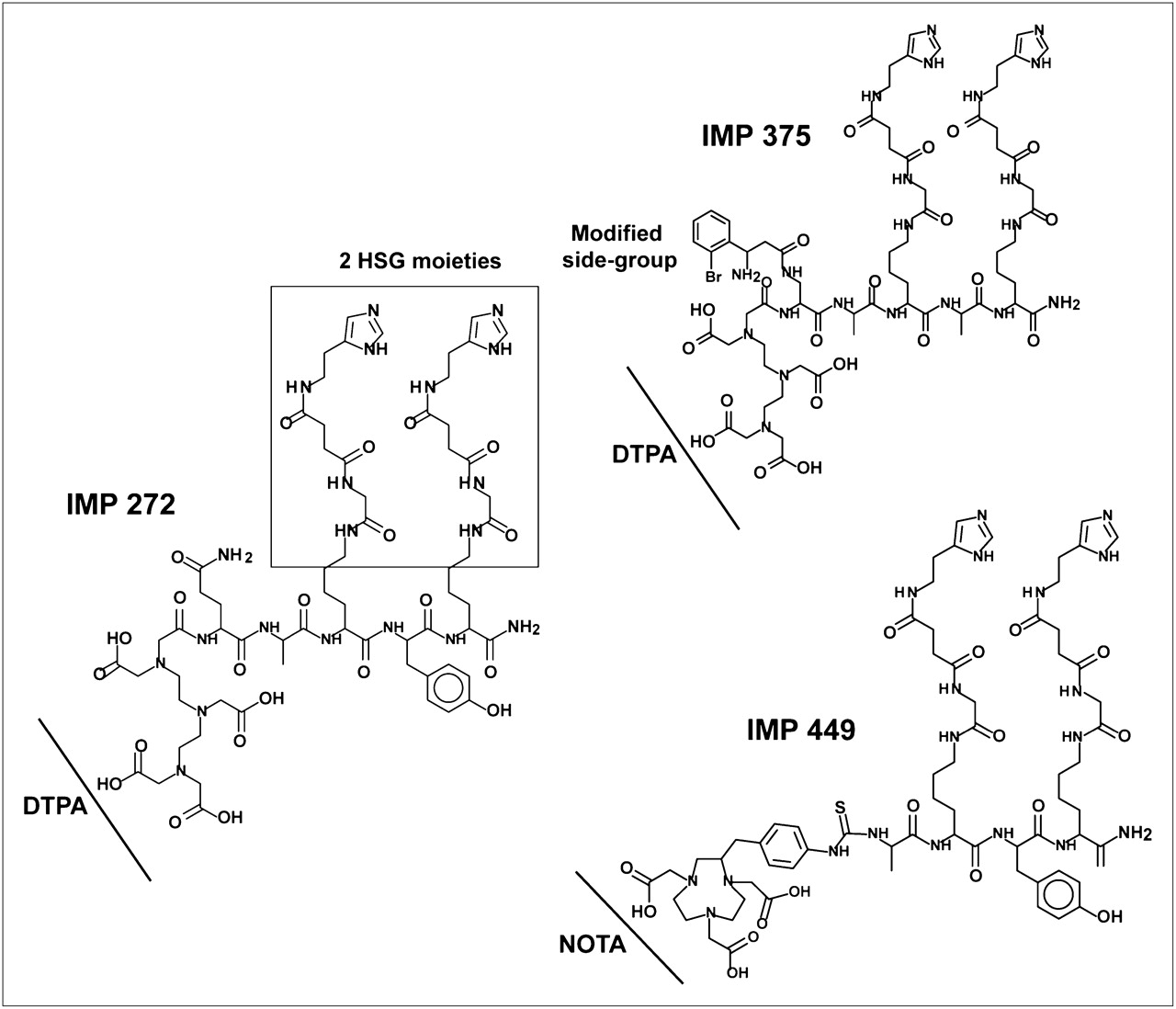
DTPA- and NOTA-HSG-hapten-peptides.
Because the literature indicated that Al3+ binds F− more strongly than most other metals (20,33,35), and that Al(F)n complexes are stable in vivo (35,36), the instability of Al18F on the DTPA ligand most likely is due to the weak binding of the metal complex to DTPA and not to dissociation of the AlF complex. Thus, we continued with the AlF complex but focused on new chelates to bind Al18F, recognizing that the AlF complex might have binding characteristics different from what has been reported for aluminum. We tested several different chelates, such as deferoxamine, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, ethylenediaminetetraacetic acid, symmetric DTPA, phosphonates, phosphates, and 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA). Screening was performed by mixing 18F with 6 nmol of Al3+, adding 40 nmol of peptide, and heating for 15 min at 100°C. The peptides were then examined by RP-HPLC to determine the labeling yield. Labeling yields with all the other chelate hapten-peptides were lower (or not bound at all) than with DTPA, and all, except the Al18F NOTA complex, were unstable in serum. Because Andre et al. (34) had reported that NOTA formed stable complexes with aluminum, this chelate was investigated further.
18F Labeling of IMP 449
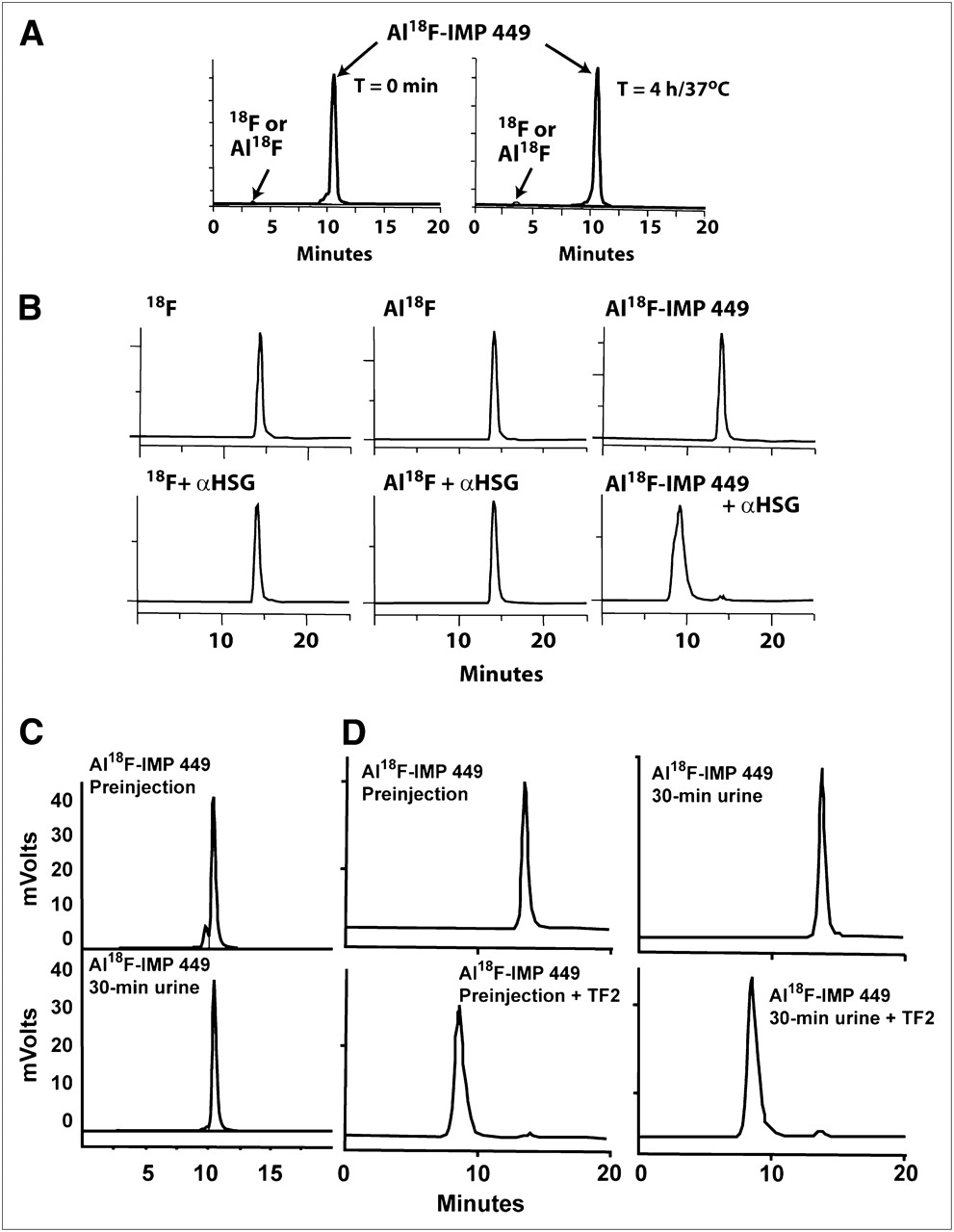
Initial yields of the Al18F NOTA-peptide (IMP 449; Fig. 1) were only about 5% under the standard screening conditions (i.e., with 40 nmol of peptide); however, when 522 nmol of peptide were used, labeling yields increased to a range of 5%−20% (without correcting for decay) after HPLC purification. The 18F-labeled peptide was produced without a drying step and with a single HPLC purification within 60 min to obtain a labeled peptide suitable for in vivo use. The purified product was stable in serum at 37°C for 4 h (Fig. 2A), and immunoreactivity testing also confirmed that the Al18F complex was bound firmly to the peptide. As shown in Figure 2B, when mixed with an HSG-binding BsmAb, TF2, and analyzed by SE-HPLC, only the purified Al18F-IMP 449 eluted at a molecular size consistent with complexes formed between TF2 and the Al18F-IMP 449 di-HSG peptide, whereas 18F alone and the nonchelated Al18F complex continued to elute in the included volume. Thus, Al18F-IMP 449 met our primary criteria that the compound should be prepared in a timely manner and have suitable stability in serum to proceed with animal testing.
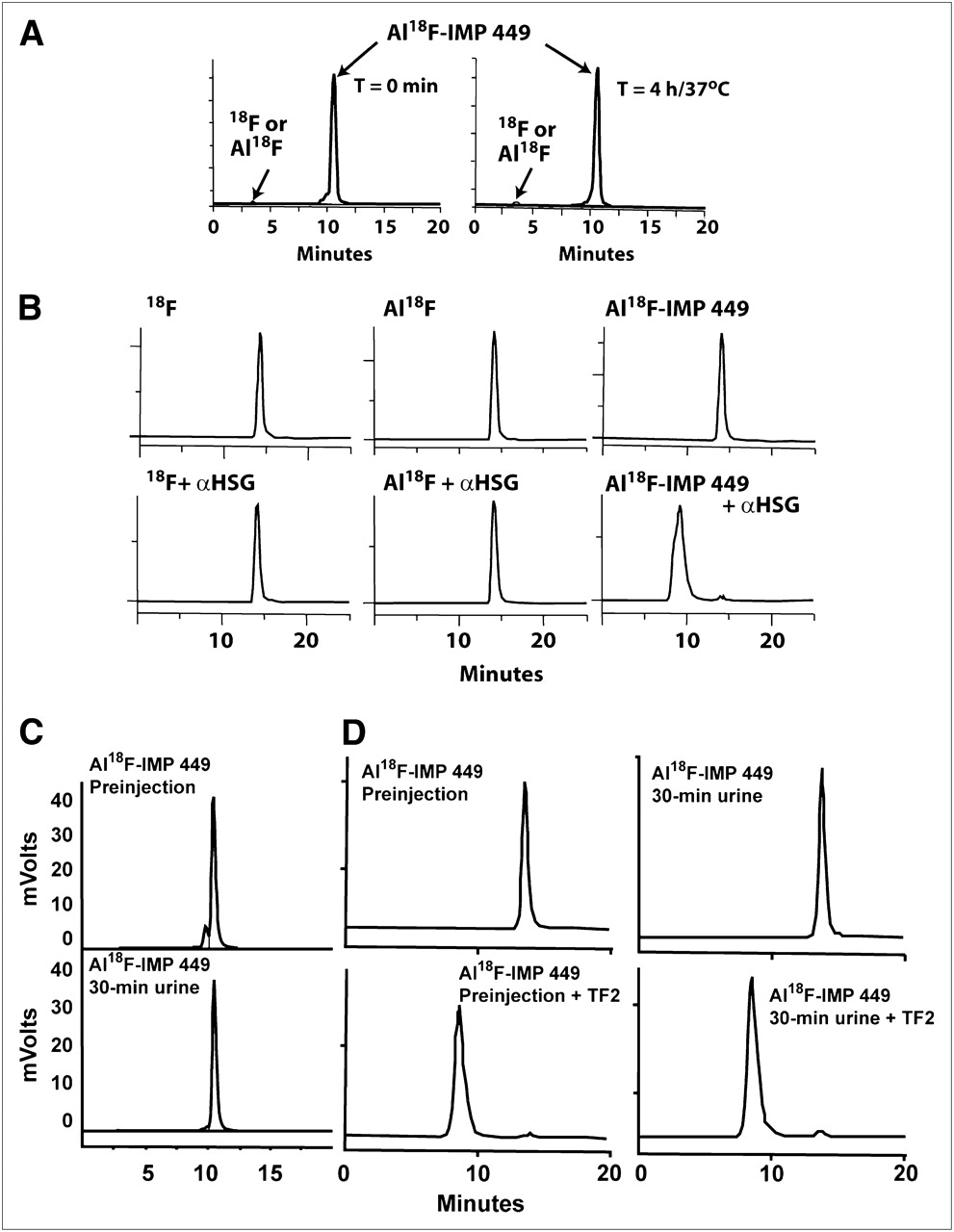
Serum stability and immunoreactivity of Al18F-IMP 449. (A) RP-HPLC showing elution of purified product at start of study (left) and then 4 h later after incubation at 37°C in fresh human serum (right). (B) SE-HPLC illustrates retention of binding of di-HSG containing IMP 449 peptide after radiolabeling with Al18F. Top graphs show 18F alone, nonchelated Al18F complex, and Al18F IMP 449 eluting in total included volume of column. Bottom graphs show elution of each product after being incubated with excess amount of anti-HSG antibody for 30 min at 37°C. Only Al18F IMP 449 elution profile shifted to migrate with antibody, and 100% of activity eluted at this earlier time. (C) RP-HPLC profile of Al18F-IMP-449 before injection in animals (top) and then in 30-min urine sample (bottom). (D) SE-HPLC of same labeled product as in C before animal injection (left), with and without addition of TF2 to show binding to HSG. Right graphs show profiles from urine sample taken 30 min after injection.
Al19F-IMP 449 Mass Spectroscopy Analysis of AlF Binding to IMP 449
The aluminum and Al19F complexes of the peptide were prepared so that the complexes could be analyzed by HPLC and by mass spectroscopy to help determine the nature of the complexes formed. Two Al19F complexes were formed with retention times that matched the 18F complexes when examined under similar conditions. The mass of the Al19F-IMP 449 MH+ 1,502.6588 (C66H93N19O17S1Al1F1, theoretic 1,502.6589) is consistent with a complex in which the AlF is binding 2 of the NOTA carboxyl groups and the third carboxyl is still protonated (Fig. 3). Aluminum is known to bind NOTA to form hexadentate bonds to the 3 nitrogens and 3 carboxyls (34). Thus, it appears that the AlF complex has pentadentate binding to NOTA, with the sixth binding site of the aluminum filled with the fluoride ion.
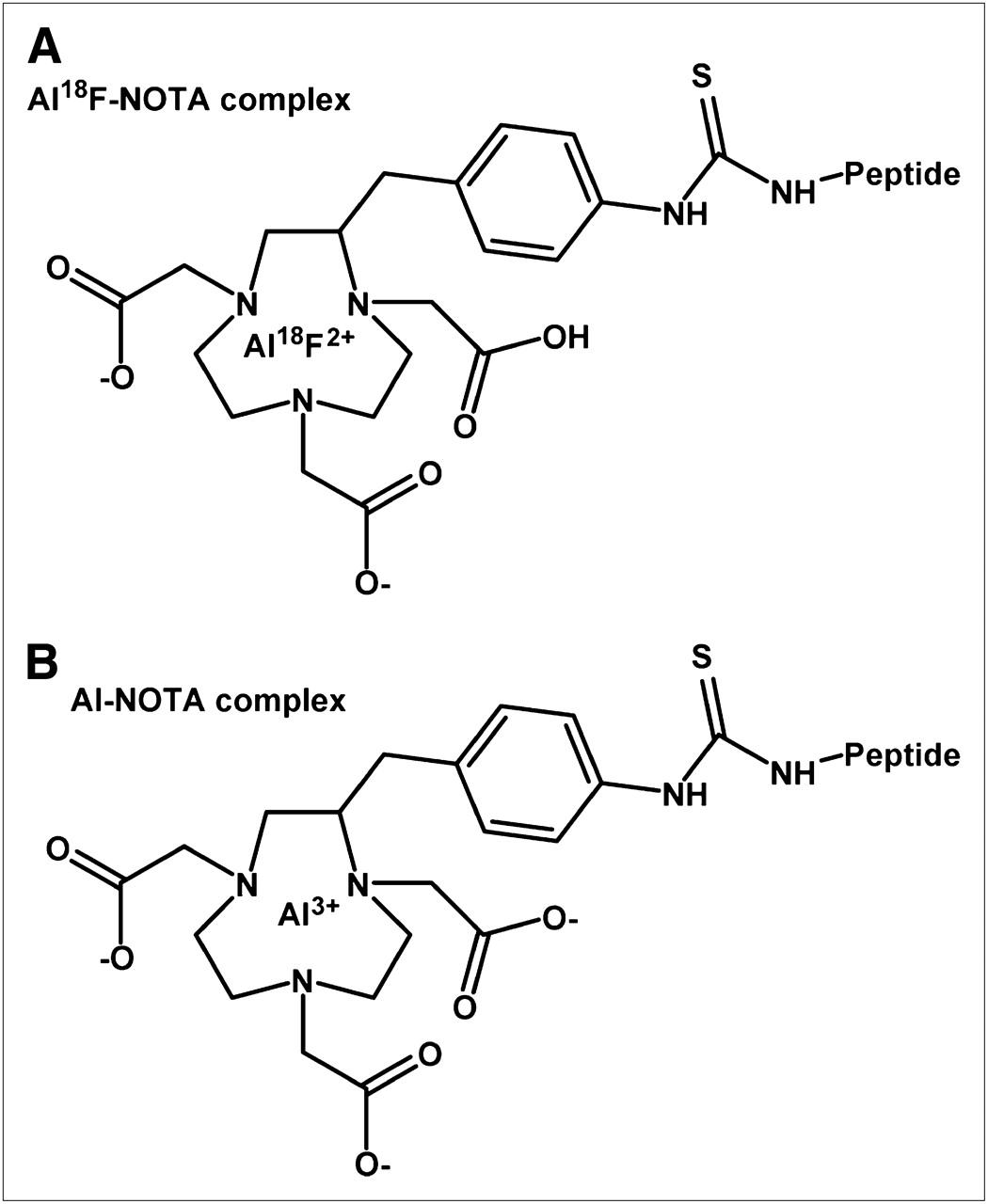
Schematic representation of Al18F-NOTA (A) and Al-NOTA (B) binding based on mass spectroscopy data of 19F-IMP 449 complex.
In Vivo Studies
To assess the stability of the Al18F-IMP 449 in vivo, animals were given 18F alone and Al18F so that the tissue distribution of each component would be known. 18F alone and Al18F complexes had similar uptake in all tissues, but considerable differences were found when the Al18F complex was chelated to IMP 449 (Table 1). The most striking differences were found in the uptake in bone, where the nonchelated 18F was 60- to nearly 100-fold higher in the scapula and approximately 200-fold higher in the spine. This distribution is expected, since 18F, or even a metal-fluoride complex, is known to accrete in bone (37). Higher uptake was also observed in the tumor and intestines, as well as in muscle and blood. The chelated Al18F-IMP 449 had significantly lower uptake in all the tissues except the kidneys, illustrating the ability of the chelate complex to be removed efficiently from the body by urinary excretion. Pretargeting the Al18F IMP 449 using the TF2 anti-CEACAM5 BsmAb shifted uptake to the tumor, increasing it from 0.20% ± 0.05% to 6.01% ± 1.72% injected dose per gram at 1.5 h, whereas uptake in the normal tissues was similar to the Al18F-IMP 449 alone. Tumor-to-nontumor ratios were 146 ± 63, 59 ± 24, 38 ± 15, and 2.0 ± 1.0 for the blood, liver, lungs, and kidneys, respectively, with other tumor-to-tissue ratios being greater than 100:1 at this time. Although both 18F alone and Al18F alone had higher uptake in the tumor than did the chelated Al18F-IMP 449, yielding tumor-to-blood ratios of 6.7 ± 2.7 and 11.0 ± 4.6 vs. 5.1 ± 1.5, respectively, tumor uptake and tumor-to-blood ratios were significantly increased with pretargeting (all P values < 0.01).
Biodistribution of TF2-Pretargeted Al18F-IMP 449 and Other Control 18F-Labeled Agents in Nude Mice Bearing LS174T Human Colonic Cancer Xenografts
Tissue distribution of the Al18F-IMP 449 was also compared with 18F-FDG (Table 1). 18F-FDG uptake was appreciably higher than Al18F-IMP 449 uptake in all normal tissues, except the kidneys. Uptake in tumor was similar for both the pretargeted Al18F-IMP 449 and 18F-FDG, but because of the higher accretion of 18F-FDG in most normal tissues, tumor-to-nontumor ratios with 18F-FDG were significantly lower than those in the pretargeted animals (all P values < 0.001).
We attempted to examine the stability of the Al18F-IMP 449 in the blood from several animals given the peptide alone, but because the peptide cleared so quickly, there was insufficient activity for HPLC analysis with samples collected just 30 min after injection. However, analysis of the activity eliminated in the urine of these animals showed that all of it was associated with the peptide, and in fact, the activity in the urine also shifted on a SE-HPLC column when TF2 was added, indicating that the peptide retained at least one of its HSG moieties (Figs. 2C and 2D). Collectively, these data indicate that 18F remained firmly attached to the aluminum, and the Al18F complex bound to NOTA-IMP 449 was stable in vivo.
Several animals were imaged to analyze the biodistribution of Al18F IMP 449 alone or Al18F-IMP 449 pretargeted with TF2, as well as 18F-FDG. Static images initiated at 2.0 h after the radioactivity had been injected corroborated the previous tissue distribution data, showing uptake almost exclusively in the kidneys (Fig. 4). A 21-mg tumor was easily visualized in the pretargeted animal, whereas the animal given the Al18F-IMP 449 alone failed to localize the tumor, having only renal uptake. No evidence of bone accretion was observed, suggesting that the Al18F was bound firmly to IMP 449. This was confirmed in another pretargeted animal that underwent a dynamic imaging study monitoring the distribution of the Al18F IMP 449 at 5-min intervals over 120 min (Supplemental Fig. 1; supplemental materials are available online only at http://jnm.snmjournals.org). Coronal and sagittal slices showed primarily cardiac, renal, and some hepatic uptake over the first 5 min, but heart and liver activity decreased substantially over the next 10 min, whereas the kidneys remained prominent throughout the study. There was no evidence of activity in the intestines or bone over the full 120-min scan. Uptake in a 35-mg LS174T tumor was first observed at 15 min, and by 30 min, the signal was clearly delineated from background, with intense tumor activity being prominent during the entire 120-min scan.

Biodistribution of 18F-labeled agents in tumor-bearing nude mice by small-animal PET. Coronal slices of 3 nude mice bearing small, subcutaneous LS174T tumor on each left flank after being injected with either 18F-FDG (A), Al18F-IMP 449 pretargeted with anti-CEA × anti-HSG BsmAb (B), or Al18F-IMP 449 alone (not pretargeted with BsmAb) (C). Biodistribution data expressed as percentage injected dose per gram (%ID/g) are given for tissues removed from animals at conclusion of imaging session. Br = brain; BM = bone marrow; H = heart; K = kidney; T = tumor.
In comparison, static images from an animal given 18F-FDG showed the expected pattern of radioactivity in the bone, heart muscle, and brain observed previously (26,27), with considerably more background activity in the body (Fig. 4). Tissue uptake measured in the 3 animals necropsied at the conclusion of the static imaging study confirmed much higher tissue 18F radioactivity in all tissues. Although tumor uptake with 18F-FDG was higher in this animal than in the pretargeted one, tumor-to-blood ratios were more favorable for pretargeting, and with much less residual activity in the body, tumor visualization was enhanced by pretargeting.
DISCUSSION
The preparation of fluorinated compounds has historically required specialized chemistries that can be cumbersome and time-consuming. We hypothesized that it might be possible to bind 18F to compounds by first creating a metal-Al18F complex that could then be attached to the compound through a metal-binding ligand. Indeed, radiolabeling of chelate-conjugated peptides and proteins with radiometals has been practiced for over 25 y (38), and thus it seemed feasible that a similar approach might be possible with a metal-18F complex, particularly because 18F can form stable complexes with several metals.
Our investigation indicated that the Al18F complex is indeed stable, but the more arduous task was finding a suitable linker for stable binding to various compounds. Several of the more commonly used chelating agents were examined, but although some would capture Al18F (even quantitatively), most were not sufficiently stable for in vivo applications. NOTA provided the first indication that the Al18F complex could be bound stably. Mass spectroscopy analysis suggests that the Al18F complex is held in place by the 3 nitrogens and 2 of the carboxyl groups. It is also important to note that while the labeling process reported here added a preformed Al18F complex to the chelate-peptide, we also were able to bind 18F to aluminum that was preloaded in the NOTA-IMP 449 (not shown).
Although yields were within the range found with conventional 18F labeling procedures, further studies are needed to select the coordination chemistries that optimize yields while retaining the stability found with this lead NOTA derivative. Nevertheless, these studies lay the foundation for a new, simplified 18F-labeling method that could allow many more compounds to be prepared with 18F PET tracer.
The labeling method described here does not require a dry-down for the 18F and is a major time-saving advantage over existing methods. The yields reported here are similar to many of the reported procedures but less than those reported for the click chemistry method (17). However, we believe that we will be able to enhance the radiolabeling yield by modifying the NOTA ligand to improve the binding kinetics of the ligand, thus possibly increasing the labeling yield while reducing the amount of peptide needed for the labeling. If the binding kinetics were to be improved sufficiently, it might be possible to eliminate the need for HPLC purification and make an 18F kit that labels in the same manner as the radiometals.
CONCLUSION
To our knowledge, this is the first report describing a direct, facile, and rapid method (60 min of total preparation time) of binding 18F via an aluminum conjugate. Although the feasibility of this approach was shown with a hapten-peptide used for pretargeting, we have recently extended our findings to include other receptor-binding peptides and other binding ligands with improved yields, which will be reported in the future.
Acknowledgments
We thank Dion Yeldell, Jayson Jebsen, Christine Johnson, Ali Mostafe, Maarten Brom, and Eric P. Visser for their technical assistance. This work was funded in part by NIH grant 1 R43 EB003751-01A1 from the National Institute of Biomedical Imaging and Bioengineering. All authors except Drs. Karacay, Sharkey, Boerman, and Laverman declare a financial interest in Immunomedics, Inc., either through employment, stock, or patents. This work was presented in part at the 55th annual meeting of the Society of Nuclear Medicine, New Orleans, Louisiana, June 15–18, 2008.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication November 18, 2008.
- Accepted for publication March 18, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Anti-CEA Pretargeted Immuno-PET Shows Higher Sensitivity Than DOPA PET/CT in Detecting Relapsing Metastatic Medullary Thyroid Carcinoma: Post Hoc Analysis of the iPET-MTC Study
- FAPI-74 PET/CT Using Either 18F-AlF or Cold-Kit 68Ga Labeling: Biodistribution, Radiation Dosimetry, and Tumor Delineation in Lung Cancer Patients
- Clinical Translation of a 68Ga-Labeled Integrin {alpha}v{beta}6-Targeting Cyclic Radiotracer for PET Imaging of Pancreatic Cancer
- Radiohybrid Ligands: A Novel Tracer Concept Exemplified by 18F- or 68Ga-Labeled rhPSMA Inhibitors
- 18F-AlF-Labeled Biomolecule Conjugates as Imaging Pharmaceuticals
- Recent Advances in 18F Radiochemistry: A Focus on B-18F, Si-18F, Al-18F, and C-18F Radiofluorination via Spirocyclic Iodonium Ylides
- Immuno-PET Using Anticarcinoembryonic Antigen Bispecific Antibody and 68Ga-Labeled Peptide in Metastatic Medullary Thyroid Carcinoma: Clinical Optimization of the Pretargeting Parameters in a First-in-Human Trial
- Clinical Translation of an Albumin-Binding PET Radiotracer 68Ga-NEB
- In Vivo Evaluation of 18F-SiFAlin-Modified TATE: A Potential Challenge for 68Ga-DOTATATE, the Clinical Gold Standard for Somatostatin Receptor Imaging with PET
- Preclinical Comparison of Al18F- and 68Ga-Labeled Gastrin-Releasing Peptide Receptor Antagonists for PET Imaging of Prostate Cancer
- In Vivo Labeling of Serum Albumin for PET
- Effect of Chelate Type and Radioisotope on the Imaging Efficacy of 4 Fibrin-Specific PET Probes
- Al18F Labeling of Affibody Molecules
- A Comparative Study of Radiolabeled Bombesin Analogs for the PET Imaging of Prostate Cancer
- Three Methods for 18F Labeling of the HER2-Binding Affibody Molecule ZHER2:2891 Including Preclinical Assessment
- Al18F: A New Standard for Radiofluorination
- Phase II Trial of Anticarcinoembryonic Antigen Pretargeted Radioimmunotherapy in Progressive Metastatic Medullary Thyroid Carcinoma: Biomarker Response and Survival Improvement
- PET of Tumors Expressing Gastrin-Releasing Peptide Receptor with an 18F-Labeled Bombesin Analog
- Imaging of Human Epidermal Growth Factor Receptor Type 2 Expression with 18F-Labeled Affibody Molecule ZHER2:2395 in a Mouse Model for Ovarian Cancer
- Assessing Antibody Pharmacokinetics in Mice with In Vivo Imaging
- Pretargeted Immuno-Positron Emission Tomography Imaging of Carcinoembryonic Antigen-Expressing Tumors with a Bispecific Antibody and a 68Ga- and 18F-Labeled Hapten Peptide in Mice with Human Tumor Xenografts
- A Novel Facile Method of Labeling Octreotide with 18F-Fluorine