


Abstract
Although the herpes simplex virus thymidine kinase gene has been frequently applied as a reporter gene for monitoring gene transfection in animals, it has some intrinsic limitations for use in humans. In our search for a reporter gene that lacks these limitations, we have evaluated the feasibility of the human norepinephrine transporter (hNET) as a reporter gene in combination with the reporter probe 11C-m-hydroxyephedrine (mHED) for PET. Methods: An adenoviral vector (AdTrack-hNET) containing the hNET gene as reporter gene and the enhanced green fluorescent protein (EGFP) as a substitute for a therapeutic gene was constructed. After COS-7, A2780, and U373 cells were transiently transduced with AdTrack-hNET, hNET protein expression, EGFP fluorescence, and cellular uptake of 11C-mHED were determined. In rats, U373 tumor xenografts were grown and transiently transduced with either AdTrack-hNET or an AdTrack-Luc control adenovirus. Intratumoral accumulation of 11C-mHED was determined by PET and ex vivo biodistribution. The tumors were subsequently examined for EGFP fluorescence. Results: 11C-mHED uptake was positively correlated with AdTrack-hNET viral titer and hNET protein expression. However, large differences in transfection efficiency between cell lines were observed. The highest 11C-mHED uptake was found in hNET transfected U373 cells, in which tracer uptake was >70-fold higher than that in control cells. 11C-mHED accumulation could be inhibited by desipramine, a potent inhibitor of hNET. In all cell lines, 11C-mHED uptake was positively correlated with EGFP fluorescence, implying that imaging of hNET with 11C-mHED would enable monitoring of a coexpressed therapeutic gene. In the animal model, gene transfection efficiencies were very low, as determined by EGFP fluorescence. Still, a significantly higher 11C-mHED uptake in hNET transduced tumors than that in control tumors was demonstrated by ex vivo biodistribution studies. PET with a clinical camera could visualize 1 of 3 hNET transduced tumors, indicating that the transfection efficiency was near the detection limit. Conclusion: These results indicate that monitoring of gene therapy using the hNET/11C-mHED reporter gene/probe is feasible, but further investigation with regard to the sensitivity of the technique is required.
Several clinical protocols for gene therapy are being evaluated as treatment for human disease. However, thus far, gene therapy has not come up to the expectations, as difficulties to accomplish controlled, selective, and effective delivery of genes to target cells have limited its success. To evaluate and improve gene delivery strategies, measurement of therapeutic gene expression is of major importance. For some therapeutic genes, direct imaging of the gene of interest is possible, but for the majority of therapeutic genes, no radiotracers are available for imaging. In these cases, a reporter gene can be coupled to the therapeutic gene of interest. In this way, the expression of the therapeutic gene can be monitored indirectly via imaging of the linked reporter gene with a suitable reporter probe (1,2). In principle, the expression of any therapeutic gene can be evaluated by this indirect imaging approach.
The herpes simplex virus thymidine kinase (HSVtk) gene has been studied most extensively as a reporter gene for gene therapy. Several thymidine- and acycloguanosine-derived tracers have been investigated as reporter probes for imaging of HSVtk (2). The 124I-labeled thymidine-derived tracer 5-124I-iodo-2′-fluoro-2′-deoxy-1-β-d-arabinofuranosyl-5-iodouracil (124I-FIAU) and the 18F-labeled acycloguanosine derivative 9-(4-18F-fluoro-3-hydroxymethylbutyl)guanine (18F-FHBG) have already been studied in humans (3,4). The advantage of an enzyme-based reporter gene such as HSVtk is that the gene product can induce signal amplification. An important limitation of HSVtk as a reporter gene is the fact that it is a foreign gene and could therefore provoke an immune response. Another disadvantage of an intracellularly expressed enzyme such as HSVtk is that the transport of the reporter probe into the cell can be rate limiting (5–7).
Other reporter genes that have been previously studied include the human dopamine D2 receptor (D2R) gene, the human somatostatin receptor subtype 2 (hSSTr2), and the sodium iodide symporter (NIS) gene (8). The advantage of the receptor-based systems is that the genes are expressed on the cell surface and, therefore, can be reached easily by the reporter probes. Furthermore, the D2R and hSSTr2 are encoded by endogenous genes and, therefore, do not elicit an immune response. However, this characteristic can also lead to high background levels due to reporter probe accumulation in host tissues. Furthermore, the receptor-based systems do not have the advantage of signal amplification: In general, only one reporter probe molecule can be retained per receptor molecule. In the case of low gene transfer, the produced reporter probe signal may not be high enough for imaging of the reporter gene.
The NIS can be visualized with radioactive iodide by SPECT and PET (9). Although the NIS can induce signal amplification, rapid release of radioactive iodide from NIS transfected cells has also been observed (10). Consequently, cells that are transfected with the NIS gene do not always sufficiently retain the radiotracer (11).
Thus, the currently available reporter genes for imaging of gene therapy have not only their specific advantages but also their own intrinsic disadvantages. Therefore, other reporter genes for imaging of gene therapy in humans are still required.
We investigated the potential of the human norepinephrine transporter (hNET) gene as reporter gene and 11C-m-hydroxyephedrine (mHED) as reporter probe for imaging of gene therapy. hNET is a transmembrane protein that is found at noradrenergic nerve terminals and mediates the active reuptake of norepinephrine that is released by neurons. The norepinephrine is subsequently stored in neuronal vesicles by the vesicular monoamine transporter (12–14). The expression of hNET is almost completely restricted to structures of the sympathetic nervous system (heart, brain). This allows the imaging of hNET as a reporter gene in all organs without sympathetic innervation. Other advantages of the hNET gene as reporter gene is that hNET is expressed in the cell membrane, so limited membrane transport of the reporter probe will not be a problem. 11C-mHED is a good substrate for the hNET (uptake-1) carrier and several tracer molecules can be transported by a single carrier protein molecule, thus resulting in signal amplification. Furthermore, hNET is an endogenous protein and, as a consequence, the reporter gene product will not induce an immunologic reaction. The small size (<2 kb) of the hNET gene allows it to be easily incorporated in the expression cassette of the delivery vehicle when the hNET gene is linked to the therapeutic gene.
11C-mHED is structurally closely related to norepinephrine (Fig. 1A) and has been applied as a PET tracer for assessment of the integrity of presynaptic sympathetic nerve terminals (15). 11C-mHED has demonstrated highly specific uptake and retention in sympathetic nerve terminals of the heart. The radiotracer shares the same neuronal uptake mechanism as norepinephrine, but it is resistant to metabolism by monoamine oxidase and catechol-O-methyltransferase (16,17). Retention of 11C-mHED in hNET-expressing tissue is the result of rapid hNET-mediated reuptake of the tracer. 11C-mHED is rapidly cleared from plasma and whole blood. There is a rapid excretion of 11C-mHED from kidney to the urinary tract (18). 11C-mHED is relatively polar and, therefore, nonspecific uptake is low. High target-to-background ratios can be obtained early after injection of 11C-mHED (19). The use of hNET as a reporter gene has been studied previously using radiolabeled metaiodobenzylguanine (MIBG) as the reporter probe. For PET, MIBG can be labeled with 124I. However, the clinical usefulness of 124I is rather limited. The synthesis of MIBG analogs, labeled with the PET nuclide 18F, has also been described; however, these tracers have not yet been applied in humans. 11C-mHED on the other hand, is a well-established PET tracer and has already been safely applied in humans.
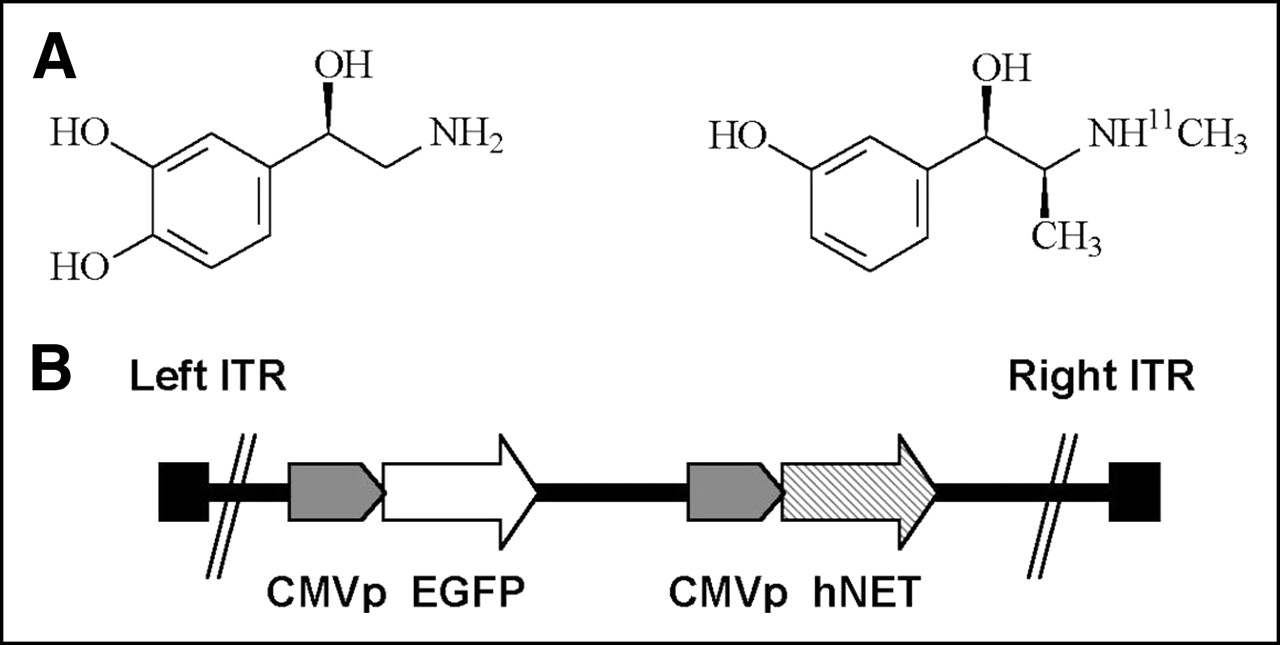
(A) Chemical structures of norepinephrine (left) and 11C-mHED (right). (B) Genome of the first-generation adenoviral vector, containing transgenes EGFP (enhanced green fluorescence protein) and hNET, located in region of the E1 deletion. The 2 transgenes are expressed from identical but separate expression cassettes. CMVp = cytomegalovirus promoter; ITR = inverted terminal repeat.
In this study, the feasibility of hNET as a reporter gene for monitoring therapeutic gene expression was investigated. To this end, cells were transduced with an adenoviral vector containing both hNET as the reporter gene and the enhanced green fluorescent protein (EGFP) as a substitute for a therapeutic gene. Both in vitro and in vivo, the uptake of 11C-mHED in hNET transduced cells was studied and the correlations with hNET expression and EGFP fluorescence were examined.
MATERIALS AND METHODS
Materials
RPMI 1640 medium, trypsin (2.5%, w/v), restriction enzymes, and Lipofectamine PLUS reagent were purchased from Invitrogen. Fetal calf serum (FCS) and Matrigel were obtained from PAA Laboratories. Dulbecco’s minimum essential medium (DMEM), penicillin, and streptomycin were purchased from GibcoBRL, Life Technologies BV. The pcDNA3-hNET plasmid was a generous gift from Randy D. Blakely (20). The pcDNA3.1(−)mychisLacZ plasmid was acquired from Invitrogen. The hNET antibody (NET17-1) was purchased from MAB Technologies Inc. The rabbit anti-mouse IgG antibody, chemically conjugated to horseradish peroxidase, was purchased from DakoCytomation BV. Metaraminol bitartrate salt was obtained from Sigma.
Cell Lines
The green monkey kidney cell line COS-7, the human glioma cell line U373, and the human kidney cell line HEK293 were obtained from the American Type Culture Collection. The human ovarian carcinoma cell line A2780 was obtained from Dr. Robert Ozols (Philadelphia, PA). COS-7 cells were cultured in DMEM supplemented with 5% FCS, 50 IU/mL penicillin, and 50 μg/mL streptomycin. U373 and HEK293 cells were cultured in DMEM/F12 medium supplemented with 10% FCS, 50 IU/mL penicillin, and 50 μg/mL streptomycin. A2780 cells were cultured in RPMI 1640 medium supplemented with 10% FCS. All cells were cultured in a humidified atmosphere with 5% CO2 at 37°C.
Construction of Adenoviral Vector
First-generation (both E1 and E3 regions are deleted from the viral genome) adenoviral vectors were constructed using the AdEasy system (21). The adenovirus (AdTrack-hNET) contains the 2 transgenes EGFP and hNET, which are expressed from identical but separate expression cassettes (Fig. 1B). The control virus (AdTrack-Luc) contains EGFP and luciferase (Luc). Both viruses are based on the shuttle plasmid pAdTrack-CMV. The hNET gene was inserted as XhoI–XbaI fragment of the plasmid pcDNA3-hNET into the corresponding sites in pAdTrack-CMV. The resulting shuttle plasmid was recombined with the plasmid pAdEasy-1 containing part of the adenoviral genome to form viral DNA, which was subsequently transfected into HEK293 cells to produce viral particles. Virus was purified by CsCl gradient.
Preparation of 11C-mHED
Metaraminol bitartrate salt (Sigma; 3 mg) was dissolved in 38 μL of 0.5N NaHCO3. At 125°C, the product was azeotropically dried with 2 × 1 mL of acetonitrile using an argon flow. The residue was resuspended in 0.3 mL of acetonitrile and the clear supernatant containing the metaraminol (free base) was transferred into a clean dry minivial.
11C-Methane was produced by the 14N(p,α)11C nuclear reaction, using a Scanditronix MC17 cyclotron to irradiate the N2 + 5% H2 target gas with 17-MeV protons. 11C-Methyl iodide was prepared from 11C-methane (22) and converted into 11C-methyl triflate (23) by previously described procedures. 11C-Methyl triflate was transported by a stream of argon (20 mL/min) into the minivial containing the metaraminol solution. After the trapping of 11C-methyl triflate was complete, the solvent was evaporated at 125°C with the aid of an argon flow. The residue was dissolved in 1 mL of water and purified by semipreparative reversed-phase high-performance liquid chromatography (HPLC) using a μBondapak C18 column (Waters-Millipore; 10 μm, 300 × 7.8 mm) and 1% ethanol in 0.01 mol/L H3PO4 as the eluent at a flow of 3 mL/min. The radioactive product with a retention time of 8 min was collected (retention time of metaraminol, 5 min) and neutralized with 0.5N NaOH. In this manner, 11C-mHED with a specific activity of 11 ± 6 GBq/μmol was prepared in 65% ± 14% decay-corrected radiochemical yield (based on 11C-methyl triflate). The identity of the product was confirmed by coinjection with a reference sample on reversed-phase HPLC.
Accumulation of 11C-mHED in hNET Transfected Cells
Cells were seeded in 12-well plates at a density so that cells were 70% confluent the next day. Two days after seeding, the cells were infected with adenovirus at various multiplicities of infection (MOI) by adding AdTrack-hNET virus to the cells in fresh culture medium (MOI 30, 100, 300, and 1,000 for COS-7 cells and A2780 cells and MOI 0, 30, 50, 70, 100, and 300 for U373 cells). Control cells were infected with AdTrack-Luc. After 2 h of infection, the medium was changed to fresh medium without virus. Two days after infection, the cells were incubated with 44 ± 15 MBq of 11C-mHED for 1 h. For competition experiments, desipramine (DMI) was added 5 min before the tracer (final DMI concentration, 6 μmol/L). After tracer incubation, the culture medium was removed and the monolayers were washed 3 times with 1.0 mL of phosphate-buffered saline (PBS). The cells were harvested from the culture plates by treatment with 0.25 mL of trypsin. The cells were resuspended in 1.2 mL of culture medium to neutralize the trypsin. A 50-μL sample was taken and mixed with 50 μL of trypan blue to count the number of viable cells under a microscope. The radioactivity in the cell suspensions was measured in a γ-counter (LKB, Wallac) and normalized to the number of viable cells. Results are reported as the percentage of tracer dose per million cells (%ID/106 cells). For COS-7 cells, 3 independent experiments were performed, either in duplicate or triplicate. For A2780 cells, 1 experiment was performed in duplicate. Two experiments (each in quadruplicate) were performed for U373 cells.
hNET Protein Expression
hNET protein expression was determined in cells grown and infected parallel to the cells used for 11C-mHED accumulation experiments. EGFP expression of cells for the protein isolation was comparable to the cells used for the accumulation experiments. Cells were detached by scraping the cells in 500 μL of PBS and lysed by adding 2.5 μL of 20% (w/v) sodium dodecyl sulfate. Lysates were analyzed by dot blot and immunohistochemistry. Protein was detected with NET17-1, a mouse antibody directed against the extracellular domain of the hNET protein, and a rabbit secondary antibody linked to horseradish peroxidase. Blots were stained using enhanced chemoluminescence ([ECL] ECL plus Western Blotting Detection system (Amersham Biosciences, Ltd.). The amount of protein was quantified on the western blot using the Quantity One program (Bio-Rad Laboratories) and expressed in arbitrary units. The method was validated by determining the correlation between ECL intensity and the amount of protein and the detection range using increasing amounts of an unrelated protein.
EGFP Analysis
EGFP expression in the transfected cells was measured immediately before the start of the 11C-mHED accumulation experiments by measuring the EGFP intensity in an FL500 microplate fluorescence reader (Bio-Tek Instruments; excitation, 485 nm; emission, 508 nm). The EGFP intensity was expressed in arbitrary units.
Animal Model
U373 cells (2 × 107 cells in 0.2 mL of culture medium) were mixed 1:1 with Matrigel and injected subcutaneously into the left and right shoulder of male nude rats (HSD RH rnu, Harlan; body weight, 140–200 g). When solid tumor nodules had grown to approximately 1.0 cm in diameter (after approximately 2 wk), they were injected with AdTrack-hNET adenovirus (1010 plaque-forming units; 5.7 × 1010 viral particles in 80 μL) in 1 tumor and with AdTrack-Luc control virus (5.7 × 1010 viral particles in 80 μL) in the contralateral tumor by single injection of the adenoviruses. All studies were performed in compliance with the national law and the local ethical guidelines for animal experiments. The protocols were approved by the Animal Ethics Committee of the Groningen University.
In Vivo Accumulation of 11C-mHED
PET studies were performed using a Siemens Exact HR+ positron camera (Siemens/CTI) with a resolution of about 5 mm (full width at half maximum). Transmission and emission scans were obtained in 2-dimensional mode. Two days after virus delivery, the rats (n = 3) were anesthetized with an intraperitoneal injection of a mixture of ketamine (25 mg/kg) and medetomidine (1 mg/kg). The animals were positioned in the PET camera with their long-axis parallel to the transaxial plane of the tomograph. A transmission scan was obtained to correct for attenuation of 511-keV photons by tissue. Subsequently, 42 ± 6 MBq of 11C-mHED was injected into the tail vein and data acquisition was started. Data were acquired for 1 h using the following frames: 6 consecutive frames of 10 s, 2 of 30 s, 3 of 60 s, 2 of 120 s, 2 of 180 s, 3 of 300 s, and 3 of 600 s. Emission scans were corrected for radioactive decay, attenuation, random counts, and dead time.
After the PET scan was completed, the rats were sacrificed by extirpation of the heart. Tumors were dissected and weighed and the accumulated radioactivity was measured with a γ-counter. Tracer accumulation is expressed as percentage of injected dose per gram tissue (%ID/g). EGFP expression in tumor was visualized under a fluorescence microscope in cryosections. Series of 4-μm sections from 4 randomly chosen sites were prepared from each tumor. The sections were placed on a glass slide and examined unfixed and unstained.
Statistical Analysis
Differences in tracer accumulation were analyzed using a 2-sided unpaired Student t test for in vitro studies and a 2-sided paired Student t test for in vivo experiments. P < 0.05 was considered significant.
RESULTS
Accumulation of 11C-mHED in hNET Transfected Cells
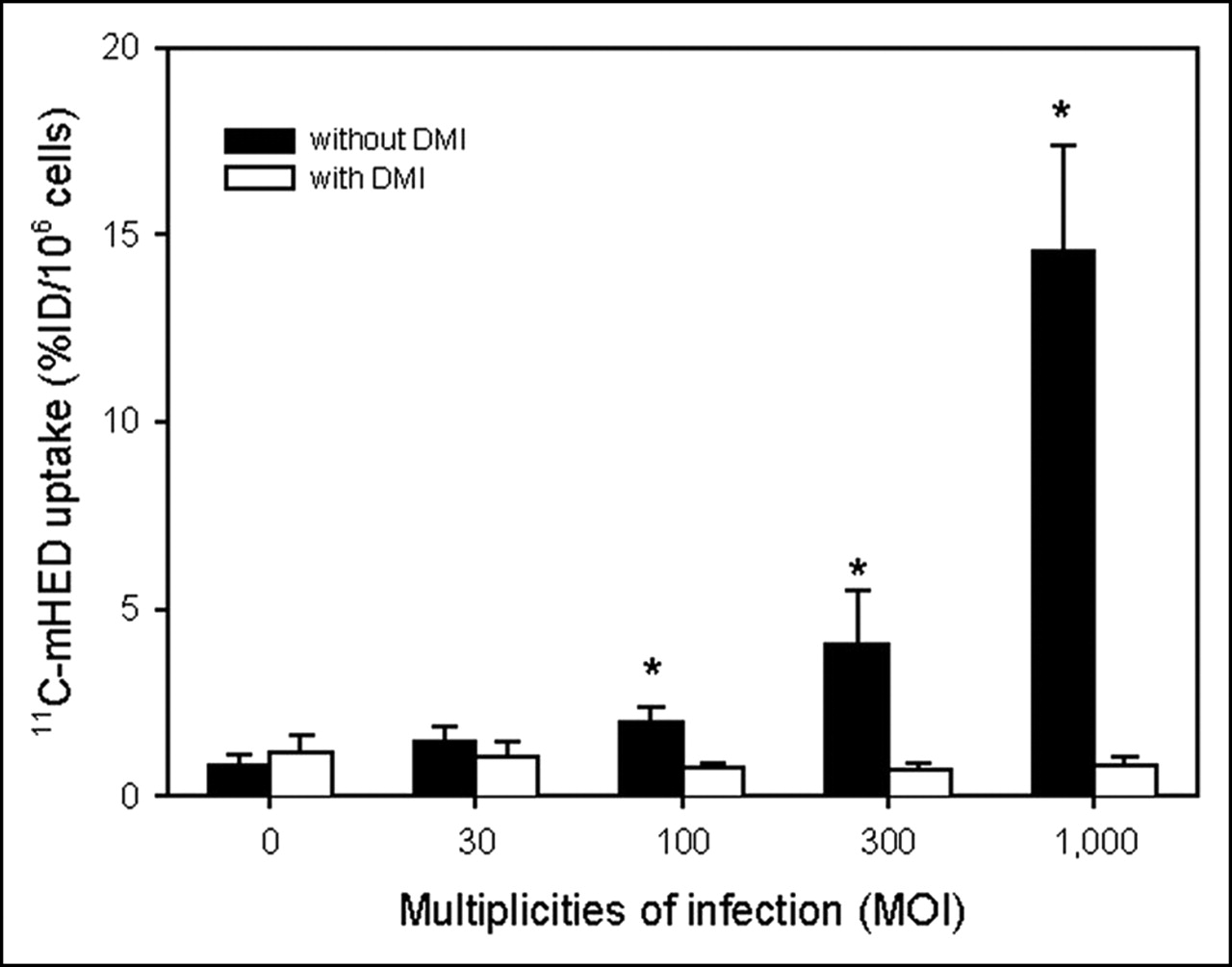
11C-mHED uptake was determined in COS-7 cells that were infected with AdTrack-hNET virus at various MOIs (MOI 30, 100, 300, and 1,000) and with AdTrack-Luc control virus (MOI 100). Figure 2 shows that the uptake of the tracer in the AdTrack-hNET transfected cells increased with increasing viral titers. After 1 h of incubation with 11C-mHED, the tracer uptake in hNET transfected cells at the highest viral titer (MOI 1,000: 14.6 ± 2.8 %ID/106 cells) was approximately 17-fold higher than that in cells transfected with control virus. Incubation with 11C-mHED in the presence of DMI (a potent antagonist of norepinephrine transport) reduced tracer uptake in hNET transfected cells to the level of control cells (cells infected with control virus; Fig. 2). The DMI-induced reduction in tracer uptake was statistically significant for cells infected with AdTrack-hNET at MOI 100 or higher (P ≤ 0.01).
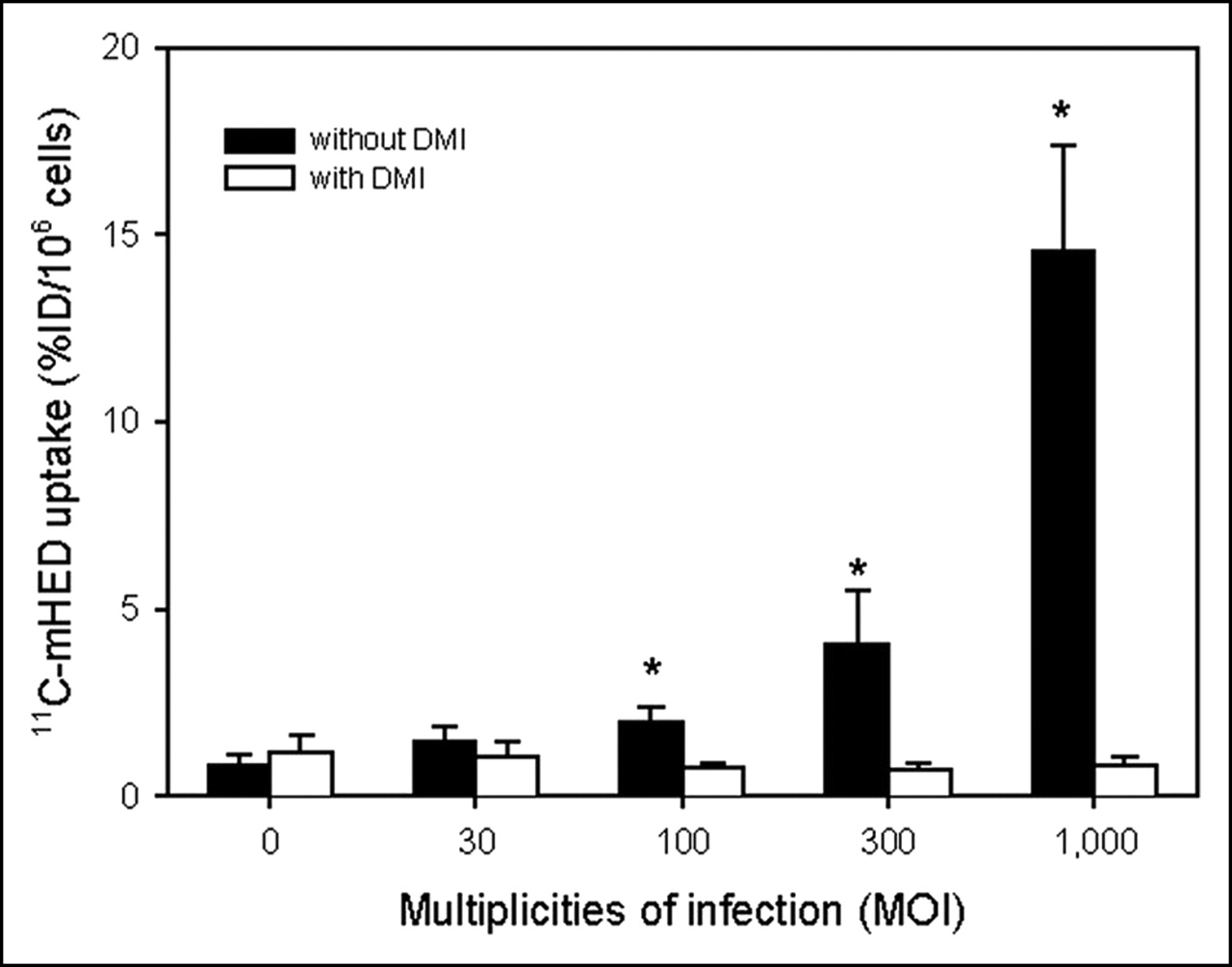
11C-mHED uptake in COS-7 cells transfected with adenoviral AdTrack-hNET at various MOI. MOI = 0 represents control (cells infected with AdTrack-Luc). Two days after transfection, cells were assayed for 11C-mHED accumulation in presence or absence of 6 μmol/L DMI. Three independent experiments were performed, either in duplicate or triplicate. Average ± SD is shown. *Significantly higher uptake than that of DMI-treated cells (P < 0.05).
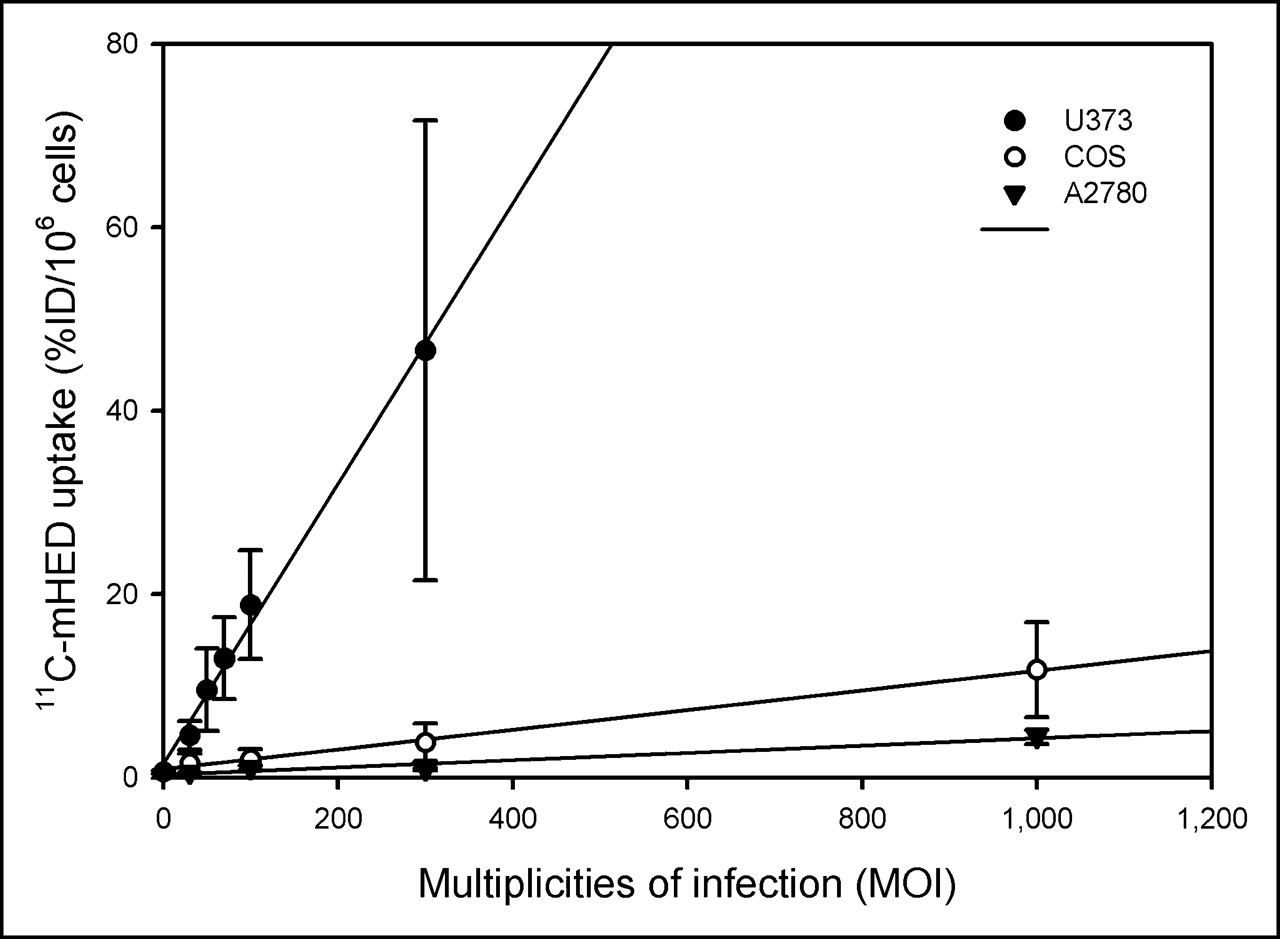
Over the range of viral titers used, 11C-mHED uptake showed an excellent linear correlation with the viral titer not only for COS-7 cells (r2 = 0.997; P = 0.0002) but also for A2780 cells (r2 = 0.949; P = 0.02) and for U373 cells (r2 = 0.994; P < 0.0001). However, the slope of the plot of the tracer uptake versus the viral titer was strongly dependent on the type of cell line used (Fig. 3). At the highest viral titer, 11C-mHED uptake in hNET transfected A2780 cells (MOI 1,000: 4.4 ± 0.8 %ID/106 cells) was approximately 3.5-fold lower than that in transfected COS-7 cells. The 11C-mHED uptake in transfected U373 cells, on the other hand, was approximately 11-fold higher than tracer uptake in transfected COS-7 cells. The tracer uptake in U373 cells transfected with the highest viral AdTrack-hNET titer (MOI 300: 47 ± 25 %ID/106 cells) was >70-fold higher than that in control cells.

11C-mHED uptake vs. viral titer in COS-7 cells, A2780 cells, and U373 cells. Cells were transduced with AdTrack-hNET at various MOI. MOI = 0 represents control (cells infected with AdTrack-Luc). Two days after infection, cells were assayed for 11C-mHED accumulation. In all cell lines, 11C-mHED uptake increased linearly with viral titer (r2 = 0.997 for COS-7 cells, r2 = 0.949 for A2780 cells, and r2 = 0.994 for U373 cells; P < 0.05). Error bars represent SD.
hNET Protein Expression
The amount of hNET protein was determined in U373 cells. Figure 4 shows a good correlation between the amount of hNET protein and the 11C-mHED uptake (r2 = 0.987; P < 0.0001). This indicates that the transduced hNET gene produces functional hNET protein and that 11C-mHED uptake correctly reflects the magnitude of hNET expression.

11C-mHED uptake vs. amount of hNET protein in U373 cells. hNET protein expression was determined in U373 cells grown and transfected parallel to cells used for 11C-mHED accumulation experiments 2 d after infection. Good correlation between amount of hNET protein and 11C-mHED uptake is evident (r2 = 0.987; P < 0.0001). Error bars represent SD.
EGFP Analysis
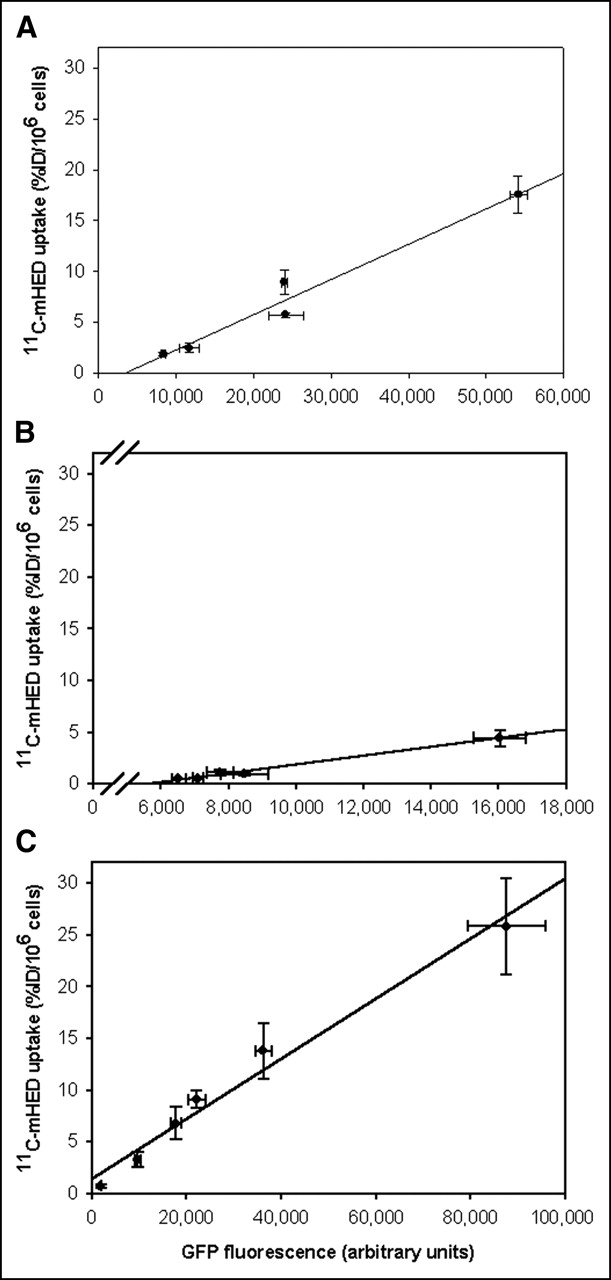
In COS-7, A2780, and U373 cells, EGFP fluorescence was measured in the cells just before addition of the tracer. The tracer uptake versus EGFP fluorescence is shown in Figure 5. EGFP fluorescence shows a good linear relationship with 11C-mHED uptake in the 3 cell lines tested (r2 = 0.965, P = 0.003 for COS-7 cells; r2 = 0.989, P = 0.0005 for A2780 cells; r2 = 0.979, P = 0.0002 for U373 cells), indicating that EGFP expression was positively correlated with hNET expression.
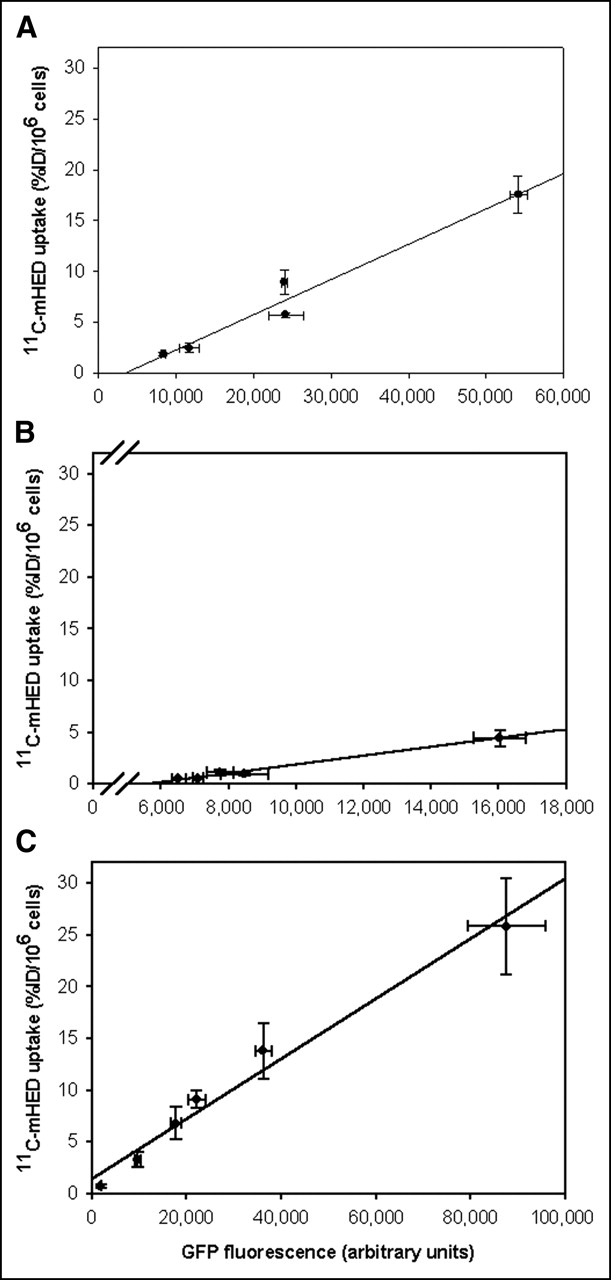
11C-mHED uptake vs. EGFP fluorescence in COS-7 cells (A), A2780 cells (B), and U373 cells (C). Cells were transduced with AdTrack-hNET. Two days after infection, EGFP intensity was measured immediately before the start of 11C-mHED accumulation experiments. In all 3 cell lines, EGFP fluorescence correlated well with 11C-mHED uptake (r2 = 0.965 for COS-7 cells, r2 = 0.989 for A2780 cells, and r2 = 0.979 for U373 cells; P < 0.005). Error bars represent SD.
Accumulation of 11C-mHED In Vivo
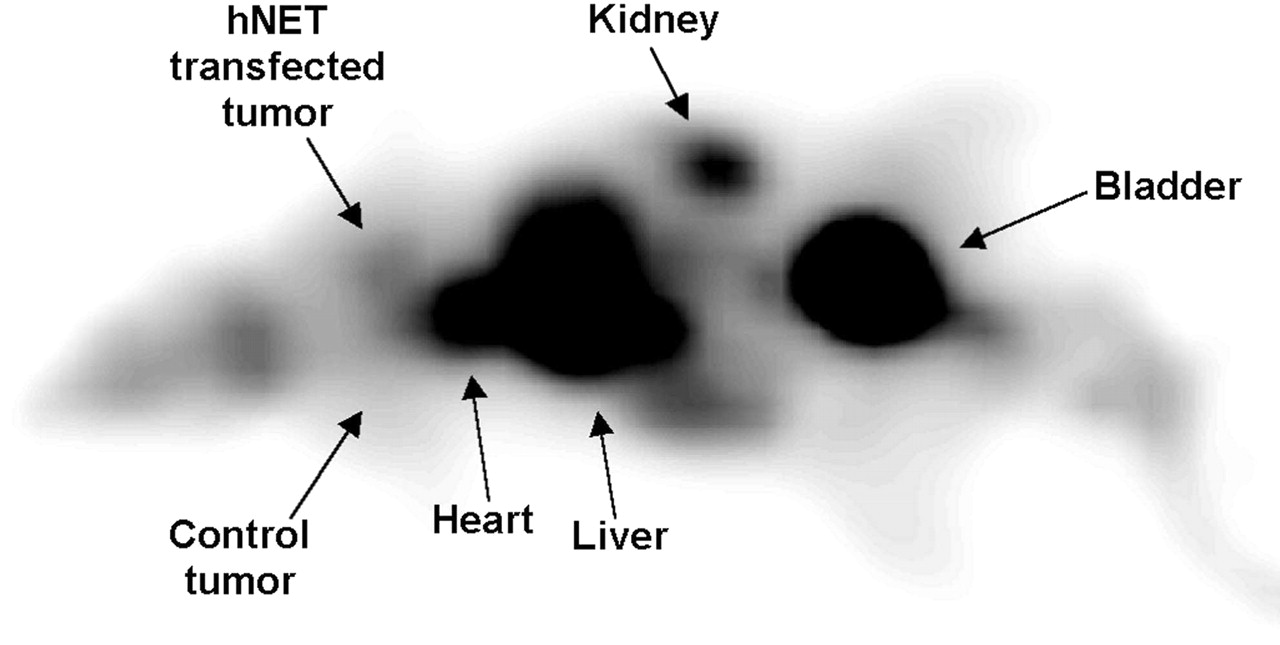
To investigate the transfection with the hNET gene in vivo, 11C-mHED uptake was studied in rats bearing 2 subcutaneous U373 tumor xenografts that were injected with either AdTrack-hNET adenovirus or AdTrack-Luc control virus 2 d before PET. 11C-mHED PET images showed high tracer uptake in heart, liver, kidney, and bladder. The hNET transfected tumor was visible in 1 of 3 animals at 1 h after injection of the tracer, whereas the control tumor could not be discerned in any of the animals (Fig. 6).

Anterior 11C-mHED PET image of HSD RH rnu rat bearing 2 U373 tumors that were either transfected with adenoviral vector AdTrack-hNET (top) or control vector (bottom) 2 d before PET study. PET image was obtained 50–60 min after injection of 42 ± 6 MBq of 11C-mHED tracer. Image is summation of all coronal planes, on which animal was visible.
After PET, tumors were excised and 11C-mHED uptake was determined by ex vivo γ-counting. In all animals, tracer accumulation was consistently 14%–27% higher in hNET transfected tumors than that in control tumors. As a result, average 11C-mHED uptake in the hNET transfected tumors (0.13 ± 0.02 %ID/g) was significantly higher (paired t test, P < 0.05) than 11C-mHED uptake in tumors transfected with control virus (0.11 ± 0.01 %ID/g).
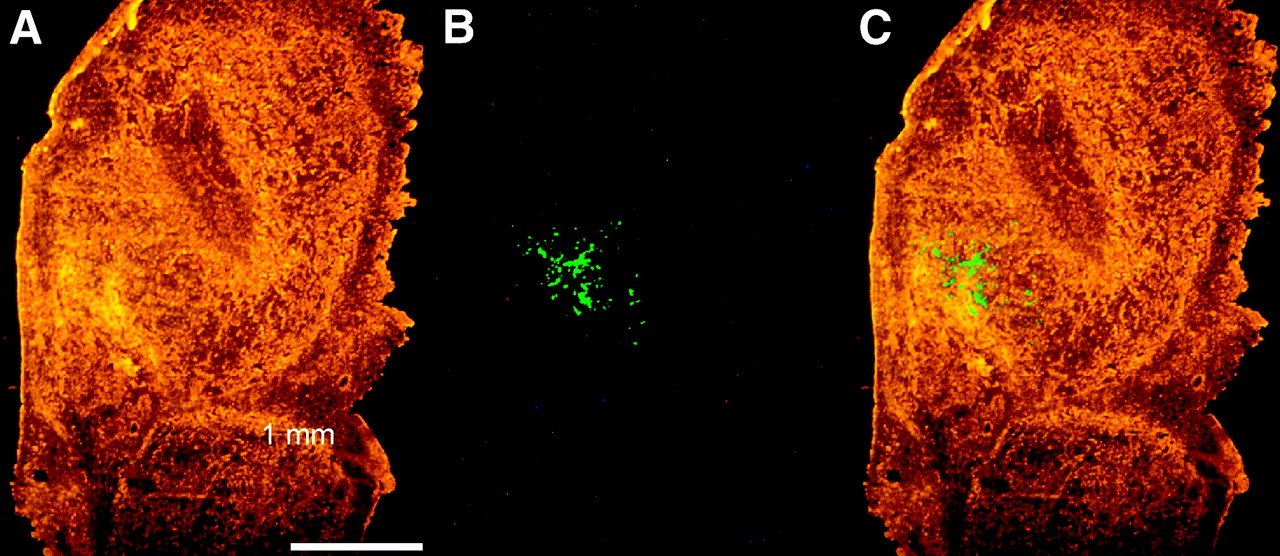
To determine the extent of transgene expression, random tumor sections were cut and EGFP expression was investigated by fluorescence microscopy. EGFP fluorescence could be visualized in only 2 of 24 series of tumor sections. In both sections, a clear patch of fluorescent cells of approximately 1 mm in diameter could be seen (Fig. 7). These results indicate that only a small fraction of the tumor cells was transfected and expressed the transgene.
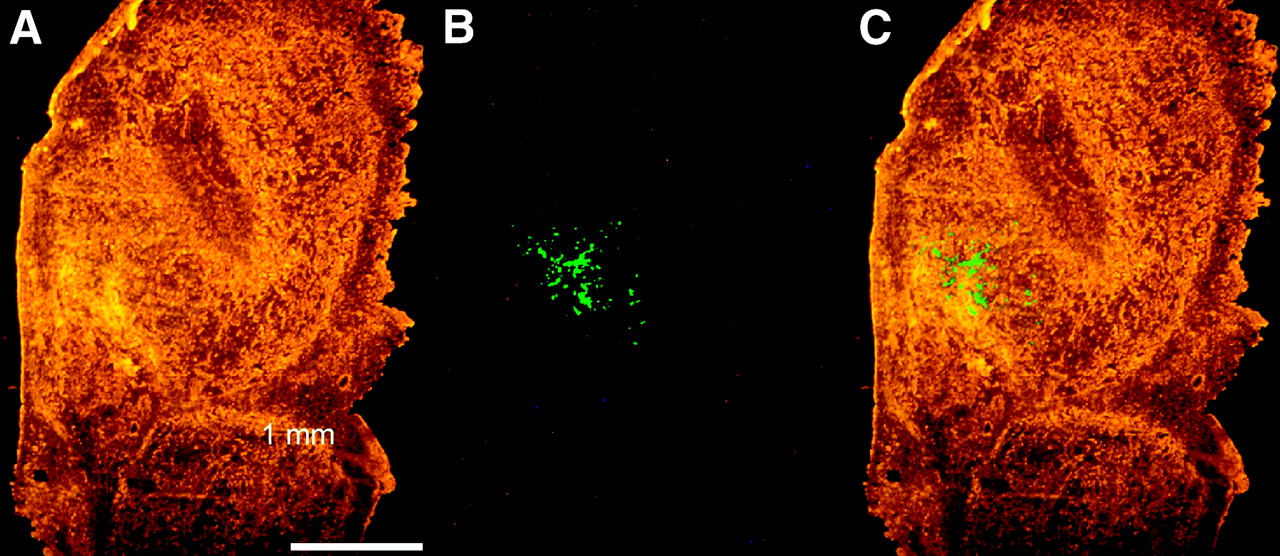
Visualization of EGFP fluorescence in tumor tissue. EGFP expression in tumor tissue was visualized under fluorescence microscope in 4-μm cryosections. (A) Phase-contrast image. (B) EGFP fluorescence. (C) Merged picture.
DISCUSSION
Monitoring the efficiency and spatial distribution of transduction of a therapeutic gene is highly desirable for the development of safe and efficient gene therapy for treatment of human disease. In our search for a reporter system with optimal characteristics, we investigated the feasibility of hNET as a reporter gene for imaging of gene therapy. The use of hNET as a reporter gene has been studied previously using radiolabeled MIBG as the reporter probe (24). Radiolabeled MIBG is a promising radiopharmaceutical for targeted radiotherapy. Tumors derived from the neural crest (neuroblastoma and phaeochromocytoma) respond to 131I-MIBG–mediated radiotherapy by actively taking up 131I-MIBG via the hNET (25,26). It has been demonstrated that 131I-MIBG can also be used as a radiotherapeutic agent in tumors other than those derived from the neural crest. Tumor cells, which do not express hNET, can be transfected with the hNET gene, which results in an enhanced 131I-MIBG uptake and related toxicity (27–30).
PET seems to be the method of choice for monitoring gene transduction because of its high sensitivity and its ability to generate quantitative results. However, for imaging reporter gene expression with PET, MIBG is not the most suitable tracer. The PET isotope 124I has a relatively long half-life (4.2 d) and emits high-energy γ-rays, and only 23% of the disintegrations produce positrons. These characteristics reduce the quality of the PET images, complicate quantification, and could limit the dose administered to patients, which makes the clinical usefulness of 124I rather limited (31,32). MIBG analogs, labeled with the more conventional PET nuclide 18F, have also been synthesized; however, none of them is used in clinical practice (33). Therefore, we have chosen to evaluate 11C-mHED as the reporter probe for PET of the reporter gene hNET. 11C-mHED is a polar compound with low nonspecific uptake. Because of its rapid accumulation in target tissue and fast clearance from plasma, high-contrast 11C-mHED PET images can be obtained soon after injection of the tracer (34). Together with the short half-life of 11C (20 min), this would allow rapid repetitive imaging. On the other hand, the short half-life may be a potential limitation as the accumulation of the tracer in the tumor may be expected to rise over time while the background signal would wash out, leading to an increased signal-to-background ratio at later time points.
In the above studies, in which MIBG was used as the reporter probe to image hNET transgene expression, stably transduced cell lines and tumors were used. This approach ensures that the percentage of transfected cells is high and, consequently, high levels of tracer uptake can be achieved. In clinical practice, however, transfection efficiencies are usually low.
To mimic the clinical situation more closely in this study, we used tumor cells and tumor xenografts that were transiently transfected with an adenoviral construct. In cell experiments, we have shown that hNET can be successfully transfected into various cell lines using the AdTrack-hNET adenoviral vector. Transfected cells produced functional hNET protein. Cellular 11C-mHED uptake correlated well with the expression of the hNET reporter gene. When the hNET transporter was inhibited with DMI, the cellular 11C-mHED uptake was reduced to the level of control cells. These results indicate that cellular 11C-mHED uptake is specific for hNET and, thus, is a suitable measure for the reporter gene expression after gene transduction.
In all cell lines tested, 11C-mHED increased linearly with increasing viral titer. However, at an established viral titer, the 11C-mHED uptake varied to a great extent between the different cell lines. Apparently, the different cell types showed variable degrees of susceptibility to adenoviral infection, resulting in variable levels of expression of the hNET gene. Adenoviral infection is mediated by the coxsackie adenovirus receptor (CAR). The differences in infection susceptibility between cell lines is probably caused by differences in expression levels of the CARs. It has been shown that a deficiency of the primary receptor for adenoviruses on tumor cells limits the efficiency of adenovirus-mediated gene transfer in vivo (35). A2780 cells have low expression levels of the CAR (36), which would explain the low transfection efficiency and low cellular 11C-mHED uptake that was observed in this cell line.
To test the feasibility of hNET as a reporter gene, we have used an adenoviral vector, which contains both the hNET reporter gene and the EGFP gene, under control of the same promoter. In this way, we hypothesized equal expression of both genes. In all cell lines, EGFP fluorescence was positively correlated with the 11C-mHED uptake, indicating a linear relationship between EGFP expression and hNET expression. Thus, the hNET/11C-mHED appears to be a suitable reporter gene/probe system to monitor the expression of a second gene in the transfection vector. This approach should be easily adaptable to clinical protocols by replacing the EGFP with any therapeutic gene of choice. Thus, imaging of hNET with 11C-mHED could enable monitoring of a coexpressed therapeutic gene.
To test the feasibility of the reporter system in vivo, the adenoviral vector was injected in U373 tumors grown in rats. In contrast to the in vitro results, in vivo gene transfection efficiency in the tumor was poor. Minor tumor transfection was only observed in the immediate area of the needle tract. Still, ex vivo biodistribution studies revealed a consistent and significant elevation of 11C-mHED uptake (19% ± 7%) in the AdTrack-hNET transfected tumors as compared with the contralateral tumors that were transfected with control virus. This increase in 11C-mHED uptake may appear small compared with the in vitro data, but it should be noted that transfection in the tumor was restricted to only a small area of 1 mm in diameter around the injection site. However, in the ex vivo biodistribution studies, the tracer uptake in the whole tumors (0.44 ± 0.10 g) was measured. From these data, it is estimated that <1% of the cells in the tumor was transfected by the viral vector. Obviously, the 11C-mHED uptake in the whole tumor strongly underestimates the actual tracer uptake in the infected region.
Despite the low in vivo transfection efficiencies, the hNET transfected tumor could still be visualized by our clinical PET camera in 1 of 3 animals. This indicates that the transfection levels that were achieved in this study were near the detection limit. Obviously, the detection limit was strongly affected by partial-volume effects, as the spatial resolution of the camera was about 4–5 mm, whereas the transfected tumor area was only approximately 1 mm in diameter. As a consequence, the hNET-derived signal was averaged out over the resolution volume. Because a small-animal PET camera is not available at our institution yet, we performed our experiments with a clinical PET camera. However, animal studies using a dedicated animal PET camera with a resolution of <2 mm or human studies could give a better indication of the sensitivity of 11C-mHED PET for monitoring of hNET reporter gene expression.
CONCLUSION
hNET in combination with 11C-mHED appears to be a promising reporter system for monitoring of gene therapy with PET and should be applicable in clinical protocols. The sensitivity of the technique, however, might be a limitation, as the low transfection efficiencies that were achieved in this study were near the detection limit. Therefore, further studies are required to investigate whether 11C-mHED PET is sensitive enough to detect clinically relevant levels of the reporter gene after in vivo gene transfection.
Acknowledgments
This work was financially supported by the Dutch Cancer Society (grant RUG 2000-2310).
Footnotes
Received Apr. 27, 2005; revision accepted Sep. 16, 2005.
For correspondence contact: Hidde J. Haisma, PhD, Department of Therapeutic Gene Modulation, University Center for Pharmacy, P.O. Box 196, 9700 AD Groningen, The Netherlands.
E-mail: h.j.haisma{at}rug.nl
↵* Contributed equally to this work.
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Norepinephrine Transporter as a Target for Imaging and Therapy
- Noninvasive Molecular Imaging Using Reporter Genes
- Noninvasive Imaging of Cell-Mediated Therapy for Treatment of Cancer
- Imaging hNET Reporter Gene Expression with 124I-MIBG
- A Human-Derived Reporter Gene for Noninvasive Imaging in Humans: Mitochondrial Thymidine Kinase Type 2