


Abstract
FK506 is an immunosuppressive agent that has been reported to have neuroprotective effects in several kinds of rodent models of stroke. The purpose of this study was to evaluate the neuroprotective effects of FK506 in a monkey model of stroke. Methods: Cynomolgus monkeys underwent 3 h of occlusion followed by 5 h of reperfusion of the right middle cerebral artery (MCA) through a transorbital approach. A single bolus dose of FK506 (0.1 mg/kg) was injected intravenously 5 or 175 min after MCA occlusion. Eight hours after ischemia, a neuropathologic study was performed and the volume of ischemic damage was determined. To measure local cerebral blood flow (CBF), the cerebral metabolic rate of oxygen (CMRO2), and the oxygen extraction fraction during the experiments, PET scans were obtained using a steady-state 15O continuous-inhalation method. Four consecutive PET scans (before and 2 h after ischemia and immediately and 3 h after reperfusion) were obtained on each monkey. Results: Treatment with FK506 (0.1 mg/kg) 5 or 175 min after ischemia significantly reduced cortical damage 8 h after ischemia by 82% (P < 0.05) and 73% (P < 0.05), respectively. In PET studies, FK506 did not affect CBF or physiologic parameters in any treatments. In the FK506-treated group, a volume of >40% CMRO2 reduction 3 h after reperfusion decreased significantly (P < 0.05). Conclusion: This study showed that FK506 showed a powerful neuroprotective effect in a nonhuman primate model of stroke. The therapeutic time window of FK506 was at least 3 h after onset. PET studies detected neuroprotective effects only in areas with >40% CMRO2 reduction 3 h after reperfusion.
An immunosuppressive agent, FK506 (tacrolimus, Prograf; Fujisawa Pharmaceutical Co. Ltd., Osaka, Japan), is used in clinical settings for the prevention of allograft rejection (1–3). Several studies have revealed its powerful neuroprotective properties in cell culture systems (4–6) and in several animal models of focal cerebral ischemia (7–10). Its mechanism of neuroprotection is unclear. However, its inhibitive effects on nitric oxide synthase (4), ryanodine, and the inositol 1,4,5-trisphosphate receptor complex (11–14), superoxide production in neutrophils (15), or antiapoptosis effects (6,16,17) have been considered. FK506 protects against ischemic damage in several models of focal and global ischemia.
Recent clinical studies on acute stroke revealed a discrepancy between the preclinical and clinical effectiveness of neuroprotective agents. This discrepancy may be attributed to the differences between human and nonhuman species in experimental stroke models. Almost all preclinical experiments have been performed on rodent models of stroke, as in the case of FK506 (7–10,14). Therefore, it is important to evaluate the neuroprotective effects of this agent in a suitable experimental model, which mimics human patients, before clinical trials. In this study, we investigated the neuroprotective effects of FK506 on a nonhuman primate model of stroke.
This monkey study allowed us to investigate brain function during cerebral ischemia by PET, which has proven to be a powerful tool with which to investigate the pathophysiology of stroke (18–26). PET studies in animal models of stroke have been useful for the characterization of the acute phase of cerebral ischemia. Although several studies showed hemodynamic and metabolic changes after ischemia or ischemia–reperfusion using a monkey or cat model of cerebral ischemia (19,21–26), little is known about whether the neuroprotective effects of potential therapeutic agents can be evaluated with PET. In this study, we also examined the neuroprotective effects of FK506 using PET.
MATERIALS AND METHODS
Animal Preparation
The studies were performed on 21 male cynomolgus monkeys (Macaca fascicularis) with body weights ranging from 5.8 to 6.7 kg (Clea Japan Inc., Tokyo Japan). All experiments were performed in accordance with the institutional guidelines of Hamamatsu University School of Medicine and the Central Research Laboratory of Hamamatsu Photonics (Shizouka, Japan). The monkeys were killed 8 h after ischemia because the animal experiment committee recommended that animals should not recover from anesthesia.
The procedure for animal preparation was the same as that of Takamatsu et al. (26). Anesthesia was induced with 10 mg/kg intramuscular ketamine hydrochloride. The monkeys were tracheostomized, immobilized with 0.05 mg/kg intramuscular pancuronium bromide every 2 h, and artificially ventilated. Anesthesia was continued with isoflurane (concentration range, 0.6%–0.8%) in a N2O:O2 gas mixture (N2O:O2 = 7:3) during the entire experiment. Catheterization of the left femoral artery was then performed for the measurement of mean arterial blood pressure (MABP), heart rate, and arterial blood sampling. The MABP, heart rate, rectal temperature, arterial Po2 and Pco2, pH, and plasma glucose levels were continuously or regularly monitored. The right middle cerebral artery (MCA) was occluded using a transorbital approach (26,27). After the administration of 0.05 mg/kg intramuscular atropine, the right globe was removed. A craniotomy was then performed superolateral to the optic nerve. After opening the dura, MCA occlusion was performed with 2 microvascular clips, 1 on the proximal part of the main MCA trunk and the other on the distal-to-orbitofrontal branch. Three hours after MCA occlusion, the microvascular clips were removed. During the experiments, body temperature was maintained within normal limits with heated blankets.
Eight hours after MCA occlusion, the monkeys were deeply anesthetized with sodium pentobarbital. The brain was fixed through transcardial perfusion with a 10% formalin neutral buffer solution, pH 7.4, after saline perfusion at 100 mm Hg. The brain was then removed, and 12 coronal sections at 2-mm intervals were made using a brain matrix (MBM-2000C; Bioresearch Center, Nagoya, Japan). Each section was embedded in paraffin wax, and 10-μm-thick sections were cut and stained with hematoxylin–eosin. The neuronal damage to each section was defined (26,28) and, after correction for brain edema (29), the area of neuronal damage was measured using a computerized image analysis system. For neuronal damage, we measured the area of necrotic damage, which could be identified as exhibiting pyknosis, karyorrhexis, karyolysis, and cytoplasmic eosinophilia or loss of hematoxylin affinity (30–32). The neuronal damage volumes were calculated from the areas of damage in the different coronal sections and their anteroposterior coordinates.
PET Studies
Four consecutive PET studies were performed on each monkey. Serial PET scanning was performed with an SHR7700 system (Hamamatsu Photonics, Hamamatsu, Japan) (33). The first scan was obtained before MCA occlusion (mean, −87 ± 7 min). The second scan was obtained 120 ± 3 min after the start of occlusion. The third and fourth scans were obtained 187 ± 8 min and 360 ± 5 min after occlusion. Assessments of cerebral blood flow (CBF), cerebral metabolic rate of O2 (CMRO2), oxygen extraction fraction (OEF), and local cerebral blood volume were done using the steady-state 15O inhalation method (21,25,26,34,35), with successive inhalation of trace amounts of C15O2 (0.8 GBq/min), 15O2 (2 GBq/min), and C15O (4 GBq/min). Each 15O gas was administered through a ventilator (15 strokes per minute; 70 mL per stroke). The C15O2 and 15O2 scans were started after saturation of radioactivity in the gantry of the PET camera; the C15O scan was started 2 min after a 20-s inhalation of C15O gas. Each scan was obtained for 5 min, consisting of five 1-min time frames. During each scan, 2 arterial blood samples were taken (1 each at the beginning and the end of acquisition) for whole-blood and plasma radioactivity measurements. The mean values of radioactivity of whole blood and plasma were used for parametric image generation (35). The calculated CMRO2 and OEF values were obtained after correction of cerebral blood volume.
Regions of Interest
Three regions of interest (ROIs) were chosen: 0%–20%, 21%–40%, and >40% CBF, OEF, and CMRO2 changes (decrease or increase) in the ipsilateral hemisphere relative to the contralateral hemisphere. To set each ROI, each PET image was divided by its mirror image, which was made using an image analysis system (Alice; Hayden Imaging Processing Group, Boulder, CO), and areas of 0%–20%, 21%–40%, and >40% CBF, OEF, and CMRO2 changes (decrease or increase) in the ipsilateral hemisphere were calculated. These calculations were performed in each plane above the orbitomeatal plane (total plane number, 10). The volume of each parameter change in the ipsilateral hemisphere was calculated from the area of change at each plane and the plane interval (3.6 mm). The total volume of the ipsilateral hemisphere using this calculation was 30,952.2 ± 1,558.4 mm3 (n = 21). In this study, a 0%–20% decrease or increase in an ROI, a 21%–40% decrease or increase in an ROI, and a >40% decrease or increase in an ROI were defined as ROIs with mild, moderate, and severe neuronal damage, respectively. The validity of the data was checked by calculation of preischemic values (theoretic value of 1).
Drug Administration
FK506 (10 mg/mL, for intravenous injection) and its vehicle (polyoxyethylene hydrogenated castor oil 60 [400 mg/mL]) were gifts from Fujisawa Pharmaceutical Co. FK506 was diluted to 0.4 mg/mL (25-fold dilution) with saline, and 0.25 mL/kg (0.1 mg/kg) was administered intravenously. The vehicle was also diluted (25-fold dilution) with saline, and 0.25 mL/kg was administered intravenously in control animals.
An intravenous bolus injection of FK506 (0.1 mg/kg) was given to each monkey 5 min after MCA occlusion (control, n = 6; FK506, n = 5) or 175 min after MCA occlusion (control, n = 5; FK506, n = 5).
Data Analysis
Data are presented as the mean ± SD. All data were evaluated by ANOVA followed by the Dunnett multiple range test. P < 0.05 was considered significant.
RESULTS
The physiologic variables during prolonged experiments in this study remained within the normal range (Table 1).
Physiologic Parameters
Neuropathologic Study
MCA occlusion and reperfusion caused necrotic damage in the cortex and the basal ganglia 8 h after ischemia. Treatment with FK506 5 min after MCA occlusion significantly (P < 0.05) reduced cortical damage by 82% (control [n = 6], 747.7 ± 509.6 mm3; FK506 [n = 5], 132.8 ± 83.0 mm3) (Fig. 1A), and treatment with FK506 175 min after ischemia also significantly (P < 0.05) reduced cortical damage by 73% (control [n = 5], 958.2 ± 281.6 mm3; FK506 [n = 5], 262.3 ± 177.1 mm3) (Fig. 1C). Damage in the basal ganglia was not affected by any treatment with FK506 (Figs. 1B and 1D).

Volume of ischemic brain damage 8 h after MCA occlusion. Intravenous bolus administration of FK506 (0.1 mg/kg) was performed 5 min (control, n = 6; FK506, n = 5) (A and B) or 175 min (control, n = 5; FK506, n = 5) (C and D) after MCA occlusion. Volume of cortical damage (A and C) and volume of damage in basal ganglia (C and D) are shown. Data are expressed as mean ± SD.
PET Study
Figure 2 shows typical PET images in a control animal and an FK506-treated animal (5 min after ischemia). MCA occlusion decreased CBF and CMRO2 and increased OEF. Reperfusion produced hyperperfusion and decreased OEF. The CMRO2 decrease during ischemia was partially recovered by reperfusion. Three hours after reperfusion, a mild hypoperfusion was observed, and OEF and CMRO2 decreased further. In an FK506-treated animal, the OEF and CMRO2 decrease after reperfusion seemed to be mild.

Typical PET images of CBF (A), OEF (B), and CMRO2 (C) in control animal and FK506-treated animal 5 min after ischemia. T1, T2, T3, and T4 indicate before ischemia, 2 h after ischemia, immediately after reperfusion, and 3 h after reperfusion, respectively. Each plane was 18 mm above orbitomeatal plane.
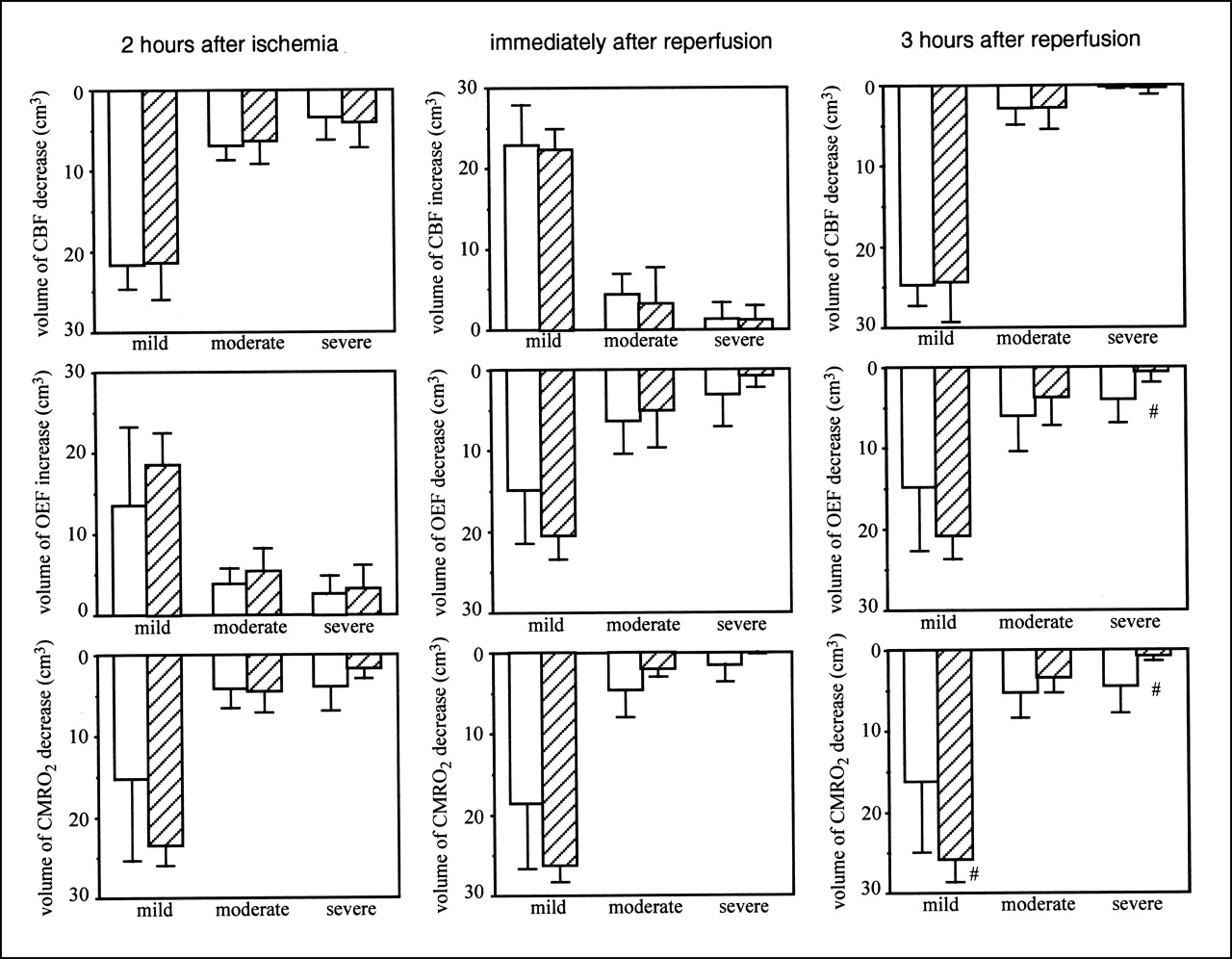
Figure 3 shows the volumes of CBF, OEF, and CMRO2 changes after MCA occlusion, when FK506 was administered 5 min after ischemia. Before ischemia, no differences in CBF, OEF, and CMRO2 were found between the ipsilateral and contralateral hemispheres or between the control group and the FK506-treated group (data not shown). FK506 did not affect the CBF decrease, OEF increase, or CMRO2 decrease during ischemia. Reperfusion caused postischemic hyperperfusion and decreased OEF and CMRO2. FK506 did not affect these changes. Postischemic hypoperfusion was observed 3 h after reperfusion. However, FK506 did not affect it. FK506 significantly (P < 0.05) attenuated the OEF decrease in severe ROIs. A volume of >40% CMRO2 decrease (a severe ROI) was also significantly (P < 0.05) decreased by FK506 and, in contrast, the volume of CMRO2 decrease in a mild ROI significantly (P < 0.05) increased with FK506 treatment.
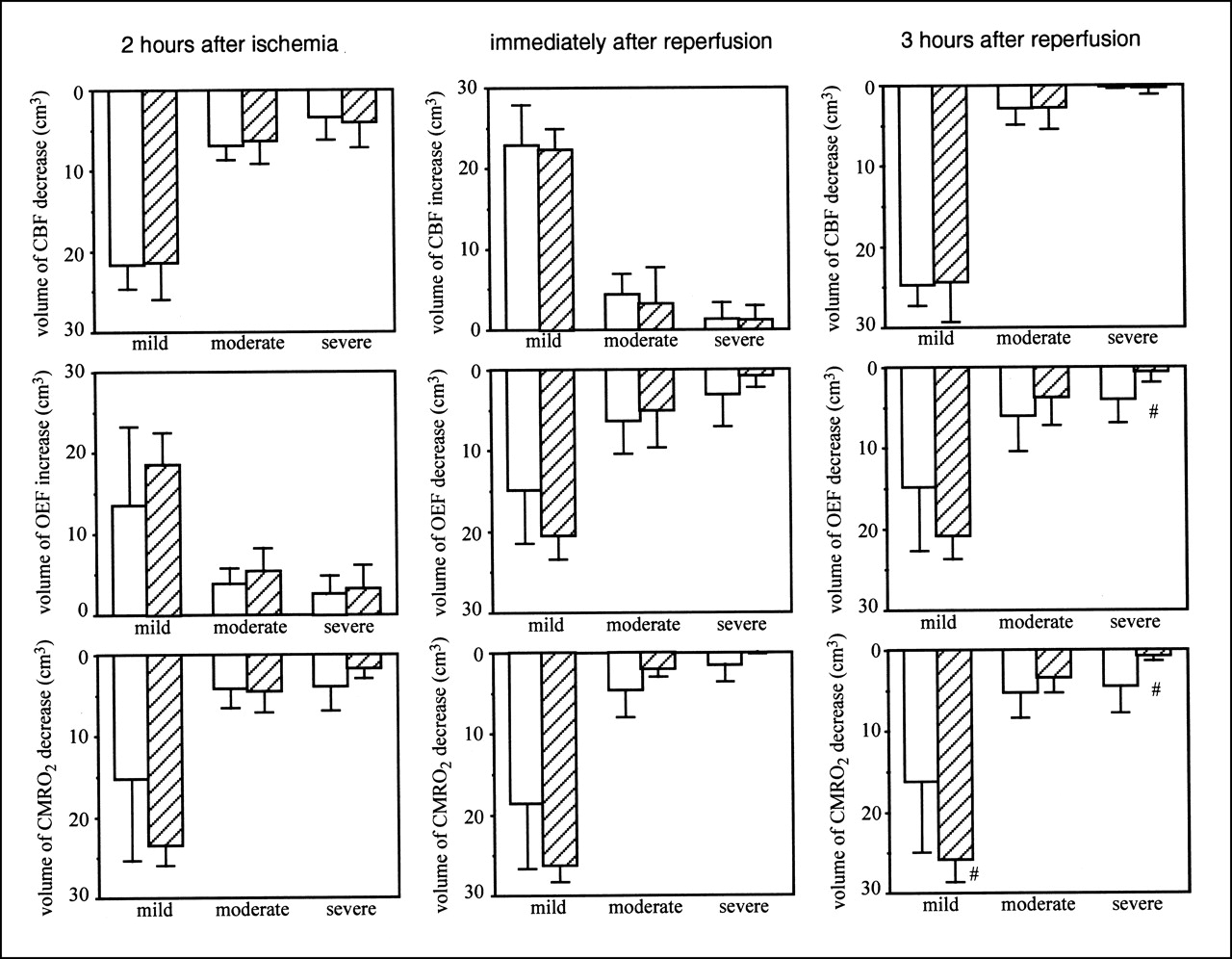
Volume of CBF (top), OEF (middle), and CMRO2 (bottom) changes in case of FK506 administration 5 min after ischemia. Left, center, and right show results 2 h after MCA occlusion, immediately after reperfusion, and 3 h after reperfusion, respectively. Results of control group (white bars; n = 6) and FK506-treated group (hatched bars; n = 5) are shown. Intravenous bolus administration of FK506 (0.1 mg/kg) or vehicle was performed 5 min after MCA occlusion. Mild, moderate, and severe mean ROIs were from 0% to 20%, from 21% to 40%, and >40% decrease or increase in each parameter in ipsilateral hemisphere relative to that in contralateral hemisphere, respectively. Data are expressed as mean ± SD. #Significantly different (P < 0.05, ANOVA; followed by Dunnett multiple range test) from value of control group.
Figure 4 shows the volumes of CBF, OEF, and CMRO2 changes after MCA occlusion, when FK506 was administered 175 min after reperfusion. Before ischemia, no differences in CBF, OEF, and CMRO2 were found between the ipsilateral and contralateral hemispheres or between the control group and the FK506-treared group (data not shown). During MCA occlusion (before FK506 treatment), no significant difference was found in the decrease in CBF or CMRO2 or in the increase in OEF. Immediately after reperfusion, FK506 did not affect CBF, OEF, or CMRO2. Three hours after reperfusion, FK506 did not affect any changes in CBF or OEF. However, FK506 significantly (P < 0.05) decreased the volume of >40% CMRO2 decreases.
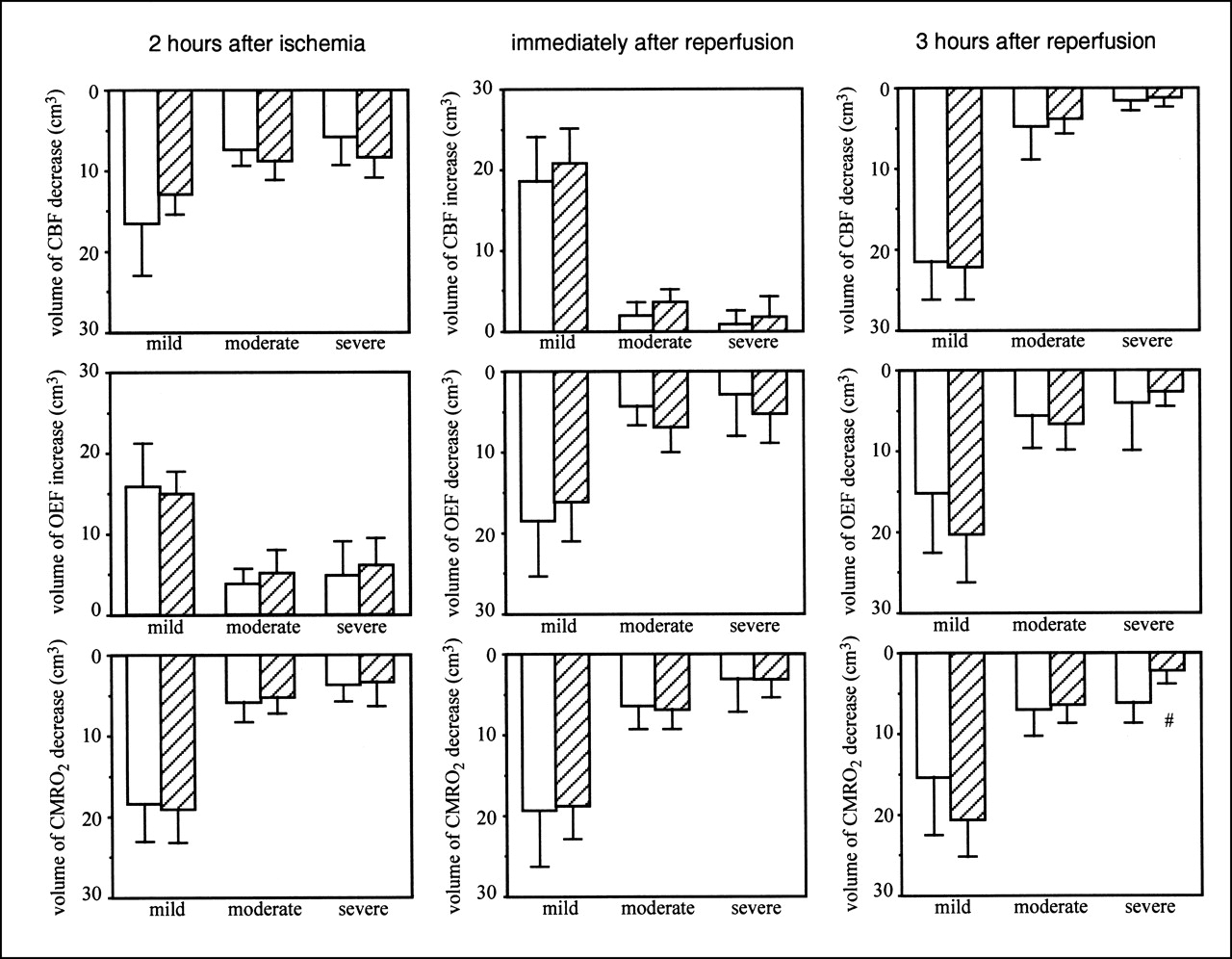
Volume of CBF (top), OEF (middle), and CMRO2 (bottom) changes in case of FK506 administration 175 min after ischemia. Left, center, and right show results 2 h after MCA occlusion, immediately after reperfusion, and 3 h after reperfusion, respectively. Results of control group (white bars; n = 5) and FK506-treated group (hatched bars; n = 5) are shown. Intravenous bolus administration of FK506 (0.1 mg/kg) or vehicle was performed 175 min after MCA occlusion. Mild, moderate, and severe mean ROIs were from 0% to 20%, from 21% to 40%, and >40% decrease or increase in each parameter in ipsilateral hemisphere relative to that in contralateral hemisphere, respectively. Data are expressed as mean ± SD. #Significantly different (P < 0.05, ANOVA; followed by Dunnett multiple range test) from value of control group.
DISCUSSION
In this study, we showed that FK506 has neuroprotective effects in a nonhuman primate model of stroke. We evaluated the neuroprotective effects of FK506 in a 3-h, temporal MCA occlusion model in the monkey because recent pilot clinical studies of ischemic stroke suggested that intravenous recombinant tissue plasminogen activator therapy is beneficial only when it is performed within 3 h of onset (36). The purpose of this study was to test the neuroprotective effects of FK506 in an animal model that mimics human stroke. The amount of FK506 (0.1 mg/kg) given to the animal was based on the study of Sharkey et al. (7), who reported that this amount was sufficient to show neuroprotective effects. Drug administration was performed 5 min (beginning of ischemia) or 175 min (just before end of ischemia) after ischemia. FK506 significantly reduced cortical damage by 82% when it was administered 5 min after ischemia and by 73% when its administration was started 175 min after ischemia. In PET studies, MCA occlusion decreased CBF and CMRO2 and increased OEF, whereas reperfusion caused postischemic hyperperfusion and then caused delayed hypoperfusion while it decreased OEF and CMRO2. These observations are in agreement with previous reports (19,21–26). FK506 did not affect CBF changes in any type of treatment. These results indicate that FK506 showed powerful neuroprotective effects against cortical ischemic damage, and its therapeutic time window was at least 3 h after the onset of stroke. It is well known that FK506 has neuroprotective properties (4–14,16,17) and shows neuroprotective effects in several kinds of rodent models of stroke (7–10,14). This study shows the effectiveness of FK506 in monkeys.
We anticipated that the PET study would reveal the neuroprotective mechanism of FK506. However, this study showed only that FK506 attenuated CMRO2 reduction 3 h after ischemia, and this result suggests that the FK506-treated group had more living cells as a result of neuroprotection. In this study, FK506 attenuated cortical damage but not striatal damage. In our previous study, which compared histology and oxygen metabolism using PET between a transient MCA occlusion model and a permanent MCA occlusion model in the monkey, cortical damage in the transient model was significantly larger than that in the permanent model, and postischemic hyperperfusion was observed in the cortex but not in the striatum (26). Thus, it seems that the striatum and the cortex behave differently in the acute occlusion–reperfusion model. At the same time, OEF and CMRO2 in the cortex, in which hyperperfusion and histologic changes were observed 8 h after ischemia, were reduced (26). In this study, events that were similar to those of the previous study were also likely to be observed (Fig. 2). The observations of the previous study suggest that these PET findings (i.e., reduction in OEF and CMRO2) may serve as an indicator of reperfusion injury. Thus, reperfusion injury appears to play an important role in the genesis of cortical damage in our monkey model. Because the neuroprotective effects of FK506 were significant only in the cortex but not in the striatum, it would be reasonable to speculate that the effects of FK506 may include the attenuation of reperfusion injury, which is also supported by the PET results. Bochelen et al. (14) also showed in rat models of cerebral ischemia that FK506 attenuated ischemic damage in a transient MCA occlusion model but not in a permanent MCA occlusion model. Although the exact neuroprotective mechanisms of FK506 remain to be elucidated, the neuroprotective effect of FK506 only against cortical damage suggests that it is an inhibitor of reperfusion injury.
This study had several limitations in its use of a higher- order animal species, the monkey. The most serious limitation was that we could not awaken the animals after ischemia because of the severity of the ischemic preparation. Therefore, we could not examine neurologic outcome data and mature infarction data. Furthermore, we recognized the influence of anesthesia on the ischemic condition. In general, to evaluate the neuroprotective effects of agents, histologic determination is required at the time point of mature infarction formation. Therefore, although FK506 attenuated early ischemic damage in the monkey, it remains unclear whether it attenuates final infarction, which must be addressed in further studies. The other problem with using monkeys was individual differences. There was a difference in the amount of ischemic damage between 2 control groups, which may not be associated with the differences in ischemic conditions because physiologic monitoring did not show any differences (Table 1). These animals were bred on Deli Island, Indonesia, not in a closed colony, and they could move freely in the island colony. Because we performed 2 separate experiments and used different lots of animals, the difference in the amount of ischemic damage may have depended on the colonies used.
CONCLUSION
This study showed the neuroprotective effect of FK506 using a nonhuman primate model of stroke. FK506 showed powerful neuroprotective effects, and its therapeutic time window was at least 3 h after the onset of stroke. Although there were several limitations to using a nonhuman primate model and the exact mechanisms of neuroprotection with FK506 still remain to be elucidated, the primary purpose of this study was to show the neuroprotective effects of FK506 in a primate model of acute stroke.
Acknowledgments
This work was supported in part by the Strategic Promotion System for Brain Science of the Science and Technology Agency of the Japanese Government.
Footnotes
Received Apr. 18, 2001; revision accepted Aug. 20, 2001.
For correspondence or reprints contact: Hiroyuki Takamatsu, PhD, The Medical and Pharmacological Research Center Foundation, Wo 32, Inoyama-chou, Hakui, Ishikawa, 925-0613, Japan.




{kind=link}
{kind=link}
{kind=link}
{kind=link}