


Abstract
Our goal was to design and manufacture a kit under good manufacturing practices (GMP) for the preparation of 111In-DTPA-hEGF Injection, a novel targeted radiotherapeutic agent for advanced epidermal growth factor receptor (EGFR)-positive breast cancer. Methods: Human EGF (hEGF) was derivatized with diethylenetriaminepentaacetic acid (DTPA) and then purified by size-exclusion chromatography and ultrafiltration. Kits were prepared by dispensing 0.25 mg (1 mL) of DTPA-hEGF in 1 mol/L sodium acetate buffer [pH 6.0] into single-dose glass vials. Raw materials were pharmacopoieal or reagent grade according to the American Chemical Society and were tested for identity and purity. Kits were tested for protein concentration, purity and homogeneity (sodium dodecyl sulfate polyacrylamide gel electrophoresis and size-exclusion high-performance liquid chromatography), pH, clarity and color, volume, DTPA substitution, labeling efficiency, receptor binding to MDA-MB-468 human breast cancer cells, and sterility and apyrogenicity. 111In-DTPA-hEGF Injection was tested for pH, radionuclidic and radiochemical purity, clarity and color, and sterility and apyrogenicity. Results: Four lots of kits and 8 lots of 111In-DTPA-hEGF Injection passed all quality specifications. The labeling efficiency was 94%–99% with 115–773 MBq 111In chloride added to a single kit. 111In-DTPA-hEGF exhibited preserved receptor binding against MDA-MB-468 cells (affinity constant [Ka], 0.9–1.1 × 107 L/mol; maximum number of binding sites per cell [Bmax], 1.1–2.2 × 106 sites per cell). In addition, labeling of aliquots of the kit suggested that a single vial could be labeled with up to 3,083 MBq 111In while maintaining a radiochemical purity of >90%. Kits were stable for >90 d and 111In-DTPA-hEGF Injection was stable for >24 h stored at 4°C. Conclusion: The kit formulation is suitable for preparing 111In-DTPA-hEGF Injection for a phase I clinical trial in patients with advanced EGFR-positive breast cancer. Establishment of the GMP processes for 111In-DTPA-hEGF Injection provides a useful example of manufacturing biotechnology-based investigational radiopharmaceuticals in an academic environment for early phase I clinical trials.
Radiopharmaceuticals labeled with low-energy Auger electron-emitting radionuclides (e.g., 111In or 125I) are receiving considerable interest as targeted radiotherapeutic agents for cancer (1). Theoretically, the micrometer range of the electrons should restrict their radiotoxicity mainly toward cells that internalize the radiopharmaceuticals into the cytoplasm and especially in cases in which they are translocated to the cell nucleus (2,3). Our laboratory has discovered a novel targeted Auger electron-emitting radiopharmaceutical, 111In-labeled human epidermal growth factor (111In-DTPA-hEGF) (where DTPA is diethylenetriaminepentaacetic acid) (4), which exploits the overexpression of epidermal growth factor receptors (EGFRs) present on almost all estrogen receptor-negative, hormone-resistant, and poor-prognosis breast cancers (5).
111In-DTPA-hEGF was rapidly internalized into the cytoplasm and translocated to the nucleus of EGFR-positive human breast cancer cells, where the Auger electron emissions were highly damaging to DNA, causing cell death (4). The radiopharmaceutical was highly cytotoxic to MDA-MB-468 human breast cancer cells overexpressing EGFR (1–2 × 106 receptors per cell) with >95% cell killing achieved at <111–148 mBq per cell (4). Furthermore, the radiopharmaceutical was 85–300 times more potent at inhibiting the growth of MDA-MB-468 cells in vitro than the chemotherapeutic agents methotrexate, doxorubicin, and paclitaxel and several orders of magnitude more effective than 5-fluorouracil (6). Administration of 5 weekly doses (18.5 MBq; 3 μg) of 111In-DTPA-hEGF to athymic mice caused growth arrest of established subcutaneous MDA-MB-468 xenografts with minimal normal tissue toxicity (modest decrease in leukocyte and platelet counts) (7). Early treatment of mice bearing smaller “nonestablished” MDA-MB-468 xenografts with 111In-DTPA-hEGF achieved tumor regression.
To translate 111In-DTPA-hEGF from preclinical investigation to a phase I clinical trial, it is necessary to create a pharmaceutical quality formulation manufactured under current Good Manufacturing Practices (GMP) and obtain regulatory approval from Health Canada in the form of a Clinical Trial Application (CTA). GMP are comprehensive quality processes that ensure the suitability of the product for its intended use in humans (8). Meeting GMP requirements is one of the major challenges facing radiopharmaceutical scientists who conduct translational research and work at a university or hospital setting with limited resources. In this study, we describe our approach to manufacturing a kit for the preparation of 111In-DTPA-hEGF Injection under GMP in the clinical radiopharmaceutical research laboratory at the University Health Network, a University of Toronto-affiliated hospital. We propose that the strategy for establishing GMP for 111In-DTPA-hEGF Injection provides a useful example of manufacturing biotechnology-based investigational radiopharmaceuticals in an academic environment for early phase I clinical trials.
MATERIALS AND METHODS
Raw Materials
hEGF (>98%) was obtained as hEGF1–53 from Upstate Biotechnology Inc. or as hEGF1–51 from Viral Therapeutics Inc. DTPA dianhydride (>98%) and chloroform (reagent grade, >99.9% meeting specifications of the American Chemical Society) (9) were purchased from Sigma-Aldrich Canada Ltd. Sodium acetate dihydrate USP and sodium bicarbonate USP were obtained from EM Science. Sterile Water for Injection USP and Sodium Chloride Injection USP were obtained from BaxterTravenol Inc. Nitrogen NF was obtained from Praxair Canada, Inc. All other chemicals and reagents were purchased in analytic grade with a minimum purity of >95%. Sterile, apyrogenic type 1 glass multidose vials (10 mL) with a gray butyl rubber septum and aluminum seal were obtained from Hollister-Stier Laboratories Inc. 111In chloride (>3.7 GBq/mL; <0.1% 114mIn and 65Zn) was purchased in radiochemical quality from MDS Nordion Inc. or PerkinElmer Life Sciences Inc.
Identity Testing and Purity Assessment of Raw Materials
Certificates of analysis were obtained from the vendor for each lot of raw materials. Identity testing of sodium bicarbonate USP and sodium acetate USP was performed by pharmacopoeial methods (10,11). The purity of nonpharmacopoeial materials was confirmed by in-house analytic techniques. Proton (1H) NMR (500 MHz) spectroscopy was used to confirm the identity of chloroform (neat) and DTPA dianhydride (dissolved in D2O). The purity of DTPA dianhydride was measured by adapting the assay for Edetic Acid NF (12) using a 10.0 mg/mL solution of the raw material to titrate a known amount of chelometric standard calcium carbonate (100.00%; Fisher Scientific Ltd.). The identity and radionuclidic purity of 111In chloride was confirmed by γ-spectroscopy on a Captus model 2000 multichannel analyzer (Capintec, Inc.) checked using radionuclide disk reference sources (133Ba, 22Na, 137Cs, and 60Co) and with a certified primary reference standard for 111In (National Institute of Standards and Technology).
Characterization and Purity Evaluation of hEGF
Amino acid analysis, ultraviolet (UV) spectroscopy, and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)/Western blot were used to characterize hEGF, and size-exclusion high-performance liquid chromatography (HPLC) was used to measure its purity and homogeneity. UV spectroscopy was performed for hEGF1−53 (0.25 mg/mL in 50 mmol/L sodium bicarbonate buffer, pH 7.5). SDS-PAGE was conducted on a 4%–20% Tris HCl gradient minigel (Bio-Rad Laboratories, Inc.) stained with Coomassie R-250 brilliant blue. Western blot was performed by transferring electrophoresed proteins onto a nitrocellulose membrane (TransBlot; Bio-Rad Laboratories) and probing with a rabbit polyclonal anti-hEGF antibody (provided by Dr. Jean Gariépy, Ontario Cancer Institute). Reactive bands were detected with a goat antirabbit IgG-horseradish peroxidase conjugate (Sigma-Aldrich Canada Ltd.) and diamidobenzidine/0.03% H2O2. Size-exclusion HPLC was performed on a BioSep SEC-S2000 column (Phenomenex Inc.) eluted with 100 mmol/L NaH2PO4 buffer (pH 7.0) at a flow rate of 1.0 mL/min using a Beckman System Gold model 125 HPLC interfaced with a model 166 UV detector (Beckman Coulter) set at 280 nm.
Pharmaceutical Buffers
Sterile, nonpyrogenic 50 mmol/L sodium bicarbonate (pH 7.5) in Sodium Chloride Injection USP and 1 mol/L sodium acetate buffer (pH 6.0) (in Sterile Water for Injection USP) buffers were prepared from pharmacopoeial-quality raw materials. Trace metals were stripped from the buffers by passage through a cation-exchange column consisting of a 60-mL sterile syringe plugged with glass wool and filled with 30 mL of Chelex-100 resin (Bio-Rad Laboratories) prehydrated overnight in Sterile Water for Injection USP. After removal of trace metals, the pH was readjusted to the desired value using sterile 1N HCl and the buffers were sterilized by filtration through a 0.22-μm pore size Millex-GV filter (Millipore Corp.). Quality control testing included USP Sterility and Pyrogen Tests as well as an assay for the concentration of sodium acetate or sodium bicarbonate by USP methods (10,11). The assay for sodium acetate consisted of titration of the buffer with standardized 0.1N perchloric acid (Fisher Scientific Ltd.). The assay for sodium bicarbonate consisted of titration with standardized 0.1N sulfuric acid (Fisher Scientific Ltd.). The stability of the buffers stored at 4°C was determined by reassaying the concentration of sodium acetate or sodium bicarbonate up to 11 mo after preparation.
Radiopharmaceutical Kits
A kit for the preparation of 111In-DTPA-hEGF Injection was constructed by derivatizing hEGF with a 20-fold molar excess of DTPA dianhydride. Briefly, DTPA dianhydride (50 mg) was suspended in 5.0 mL of chloroform in a sterilized 10-mL glass scintillation vial, and a 600-μL aliquot (17 μmol) was dispensed into a sterilized 10-mL glass Reacti-Vial (Pierce Biotechnology, Inc.). Additional chloroform was added to a final volume of 1.0 mL; then the chloroform was evaporated to dryness using a gentle stream of nitrogen NF. Approximately 1.0 mL (5 mg; 0.83 μmol) of hEGF in 50 mmol/L sodium bicarbonate buffer (pH 7.5) was added and the vial was vortexed for 1 min. The reaction mixture was allowed to incubate at room temperature for a further 30 min. A 10-μL aliquot of the reaction mixture was removed for measurement of DTPA conjugation efficiency while the remainder was transferred to the top of a 1 × 20 cm P-2 size-exclusion chromatography column (exclusion limit, 1.8 kDa; Bio-Rad). The column was eluted with 20 × 0.5-mL aliquots of 50 mmol/L sodium bicarbonate buffer (pH 7.5), and the fractions were collected in sterile, polystyrene tubes (VWR International). The absorbance of each fraction was measured at 280 nm. The fractions containing the partially purified DTPA-hEGF (usually fractions 5–12) were combined. The pooled fractions were transferred in 2 equal portions to Centricon YM-3 ultrafiltration devices (molecular weight [Mr] cutoff = 3 kDa; Millipore Corp.), and the solution in each device was diluted to 2.0 mL with 1 mol/L sodium acetate buffer (pH 6.0). The Centricon YM-3 devices were centrifuged at 4,500 rpm (2,875g) for 45 min in a fixed-angle centrifuge (model Centra-4B; IEC). The solutions were rediluted to 2.0 mL with 1 mol/L sodium acetate buffer (pH 6.0) and the devices were recentrifuged. A total of 8 dilution and ultrafiltration steps were performed. Finally, the pure DTPA-hEGF solutions were recovered in 0.5-mL volume and combined. The concentration of DTPA-hEGF was assayed spectrophotometrically at 280 nm by reference to a calibration curve created using hEGF1–53 standards (0–0.5 mg/mL). DTPA-hEGF was diluted to a final concentration of 0.25 mg/mL with 1 mol/L sodium acetate buffer (pH 6.0) and sterilized by filtration through a 0.22-μm Millex-GV filter. Unit-dose radiopharmaceutical kits were prepared by aseptically dispensing 1.0-mL (0.25 mg) aliquots into sterile, apyrogenic 10-mL glass unit-dose vials using a 1-mL sterile syringe and needle in a laminar flow hood.
Quality Control Testing of Kits
The pharmaceutical quality of the kits was evaluated by determining the protein concentration, protein homogeneity and polymerization, pH, clarity and color, volume contained in each vial, DTPA substitution level, labeling efficiency with 111In, receptor-binding properties, and sterility and apyrogenicity. The concentration of hEGF was measured spectrophotometrically at 280 nm. Protein homogeneity and polymerization were evaluated by SDS-PAGE and size-exclusion HPLC. The pH was measured using narrow-range pH paper (range, 4.5–7.5 in 0.5-unit increments; Fisher Scientific Ltd.). Clarity and color were evaluated by inspection against a light or dark background under bright light. The volume of solution contained in each vial was determined by weighing the vials before and after filling, assuming a density of 1 g/mL at 20°C. DTPA conjugation efficiency was determined by trace labeling a 10-μL aliquot (50 μg) of the unpurified reaction mixture with 1 MBq 111In and determining the proportion of 111In-DTPA-hEGF and 111In-DTPA by instant thin-layer silica gel chromatography (ITLC-SG; Pall Corporation) developed in 100 mmol/L sodium citrate (pH 5.0). Rf values for 111In-DTPA-hEGF and 111In-DTPA in this system were 0.0 and 1.0, respectively. The DTPA substitution level was calculated by multiplying the conjugation efficiency by the molar ratio of DTPA dianhydride to hEGF used in the reaction (i.e., 20:1).
The labeling efficiency of the kits was determined by adding 185 MBq 111In chloride to a single vial, incubating for 30 min, and determining the percentage of 111In-DTPA-hEGF by ITLC-SG. The labeling efficiency of the kits using 111In chloride from 2 different suppliers (MDS Nordion Inc. and PerkinElmer Life Sciences Inc.) was compared. The maximum amount of radioactivity that could be added to the kits while maintaining a radiochemical purity of >90% was studied by labeling 25 μL of kit solution containing 6 μg DTPA-hEGF with increasing amounts of 111In chloride (1.1–74 MBq) corresponding to the addition of 46–3,083 MBq to a single vial. The stability of the kits stored at 4°C was evaluated by retesting against all specifications (except sterility and apyrogenicity) at up to 90 d after manufacture.
Measurement of Receptor-Binding Properties
The equivalence of hEGF1–51 and hEGF1−53 raw materials was evaluated by comparing their ability to displace the binding of 123I-hEGF1–53 to MDA-MB-468 human breast cancer cells (1–2 × 106 EGFRs per cell; American Type Culture Collection). 123I-hEGF was prepared as previously described (13). Briefly, 123I-hEGF1–53 (3 ng; 3.7 mBq) was incubated for 30 min at 37°C with 1 × 106 MDA-MB-468 cells in the presence of increasing concentrations (1 nmol/L to 10 μmol/L) of hEGF1–51 or hEGF1–53 in 150 mmol/L sodium chloride. The tubes were centrifuged and the cell pellet was separated and measured in a γ-counter. The receptor-binding curve was obtained by plotting the radioactivity bound to the cells versus the concentration of competitor (hEGF1–51 or hEGF1–53). The dissociation constant (Kd) values were estimated by fitting the curve to a 1-site competition receptor-binding model using GraphPad Prism 3.0 software (GraphPad Software, Inc.). The receptor-binding properties of 111In-DTPA-hEGF Injection were evaluated in a direct receptor-binding assay using MDA-MB-468 cells as previously reported (13). The affinity constant (Ka) and maximum number of binding sites per cell (Bmax) were estimated by fitting the curve to a 1-site direct receptor-binding model using GraphPad Prism 3.0 software.
Final Radiopharmaceutical
111In-DTPA-hEGF Injection was prepared by aseptically removing the cap from a single unit-dose vial of the kit in a laminar flow hood and adding 115–960 MBq (5–20 μL) 111In chloride directly into the vial using an Eppendorf micropipette and sterile pipette tip. After an incubation period of at least 30 min, the radiopharmaceutical was diluted to 3.0 mL with Sodium Chloride Injection USP. The radiopharmaceutical was drawn up in a lead-shielded syringe and sterilized by filtration through a 0.22-μm Millex-GV filter into a 10-mL sterile, nonpyrogenic glass vial. Quality control of 111In-DTPA-hEGF Injection included measurement of total radioactivity, pH, radiochemical purity, clarity and color, and sterility and apyrogenicity. Total radioactivity was measured in a radioiosotope calibrator (Capintec model CRC-12). Radiochemical purity, pH, clarity, and color were determined as described previously. Radionuclidic purity was determined on the 111In chloride raw material. Sterility and apyrogenicity were assessed retrospectively by USP Sterility and Pyrogen Tests after allowing 30 d for radionuclide decay. The stability of 111In-DTPA-hEGF stored at 4°C was evaluated by measuring the radiochemical purity up to 24 h after preparation.
RESULTS
Raw Materials
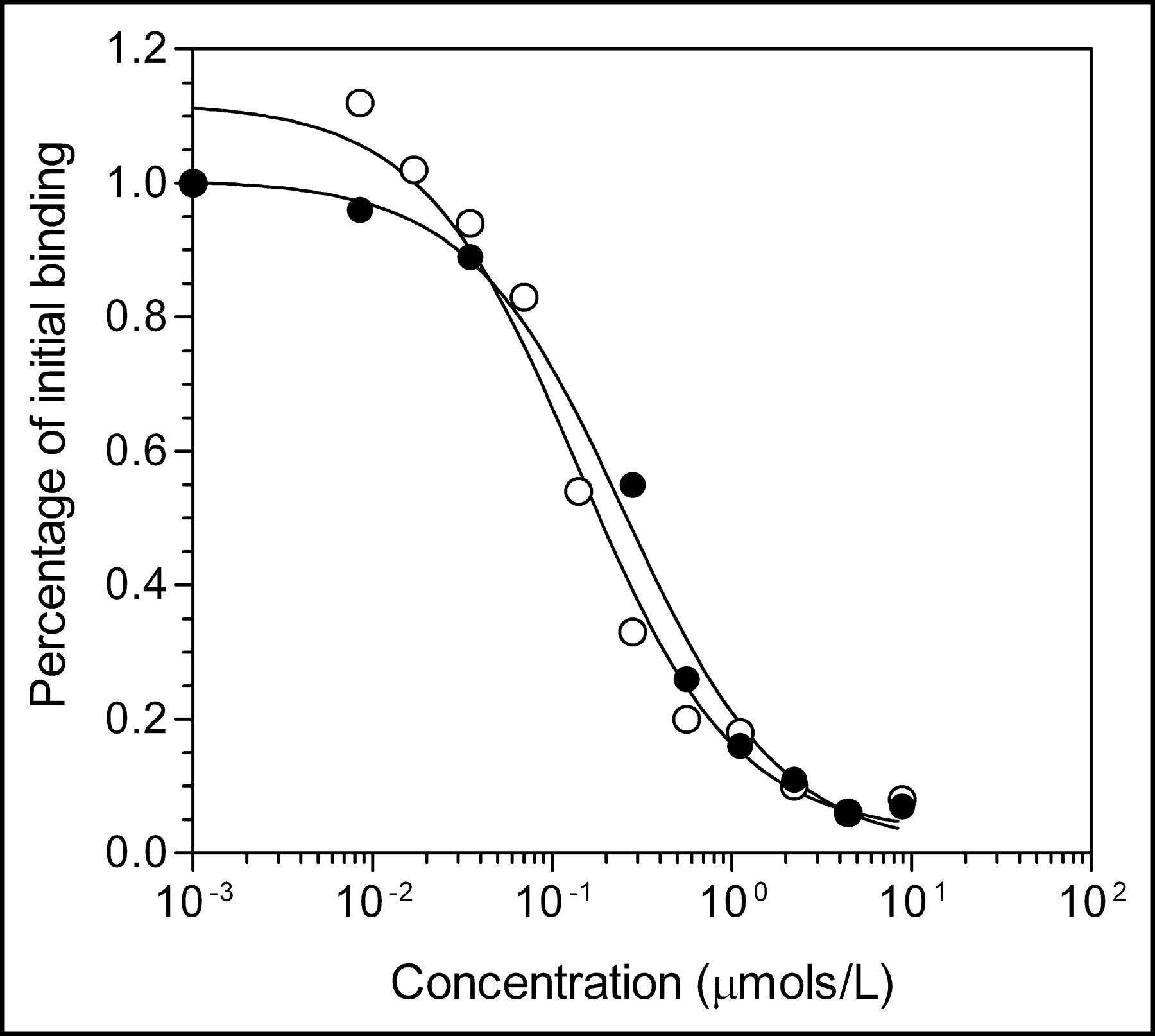
All raw materials passed tests for identity and met specifications for purity (>95%). The amino acid analyses of hEGF1−53 and hEGF1−51 were consistent with their known composition. The C-terminal residues Leu-52 and Arg-53 in hEGF1−53 are not present in hEGF1−51 nor are they required for receptor binding (14,15). The identity of hEGF was further confirmed by the absence of threonine or phenylalanine in the peptide (16). The UV spectrum of hEGF1−53 (not shown) exhibited λmax values of 220 nm (ε = 92,700 mol/L−1) and 280 nm (ε = 18,500 mol/L−1). SDS-PAGE analysis of hEGF1−53 or hEGF1−51 showed 1 major band corresponding to a protein with the expected Mr of 6 kDa (Fig. 1A) and a minor closely migrating band corresponding to a protein with slightly lower Mr. The major band was positive on Western blot when probed with a rabbit polyclonal anti-hEGF antibody (Fig. 1B). Size-exclusion HPLC of hEGF1−53 or hEGF1−51 (not shown) demonstrated 1 major peak with a retention time (tR) of 11.5 min. There were no major peaks in the HPLC analysis of hEGF associated with impurities indicating a purity of >95%. There was no difference in the ability of hEGF1−53 or hEGF1−51 to compete with 123I-hEGF1−53 for binding to MDA-MB-468 breast cancer cells (Fig. 2). Proton (1H) NMR (500 MHz) spectra of DTPA dianhydride and chloroform (not shown) were consistent with their chemical structures. The purity of DTPA dianhydride (102.9%) was within specifications (95%–105%). There were no detectable 114mIn or 65Zn radionuclidic impurities in 111In chloride. The expiry of all raw materials (except 111In chloride) was set arbitrarily at 2 y from receipt.

(A) SDS-PAGE analysis of hEGF raw materials on 4%–20% Tris HCl gradient minigel stained with Coomassie R-250 brilliant blue. MW = broad-range molecular weight markers; UB = hEGF1−53 (Upstate Biotechnology Inc.; 2 μg); VTI = hEGF1–51 (Viral Therapeutics Inc.; 2 μg). (B) Corresponding Western blot using polyclonal rabbit anti-hEGF antibody.
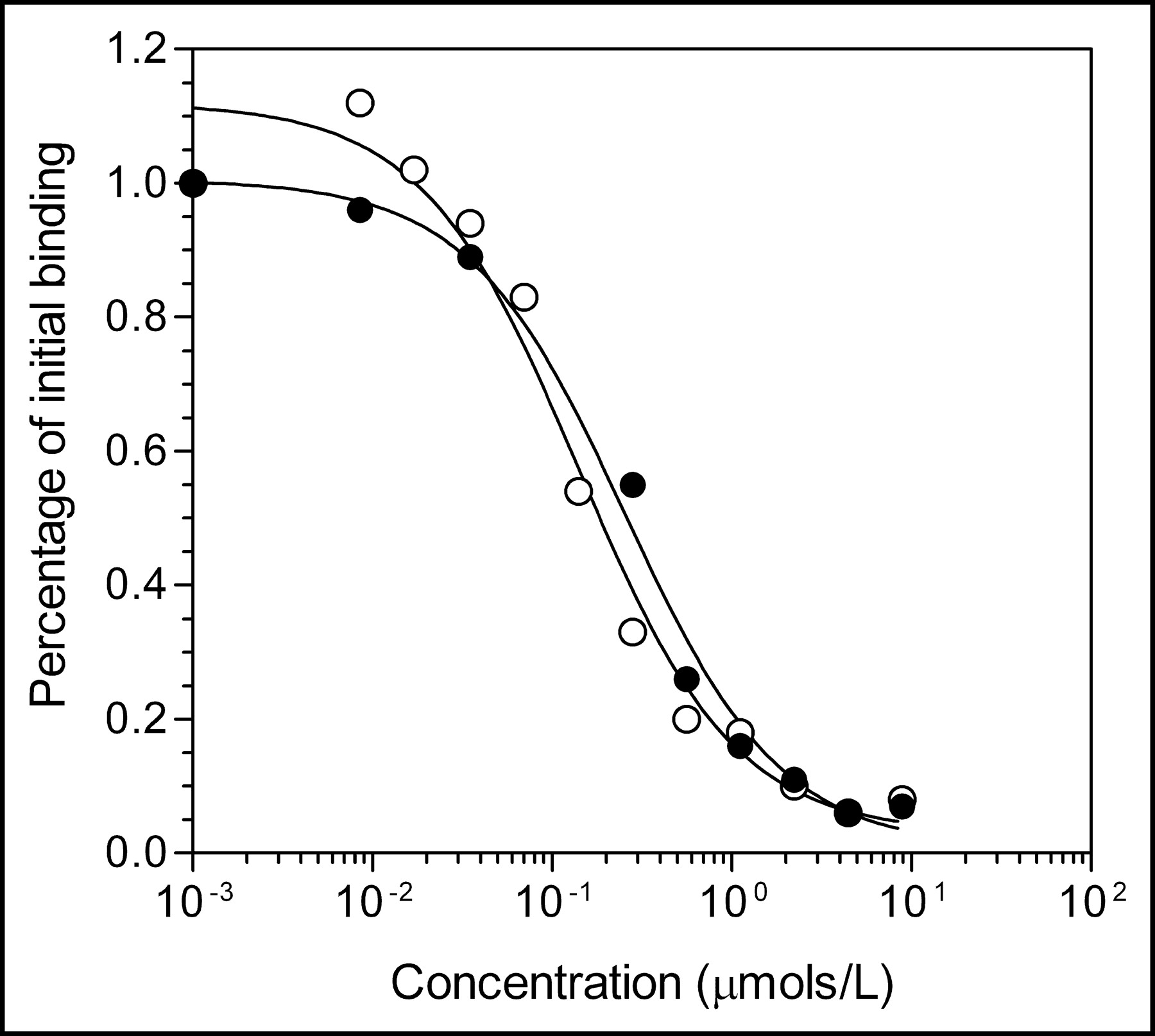
Competition receptor-binding assay curve for displacement of binding of 123I-hEGF1−53 to MDA-MB-468 human breast cancer cells by hEGF1−53 (○) or hEGF1−51 (•) raw materials. Each point represents a single determination. Kd values for hEGF1−53 and hEGF1−51 were 1.4 × 10−7 and 2.5 × 10−7 mol/L, respectively.
Pharmaceutical Buffers
Four lots of 50 mmol/L sodium bicarbonate buffer (pH 7.5) and 1 mol/L sodium acetate buffer (pH 6.0) were prepared. Each lot met specifications for sodium bicarbonate or sodium acetate, pH, clarity, and color. The buffers were sterile and pyrogen free and were stable stored at 4°C. The concentration of sodium bicarbonate in 50 mmol/L sodium bicarbonate buffer (pH 7.5) remained within ±5% of the initial assay value up to 9 mo after manufacturing. The concentration of sodium acetate in 1 mol/L sodium acetate buffer (pH 6.0) similarly remained within ±5% of the initial assay value up to 11 mo after manufacturing. Based on the stability data, the expiry of the pharmaceutical buffers was set at 9 mo.
Radiopharmaceutical Kits
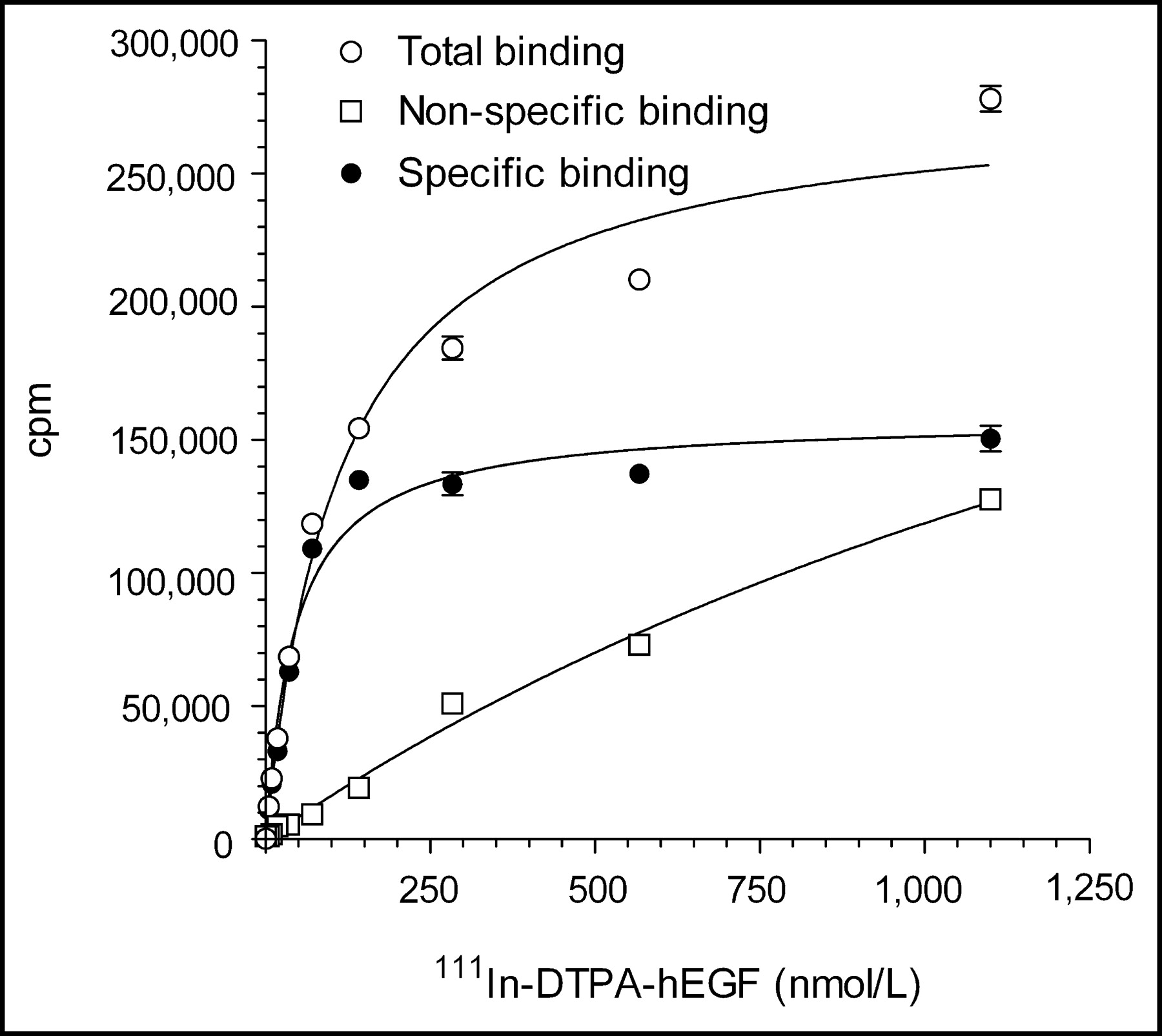
Three lots of kits for the preparation of 111In-DTPA-hEGF Injection were prepared with hEGF1−53 raw material (2F004, 2G004, and 2I002) and 1 lot (3B003) was prepared using hEGF1−51 (Table 1). Each lot of kits met specifications for protein concentration, pH, clarity and color, DTPA substitution level, purity and homogeneity, labeling efficiency with 111In, receptor binding, and sterility and apyrogenicity. SDS-PAGE analysis of DTPA-hEGF (not shown) revealed 1 major band corresponding to a protein with a Mr of 6 kDa and a second minor band corresponding to a protein with a Mr of 12 kDa, representing monomeric and dimeric DTPA-hEGF, respectively. Dimeric DTPA-hEGF is due to protein crosslinking through the DTPA moiety caused by the bifunctional nature of DTPA dianhydride. Size-exclusion HPLC (not shown) similarly demonstrated a major peak with a tR of 11.5 min representing monomeric DTPA-hEGF and a second minor peak (<5%) with a tR of 10.5 min representing dimeric DTPA-hEGF. The labeling of the kits with 111In was rapid, reproducible, and almost quantitative (94%–99%; Table 1). One lot of kits (2G004) labeled with 111In chloride (185 MBq) from 2 different suppliers (MDS Nordion Inc. and PerkinElmer Life Sciences Inc.) exhibited a labeling efficiency of 97.0% and 96.3%, respectively. The labeling efficiency for aliquots of the kit solution (25 μL; 6 μg DTPA-hEGF) incubated with 74 MBq 111In was 91.2% ± 0.3%. These results suggested that a single vial (1 mL; 250 μg DTPA-hEGF) could be labeled with up to 3,083 MBq 111In and remain within specifications for radiochemical purity (>90%). 111In-DTPA-hEGF Injection demonstrated specific and saturable binding to MDA-MB-468 breast cancer cells (Fig. 3). The mean Ka for 111In-DTPA-hEGF was 1.3 ± 0.6 × 107 L/mol and the Bmax was 1.6 ± 0.6 × 106 sites per cell. All kits retested for quality at 90 d continued to meet specifications. There was no significant decrease in labeling efficiency at 90 d compared with initial testing values (96.7% ± 1.3% vs. 97.1% ± 2.1%, respectively), and there was no change in the receptor-binding properties (mean Ka, 1.7 ± 0.6 × 107 L/mol; Bmax, 2.3 ± 0.2 × 106 sites per cell). The expiry of the kits was set at 90 d from the date of manufacture.
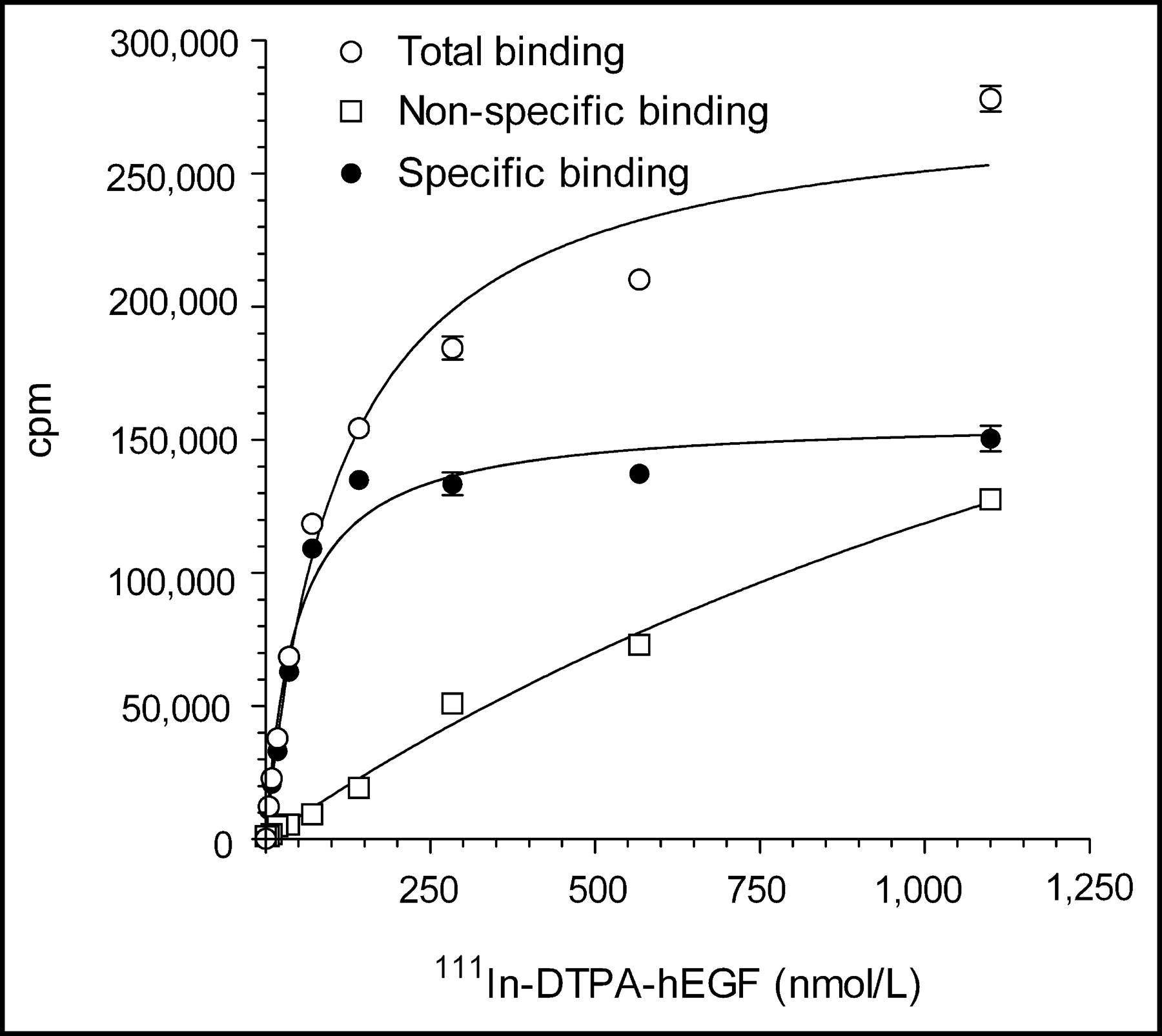
Direct receptor-binding assay curve for binding of 111In-DTPA-hEGF (prepared from kit lot 3B003) to MDA-MB-468 human breast cancer cells. Ka and Bmax values were 2.2 × 107 L/mol and 1.1 × 106 receptors per cell, respectively.
Quality Control Testing of Kits for Preparation of 111In-DTPA-hEGF Injection
Final Radiopharmaceutical
Eight lots of 111In-DTPA-hEGF Injection were prepared from the kits (Table 2). All radiopharmaceutical preparations met specifications for total radioactivity, pH, radiochemical purity, clarity and color, and sterility and apyrogenicity. 111In-DTPA-hEGF Injection was stable for 24 h stored at 4°C. The mean radiochemical purity at 24 h was 93.1% ± 4.2% (n = 3). The expiry of 111In-DTPA-hEGF Injection was set at 4 h from the time of preparation.
Quality Control Testing of 111In-DTPA-hEGF Injection
DISCUSSION
GMP are the foundation of a quality process that ensures that pharmaceuticals meet standards appropriate to their intended use. A central component of GMP is the establishment of specifications and standard operating procedures (SOPs) for pharmaceutical manufacturing extending from raw materials through intermediates to the final product. Guidelines for GMP have been standardized by the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) (8). Health Canada has adopted these guidelines in establishing the GMP regulations in Canada for pharmaceuticals (17) and modified them to include radiopharmaceuticals (18). In this study, we described our approach to designing GMP processes for manufacturing a kit for the preparation of 111In-DTPA-hEGF Injection, a novel targeted radiotherapeutic agent for advanced EGFR-positive breast cancer. Since the processes were designed and validated in the clinical radiopharmaceutical research laboratory at the University Health Network, a University of Toronto-affiliated teaching hospital, we propose that the approach represents a useful example of manufacturing an investigational biotechnology-based radiopharmaceutical in an academic setting under GMP for early phase I clinical trials in humans. The processes described are different than those of “good pharmacy practice” intended to compound radiopharmaceuticals for individual patients in a hospital setting. The unique issues in establishing GMP in a hospital setting include the low-batch sizes involved, local distribution usually only within a single institution, and the need to allocate limited resources to greatest effect. These issues were taken into consideration in designing the GMP for 111In-DTPA-hEGF Injection.
Health Canada GMP guidelines specify that all raw materials intended for pharmaceutical use be pharmacopeoeial or equivalent quality, that an identity test be performed, and that a certificate of lot analysis be obtained from the supplier confirming the purity (17). Pharmacopeoial-quality raw materials obtained for manufacturing the kits included sodium bicarbonate USP, sodium acetate USP, nitrogen NF, Sterile Water for Injection USP, and Sodium Chloride Injection USP. In addition, type 1 glass vials that met USP specifications for sterility and apyrogenicity were purchased to dispense the kits. Health Canada does not require in-house assays of materials labeled as pharmacopoieal quality (i.e., Sterile Water for Injection USP). Chloroform and DTPA dianhydride were not available in pharmacopoeial quality but were obtained in high purity (>98%). The NF assay for edetic acid was adapted to confirm the purity of DTPA dianhydride (12). Identity tests were performed on all raw materials (including those of pharmacopoeial quality) and certificates of lot analysis were obtained from the suppliers.
A major challenge in manufacturing the kits under GMP conditions was securing a source of suitable-quality hEGF. Preclinical studies of the radiopharmaceutical were conducted using hEGF1−53, a high-purity (>98%) but “research-quality” material produced in Saccharomyces cerivisiae. This material was used to establish the specifications and analytic methods for hEGF as well as to manufacture the first 3 pilot batches of the kits. Because hEGF1−53 was not recommended for human use and complete details on its production and quality control required by Health Canada were not available from the supplier, these circumstances necessitated a change in the source of hEGF raw material to hEGF1−51 obtained from an alternate supplier.
hEGF1−51 is a natural isoform of hEGF. The C-terminal amino acids Leu-52 and Arg-53 are not present in hEGF1−51 nor are they required for receptor binding (15). The hEGF1−51 material was produced in Pichia pastoris under ISO9001 standards (19) that are similar to GMP; therefore, the material was considered “pharmaceutical quality.” Complete manufacturing and quality control information was provided by the supplier. To demonstrate the receptor-binding equivalence of hEGF1−51 and hEGF1−53, the ability of the 2 materials to displace the binding of 123I-hEGF1−53 to MDA-MB-468 breast cancer cells was compared. In addition, hEGF1−51 was tested against specifications established for the identity and purity of hEGF. These tests showed that hEGF1−51 exhibited identical receptor-binding properties as hEGF1−53 and met or exceeded the specifications established for the raw material. Furthermore, we have recently determined that 111In-DTPA-hEGF prepared from hEGF1−51 exhibits identical cytotoxic properties in vitro against MDA-MB-468 cells as that prepared from hEGF1−53 (unpublished data, August 2003).
It was important to create a kit formulation for preparing 111In-DTPA-hEGF Injection because it allows rapid, simple, and reproducible preparation of the radiopharmaceutical. It also minimizes the manipulation steps involved since the very high labeling efficiency achieved (94%–99%) eliminates the need for postlabeling purification. This formulation also allows certain quality control tests (e.g., receptor-binding properties and protein purity or homogeneity) to be evaluated before patient administration and others (e.g., sterility and apyrogenicity) to be fully validated.
111In-DTPA-hEGF exhibited specific receptor-mediated binding to MDA-MB-468 breast cancer cells (Ka, 0.9–1.1 × 107 L/mol; Bmax, 1.1–2.2 × 106 sites per cell). The Ka and Bmax values were similar to those of 123I-hEGF1−53 (Ka, 1.6–3.4 × 107 L/mol; Bmax, 0.9–2.2 × 106 sites per cell [not shown]) but the Ka values were lower than those previously reported for 111In-DTPA-hEGF (Ka, 7.5 ± 3.8 × 108 L/mol) (13). The Bmax values for 111In-DTPA-hEGF were similar to those previously reported (Bmax, 1.3 ± 0.3 × 106 sites per cell) (13). Based on the similar Ka and Bmax values for 111In-DTPA-hEGF and 123I-hEGF measured using identical assay methodology, we conclude that the radiopharmaceutical exhibited preserved receptor-binding properties. There was no change in the receptor-binding properties of 111In-DTPA-hEGF Injection prepared from the kits when stored for up to 90 d at 4°C.
The labeling efficiency of the kits was almost quantitative (94%–99%) when 115–318 MBq 111In were added to each vial. It was further demonstrated by labeling aliquots of the kit solution with increasing amounts of 111In (1.1–74 MBq) that the radiochemical purity of 111In-DTPA-hEGF Injection would remain within specifications (>90%) with as much as 3,083 MBq added to each kit. Single kits were recently labeled with 740–773 MBq 111In, producing 111In-DTPA-hEGF Injection with a radiochemical purity of >94%. 111In-DTPA-hEGF Injection was prepared by aseptically decapping the vials under laminar air flow and adding 111In chloride directly into the vial using a micropipette and sterile tip. This was necessary due to the very high concentration of 111In chloride radiochemical (>3.7 GBq/mL) from MDS Nordion or PerkinElmer. Since 111In chloride was not pharmaceutical quality, 111In-DTPA-hEGF Injection was terminally sterilized by filtration through a 0.22-μm Millex GV low-protein-binding filter. Retrospective USP Sterility and Pyrogen Tests validated the method as yielding a final product that was sterile and pyrogen free. Nevertheless, it should be possible to prepare 111In-DTPA-hEGF Injection without the need for this terminal sterilization step by aseptically adding a sterile solution of 111In chloride directly into the vial using a sterile syringe and needle.
In the planned phase I clinical trial, we intend to administer escalating single doses of 111In-DTPA-hEGF ranging from 185 to 2,960 MBq to EGFR-positive breast cancer patients. It is anticipated therefore that 1 or 2 vials of the kit will be labeled with 111In, and the corresponding administered mass of DTPA-hEGF will be 0.25–0.50 mg. 131I-hEGF has been administered safely to humans for imaging squamous cell lung carcinoma in amounts up to 3.0 mg (20). Additionally, preclinical toxicology studies performed in our laboratory in mice and rabbits have shown that doses of 111In-DTPA-hEGF up to 25 μg/kg are extremely well tolerated with no evidence of significant normal tissue toxicity (unpublished data). These preclinical doses correspond to approximately 1.2–1.7 mg of 111In-DTPA-hEGF in a 50- to 70-kg human.
CONCLUSION
A kit was designed and manufactured under GMP conditions for the rapid, simple, and reproducible preparation of 111In-DTPA-hEGF Injection, a novel targeted radiotherapeutic agent for advanced EGFR-positive breast cancer. Specifications, SOPs, and quality control methods were developed for all raw materials, key intermediates (pharmaceutical buffers and kits), and the final radiopharmaceutical product. 111In-DTPA-hEGF Injection prepared from the kits was radiochemically pure, exhibited preserved receptor-binding properties toward EGFR-positive MDA-MB-468 human breast cancer cells, and was sterile and pyrogen free. We conclude that the kits are suitable for preparing 111In-DTPA-hEGF Injection for evaluation in a planned phase I clinical trial in breast cancer patients. The GMP processes were incorporated into the Chemistry and Manufacturing section of a CTA submitted by the University Health Network to Health Canada for 111In-DTPA-hEGF Injection.
Acknowledgments
This research was supported by grants from the U.S. Army Breast Cancer Research Program (DAMD17-02-1-0559), the Susan G. Komen Breast Cancer Foundation (BCTR0100840), and the Ontario Research and Development Challenge Fund. Parts of this study were presented at the 11th European Symposium on Radiopharmacy and Radiopharmaceuticals, Innsbruck, Austria, March 15–18, 2003.
Footnotes
Received Sep. 3, 2003; revision accepted Dec. 2, 2003.
For correspondence or reprints contact: Raymond M. Reilly, PhD, Leslie Dan Faculty of Pharmacy, University of Toronto, 19 Russell St., Toronto, ON, Canada, M5S 2S2.
E-mail: raymond.reilly{at}utoronto.ca.
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- ErbB-2 Blockade and Prenyltransferase Inhibition Alter Epidermal Growth Factor and Epidermal Growth Factor Receptor Trafficking and Enhance 111In-DTPA-hEGF Auger Electron Radiation Therapy
- Relationship Between Induction of Phosphorylated H2AX and Survival in Breast Cancer Cells Exposed to 111In-DTPA-hEGF
- Preclinical Pharmacokinetic, Biodistribution, Toxicology, and Dosimetry Studies of 111In-DTPA-Human Epidermal Growth Factor: An Auger Electron-Emitting Radiotherapeutic Agent for Epidermal Growth Factor Receptor-Positive Breast Cancer