


Abstract
The ibritumomab tiuxetan therapeutic regimen consists of a dose of rituximab, 250 mg/m2, followed by 111In-ibritumomab tiuxetan, for imaging, on day 1 and a dose of rituximab followed by 90Y-ibritumomab tiuxetan, for therapy, on day 7, 8, or 9. Treatment with the Food and Drug Administration–approved regimen also requires that scans be performed at 2–24 h and at 48–72 h after the 111In-ibritumomab tiuxetan, with an optional third scan at 90–120 h, to confirm appropriate biodistribution. In the clinical trials before the approval of the regimen, only 1 patient (of approximately 400) was not treated with 90Y-ibritumomab tiuxetan after imaging with 111In-ibritumomab tiuxetan, because of altered biodistribution. The Zevalin Imaging Registry was established by Biogen Idec Inc. to identify cases of potential altered biodistribution and to collect clinical information in cases in which the regimen was not completed after imaging. Methods: The registry surveyed treating physicians to verify completion of treatment with the ibritumomab tiuxetan therapeutic regimen in patients treated with 111In-ibritumomab tiuxetan between March 27, 2002, and March 31, 2003. Results: Survey data were collected on 953 of an estimated 1,144–1,192 patients in whom ibritumomab tiuxetan therapy was initiated (case capture rate of 80%–83%). Thirty-eight cases were reported in which a decision not to treat was made after imaging with 111In-ibritumomab tiuxetan (4.0% of all cases captured); 16 of these were for imaging reasons, and 22 were for medical reasons. Twelve of the 16 imaging cases met the criteria for altered biodistribution (1.3%). Of these 12 cases, 6 (0.6%) were suspected to be true altered biodistribution and 6 appeared to be due to the use of a procedure for radiolabeling 111In-ibritumomab tiuxetan that differed from that in the prescribing information. All cases of altered biodistribution were seen on the first image (2–24 h) after the administration of 111In-ibritumomab tiuxetan. The 22 cases in which decisions not to treat were made for medical reasons accounted for 2.3% of the cases. The majority of these cases (19/22) were in patients who had an expected biodistribution but had a rapid change in their clinical condition that precluded treatment. Conclusion: The rate of true altered biodistribution was 0.6% in the Zevalin Imaging Registry, which collected treatment decisions based on data from approximately 80% of all patients treated commercially in the first year after drug approval. All cases of altered biodistribution were apparent on the first image, obtained at 2–24 h after the administration of 111In-ibritumomab tiuxetan.
In February 2002, 90Y-Ibritumomab tiuxetan was approved in the United States. It is indicated for the treatment of patients with relapsed or refractory low-grade, follicular, or transformed non-Hodgkin’s lymphoma (NHL), including patients with rituximab-refractory NHL (1). Clinical data indicate that 90Y-ibritumomab tiuxetan is an effective therapy for NHL. Designed to exploit the inherent radiosensitivity of lymphomas, the regimen can produce high rates of overall response (73%–83%) and long-term responses (time to progression ≥12 mo; 37%) in patients with CD20+ B-cell NHL (2–7). Responses of >6 y duration have been observed in some patients (6). In addition, the overall response rates and durations of response are greater when 90Y-ibritumomab tiuxetan is used early in the course of treatment (≤2 prior chemotherapies or at first relapse) (8). There is evidence that the regimen is effective as a frontline therapy for follicular lymphoma, either in combination with short-course chemotherapy (9) or as a single agent followed by rituximab maintenance (10), and in relapsed aggressive lymphomas (11). Treatment with 90Y-ibritumomab tiuxetan does not seem to have any effect on the efficacy or safety of other treatments that are used after relapse (12–14).
In the United States treatment with the ibritumomab tiuxetan therapeutic regimen begins on day 1 with an infusion of rituximab, 250 mg/m2, followed within 4 h by an intravenous injection of an imaging dose of 111In-ibritumomab tiuxetan, 185 MBq (5 mCi). Two imaging scans are acquired at 2–24 h and at 48–72 h after 111In-ibritumomab tiuxetan, with an optional third scan at 90–120 h. On day 7, 8, or 9, patients are given an infusion of rituximab, 250 mg/m2, followed by an intravenous injection of 90Y-ibritumomab tiuxetan given over 10 min. The dose of 90Y-ibritumomab tiuxetan is 11.1 or 14.8 MBq/kg (0.3 or 0.4 mCi/kg) depending on platelet count, with a maximum total dose of 1,184 MBq (32 mCi).
The γ-emitting radioisotope 111In was used in the clinical development of the ibritumomab tiuxetan regimen for measuring organ-specific accumulation and for determining whether dosimetry before treatment with 90Y-ibritumomab tiuxetan was required (15,16).
Central dosimetry analysis, performed in 179 patients using regions of interest for 5 organs (liver, lungs, kidney, spleen, and sacral marrow), showed that the radiation-absorbed doses were estimated to be below the maximum allowable 2,000 cGy to uninvolved normal organs and 300 cGy to the red marrow (17). It was also determined that there was no correlation between the estimated total-body or red-marrow radiation-absorbed doses and the nadirs of the absolute neutrophil count, platelet count, or hemoglobin level.
Routine dosimetry is not required with the ibritumomab tiuxetan regimen, because of a lack of correlation between the estimated radiation-absorbed dose and hematologic toxicity, the predictable urinary clearance of 90Y-ibritumomab tiuxetan, and the absence of dehalogenation (16,17). Imaging with 111In-ibritumomab tiuxetan continues, however, as an additional measure to detect any potential altered biodistribution issues that were not seen in the clinical trials. Expected and altered biodistribution, as described in the prescribing information, are presented in Table 1. Whole-body γ-camera images of the expected biodistribution are shown in Figure 1. Only 1 of 400 patients who underwent imaging in the clinical trials was not treated with 90Y-ibritumomab tiuxetan because of altered biodistribution. A Zevalin Imaging Registry was established to identify cases of potential altered biodistribution in U.S. commercial use of the regimen, investigating the frequency of patients who were imaged with 111In-ibritumomab tiuxetan but were not then treated with 90Y-ibritumomab tiuxetan, the reasons for their not being treated, the patterns of altered biodistribution, and the utility of imaging in guiding treatment decisions. Reported here are the incidence of altered biodistribution and the reasons for not proceeding to treatment with 90Y-ibritumomab tiuxetan as collected by the Zevalin Imaging Registry.
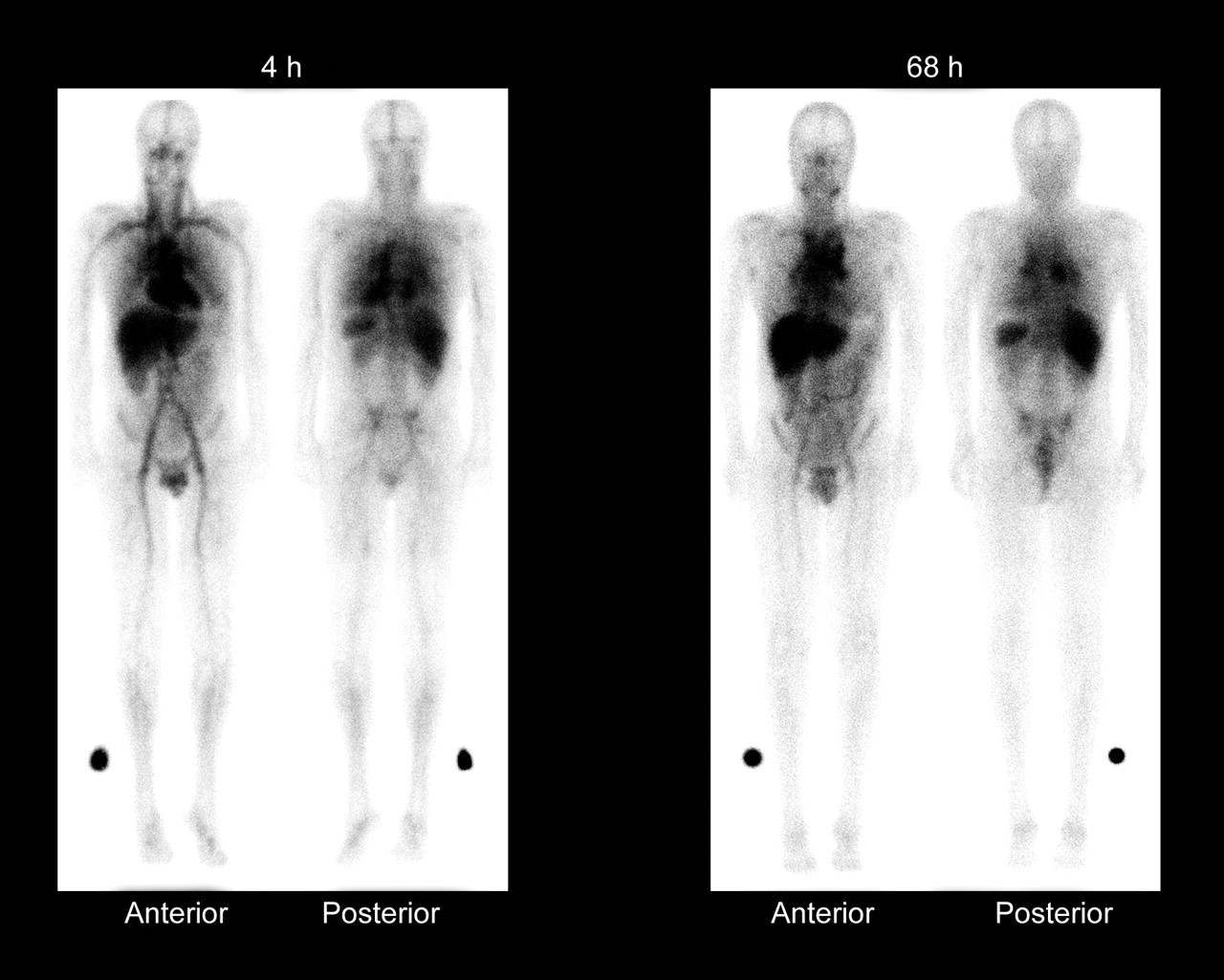
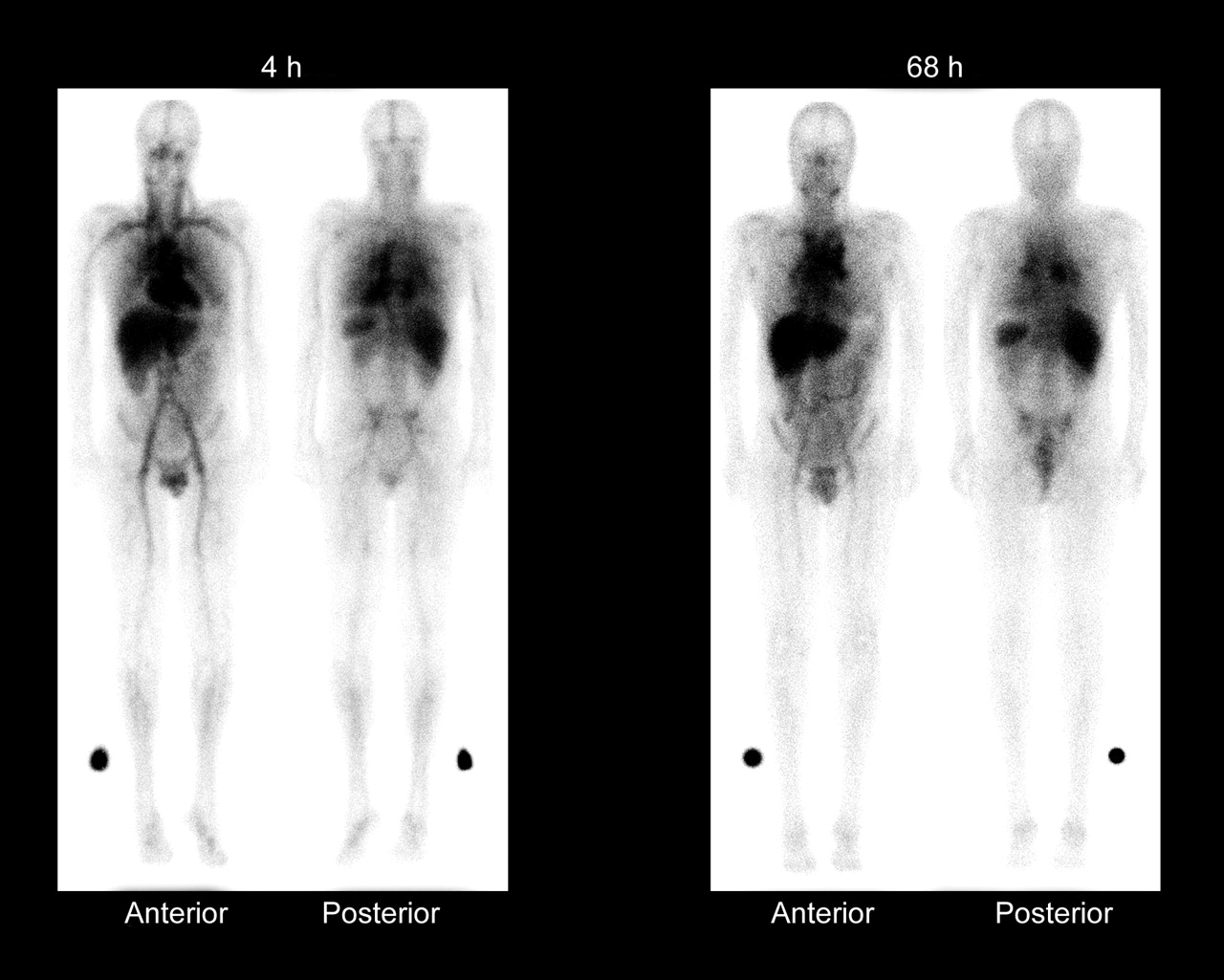
Whole-body γ-camera images obtained at 4 and 68 h after administration of 111In-ibritumomab tiuxetan, showing expected biodistribution, with the radiopharmaceutical easily detectable in the blood-pool areas on 4-h images. Moderately high or high uptake in normal liver and spleen is seen on 4- and 68-h images, and moderately low or very low uptake in normal kidneys, urinary bladder, and bowel is seen on 4- and 68-h images. Tumor uptake is seen in mediastinal lymph nodes. (Reprinted with permission of Biogen Idec Inc.)
Expected Biodistribution and Altered Biodistribution on Whole-Body γ-Images
MATERIALS AND METHODS
Registry Design
The Zevalin Imaging Registry was established by Biogen Idec Inc. to identify cases of potential altered biodistribution and to collect additional clinical information in cases in which the treatment regimen was not completed after imaging with 111In-ibritumomab tiuxetan in the commercial setting. Data were collected on patients treated from March 27, 2002, through March 31, 2003. Participation in the registry by the treating physician was voluntary, and, in cases in which clinical information was collected on no-treatment decisions, the clinical information was de-identified.
The initial mailing, consisting of a letter explaining the purpose of the registry and registry data forms, was sent to approximately 4,000 nuclear medicine physicians, radiation oncologists, and chief technicians or technologists of nuclear medicine. The mailing list was acquired from the Society of Nuclear Medicine.
Data about the shipments of ibritumomab tiuxetan kits were used to identify the medical treatment centers that ordered the therapy. Registry staff contacted all of the treating physicians and requested their participation in the registry. Registry data form 1 and the standard cover letter explaining the purpose of the registry were then sent to each treatment center. If the patient had been treated with 90Y-ibritumomab tiuxetan after imaging, no further information was requested. However, if the patient had not been treated with 90Y-ibritumomab tiuxetan after the 111In-ibritumomab tiuxetan, registry data form 2 was sent to the treatment center to query the reasons for the decision not to treat and to request copies of the whole-body images. If a treatment site did not return the data forms, verbal confirmation of the completion of therapy with the ibritumomab tiuxetan regimen and the date of administration were obtained.
Estimation of Number of Patients Treated
Biogen Idec Inc. customer service supplied the number of ibritumomab tiuxetan imaging and therapy kits that had been shipped. The maximum number of patients imaged was estimated as the number of imaging kits shipped minus the number of kits that were not used. The minimum number of patients imaged was estimated as the number of paired (imaging and therapy) kits that were shipped plus the number of no-treatment decisions captured by the registry.
RESULTS
Treatment Information
A total of 1,212 imaging kits and 1,161 therapy kits were shipped between March 27, 2002, and March 31, 2003, and 20 of the imaging kits were reported not to have been used. The estimated number of paired kits that were shipped and for which an isotope shipment could be reconciled was 1,106. The registry found 38 cases in which a patient was imaged with 111In-ibritumomab tiuxetan but was not treated with 90Y-ibritumomab tiuxetan. From these figures, the maximum number of patients who may have initiated therapy was 1,192 (1,212 imaging kits shipped minus the 20 kits that were reported not to have been used). The minimum number of patients who began therapy was 1,144 (1,106 paired kits shipped with isotopes plus the 38 cases with no-treatment decisions).
Registry Participation and Rate of No-Treatment Decisions
Approximately 340 nuclear medicine specialists participated in the registry and provided information on 953 patients. The case capture rate with an estimated 1,144–1,192 patients was 80%–83%. No-treatment decisions were made based on imaging decisions in 16 cases and on medical reasons in 22 cases. The rate of no-treatment decisions was 4.0% (Table 2).
Patients Imaged with 111In-Ibritumomab Tiuxetan But Not Treated with 90Y-Ibritumomab Tiuxetan
No-Treatment Decisions Due to Imaging Reasons
Physicians decided not to treat with 90Y-ibritumomab tiuxetan in 16 cases (15 patients) because of images that were interpreted as indicating altered biodistribution. All cases of altered biodistribution were detected on scan 1, obtained at 2–24 h after the administration of 111In-ibritumomab tiuxetan. Twelve of these 16 cases met the criteria for altered biodistribution according to the prescribing information: 6 (0.6%) of them appeared to result from the use of a procedure for radiolabeling 111In-ibritumomab tiuxetan that deviated from that in the prescribing information and 6 cases were suspected to be true altered biodistribution (Table 3).
Imaging Reasons for Not Treating Patients with 90Y-Ibritumomab Tiuxetan
Altered Biodistribution Due to a Radiolabeling Procedure that Was Inconsistent with the Prescribing Information.
Six patients (7 cases) had an indium scan pattern consistent with altered biodistribution that involved increased renal uptake. Upon investigation, it was found that treatment with 90Y-ibritumomab tiuxetan in at least 6 of the 7 cases of renal uptake would not have resulted in greater risk to the patient, because the altered biodistribution was due to a specific 111In-ibritumomab tiuxetan radiolabeling issue. This conclusion is substantiated by the following observations. First, 6 cases involved a single nuclear pharmacy chain that used an alternative radiolabeling method for 111In-ibritumomab tiuxetan. This alternative radiolabeling procedure consisted of a change in the amount of formulation buffer added after the radiolabeling reaction incubation. In addition, in some of these 6 cases, a glass syringe rather than a plastic syringe was used during the radiolabeling procedure or there was a failure to use a cool pack in the transportation of 111In-ibritumomab tiuxetan. Second, 3 of the patients who had abnormal renal uptake were subsequently given 111In-ibritumomab tiuxetan prepared at a different nuclear pharmacy. They then showed expected biodistribution and completed 90Y-ibritumomab tiuxetan treatment without reported adverse events. Finally, no further cases of altered biodistribution with a renal uptake pattern were captured in the registry after the implicated nuclear pharmacy chain began following the U.S. prescribing information method for radiolabeling 111In-ibritumomab tiuxetan. In the 1 additional case of renal uptake that did not involve the nuclear pharmacy chain, renal pathology could not be ruled out, because of a lack of clinical information from the site. However, the images collected in this case were similar to those observed in the other cases of altered renal biodistribution, and it is thought to be likely that this case was also the result of an isolated 111In-ibritumomab tiuxetan radiolabeling issue.
True Altered Biodistribution.
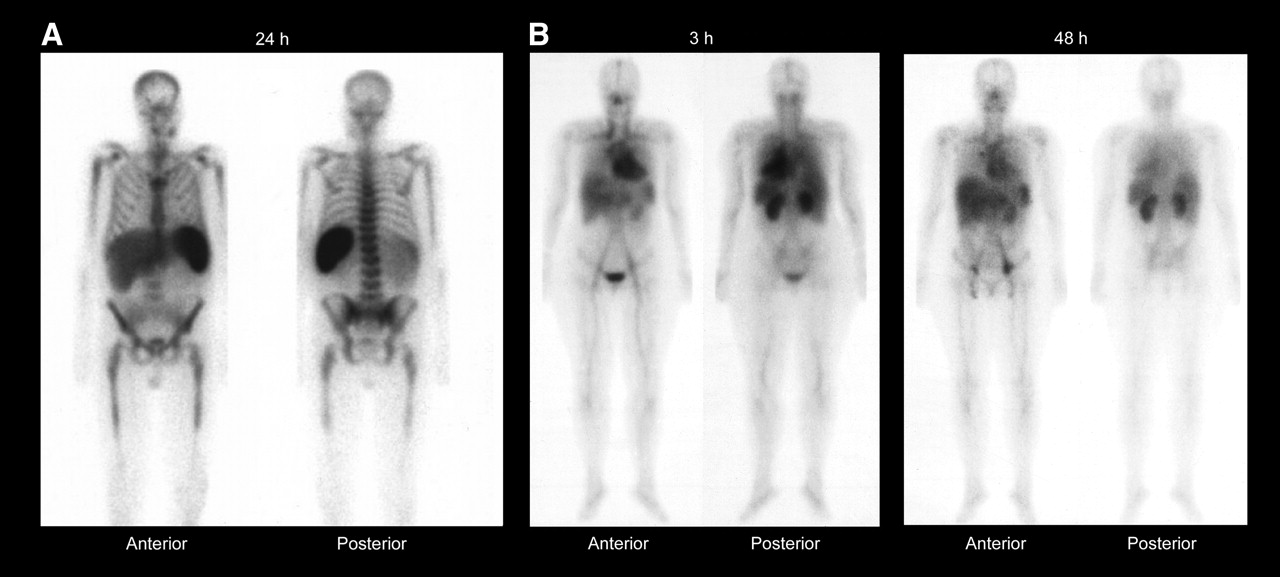
True altered biodistribution was suspected in only 6 cases (0.6%): 4 cases of prominent bone marrow uptake, 1 case of pneumonia that was seen on chest x-ray before imaging, and 1 case of increased renal uptake in which there was insufficient information to rule out a disease-related cause (described above). Images of a case of prominent bone marrow uptake and the case of increased renal uptake are shown in Figure 2. The images in the cases of prominent bone marrow uptakes were characterized by uptake in the proximal humeri, sternum, ribs, vertebral bodies, iliac crest, and proximal femurs. Two patients also had uptake in the distal long bones. The disease characteristics and treatment history varied considerably among these patients, making it difficult to determine the cause of the prominent bone marrow uptake (Table 4), and it was likely tumor related. Two patients with prominent bone marrow uptake also had prominent splenic uptake; because no splenic uptake was seen in the 2 other cases, it is likely that these patients had had splenectomies. Lymphomatous bone marrow involvement was diagnosed in 2 patients—20% involvement in 1 and not specified in the other (Table 4). No information on bone marrow involvement was available on the third patient. The documentation of bone marrow involvement in this setting is likely less rigorous than that in the clinical trial setting (bilateral biopsies required in the clinical trials) and may explain why bone marrow uptake was seen on imaging without known involvement by biopsy. Because dosimetry was not performed, it is not possible to determine the estimated radiation-absorbed doses to the red marrow and whether they may have exceeded 300 cGy had 90Y-ibritumomab tiuxetan been administered.

Whole-body γ-camera images of representative patients obtained at 24 h after administration of 111In-ibritumomab tiuxetan (A), showing prominent bone marrow and splenic uptake, and at 3 and 48 h after administration of 111In-ibritumomab tiuxetan (B), showing greater than expected renal uptake. (Reprinted with permission of Biogen Idec Inc.)
Characteristics of Patients with Prominent Bone Marrow Uptake
The final case of suspected true altered biodistribution was in a patient with a left lower lobe infiltrate that was evident on chest x-ray before the delivery of 111In-ibritumomab tiuxetan. The first posterior images showed greater uptake in the left lower lobe than in the liver or spleen. There was, however, no evidence of tumor in the left lower lobe on CT or 18F-FDG PET. Because the patient had clinical and radiographic evidence of active pneumonia, the altered biodistribution was likely related to the infection.
Other Cases of Altered Biodistribution.
The remaining 4 cases of suspected altered biodistribution were due to misinterpretation of the images or criteria for altered biodistribution. Evaluation of the images in 3 of these cases showed that they did not meet the criteria for altered biodistribution as described in the prescribing information. The decisions not to treat were incorrectly based on the appearance of 111In-ibritumomab tiuxetan in the bowel or the lack of tumor uptake.
The images in the final case showed a small pleural effusion that led to the decision not to treat. This patient’s images were not provided to the registry, and it is not clear from the clinical information provided whether the criteria for altered biodistribution were met. It is not likely, however, that treatment in this patient would have resulted in a greater risk of toxicity because the adverse events were not greater in patients with small pleural effusions treated in the clinical trials.
No-Treatment Decisions Due to Medical Reasons.
The decisions not to treat were for medical reasons in 22 cases (2.3%). These cases can be divided into 3 categories: contraindications to ibritumomab tiuxetan that were found after imaging (n = 2), expected biodistribution on imaging but a rapid change in clinical condition that precluded treatment (n = 19), and physician decision not to treat outside the labeled indication (n = 1). The medical reasons for not proceeding with treatment with 90Y-ibritumomab tiuxetan are given in Table 5.
Patients Not Treated with 90Y-Ibritumomab Tiuxetan for Medical Reasons
DISCUSSION
The Zevalin Imaging Registry captured data on 953 of an estimated 1,144–1,192 patients who began ibritumomab tiuxetan therapy between March 27, 2002, and March 31, 2003. Decisions not to treat were made in 38 cases (4.0%), for imaging reasons in 16 cases and for medical reasons in 22 cases. All cases of altered biodistribution were seen on the first images, obtained at 2–24 h after the administration of 111In-ibritumomab tiuxetan. Altered biodistribution was not reported in any of the 22 patients in whom 90Y-ibritumomab tiuxetan was withheld for medical reasons. There were only 6 cases of suspected true altered biodistribution (0.6%). These were 4 cases of prominent bone marrow uptake, 1 case of pneumonia that was seen on chest x-ray before imaging, and 1 case of increased renal uptake in which there was insufficient information to rule out a disease cause. Only the 4 cases of bone marrow uptake appear to be true altered biodistribution that could not be predicted from the clinical status of the patient. Information on the extent of bone marrow involvement in 1 of these patients was not available, however, and it is not known whether it exceeded that in the patients in the clinical trials (only patients with <25% marrow lymphoma were eligible). Consequently, bone marrow aspirate and biopsy could have excluded this patient from treatment with the ibritumomab tiuxetan therapeutic regimen. The single case of altered biodistribution detected in clinical trials occurred in a patient with hydronephrosis. Although this patient had adequate renal function (i.e., serum creatinine ≤2.0 mg/dL) at baseline, imaging scans showed more intense uptake of 111In-ibritumomab tiuxetan in the kidneys than in the liver. As a result, the patient did not proceed to the therapeutic dose.
In rare instances, the imaging component of ibritumomab tiuxetan therapy may have provided an added safety measure. The analysis of the imaging scans resulted in the detection of a 111In-ibritumomab tiuxetan radiolabeling procedure that deviated from that in the U.S. prescribing information and led to opportunities for educating physicians on interpreting the images correctly. The images also showed 4 cases of altered biodistribution that involved prominent bone marrow uptake and was possibly disease related. In the absence of a surrogate marker or a clear assessment of bone marrow involvement that would identify candidates for therapy with ibritumomab tiuxetan, the imaging component can help prevent the delivery of excessive radiation to the bone marrow. Because patients with suspected disease-related altered biodistribution did not proceed to treatment with 90Y-ibritumomab tiuxetan, however, it is not known whether such treatment would have resulted in adverse events. It is possible that effective therapy was withheld from these patients.
CONCLUSION
These findings confirm that true altered biodistribution is rare (<0.6% of cases) in patients who are treated with the ibritumomab tiuxetan therapeutic regimen in the commercial setting. It is notable that all cases of altered biodistribution documented in this registry and that the 1 case in the clinical trials was seen on the first images. Because of these findings, the current U.S. imaging requirement for therapy with ibritumomab tiuxetan could be changed to a single scan at 2–24 h after the 111In-ibritumomab tiuxetan, with optional scans at 48–72 h and at 90–120 h as necessary to resolve ambiguities. 90Y-Ibritumomab tiuxetan is routinely administered in many European countries without biodistribution studies. The European clinical trials in which the regimen was used without 111In-ibritumomab tiuxetan and imaging have not reported any new safety signals (18,19). Simplifying the imaging requirement would reduce the costs and complexity of treatment with ibritumomab tiuxetan in the United States, thereby making this important therapeutic modality available to more patients with B-cell NHL.
ADDENDUM
At the time of manuscript submission, the prescribing information for the ibritumomab tiuxetan therapeutic regimen required that 2 whole-body γ-scans be performed to confirm the biodistribution of 111In-ibritumomab tiuxetan. In August 2005, however, the Food and Drug Administration approved a revision to the prescribing information, reducing the required number of scans. This revision indicates that only 1 whole-body γ-scan is now required and should be administered 48–72 h after the imaging dose of 111In-ibritumomab tiuxetan. Optional subsequent scans may be performed to resolve ambiguities.
Acknowledgments
The authors thank Chao-Hsu John Liu, Jeffrey Feinstein, Thomas Ryskamp, Gene Menendez, V. Douglas Harrough, and Dan Matso for their assistance in preparing this article. Disclosure of financial support: Dr. Conti is a consultant to Biogen Idec Inc., Siemens, and Molecular Imaging Corp., and he has received honoraria from Biogen Idec Inc. and Schering AG. Dr. White, Dr. Pieslor, Dr. Molina, Ms. Aussie, and Dr. Foster are employees and stockholders of Biogen Idec Inc.
Footnotes
Received May 4, 2005; revision accepted Aug. 8, 2005.
For correspondence or reprints contact: Peter S. Conti, MD, PhD, Department of Radiology, University of Southern California, 1510 San Pablo St., Suite 350, Los Angeles, CA 90033.
E-mail: pconti{at}usc.edu
REFERENCES
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Observational Retrospective Study of Altered Biodistribution of Tositumomab and 131I-Tositumomab
- The Umbilical Cord: An Ally in Targeted Imaging Research?
- A re-examination of radioimmunotherapy in the treatment of non-Hodgkin lymphoma: prospects for dual-targeted antibody/radioantibody therapy
- MIRD Dose Estimate Report No. 20: Radiation Absorbed-Dose Estimates for 111In- and 90Y-Ibritumomab Tiuxetan
- 90Y-Ibritumomab Therapy in Refractory Non-Hodgkin's Lymphoma: Observations from 111In-Ibritumomab Pretreatment Imaging