


Abstract
Increasing evidence indicates that reduced intracellular drug accumulation is the parameter most consistently associated with platinum drug resistance, emphasizing the need to directly measure the intratumor drug concentration. In the era of precision medicine and with the advent of powerful imaging and proteomics technologies, there is an opportunity to better understand drug resistance by exploiting these techniques to provide new knowledge on drug–target interactions. Here, we contribute to this endeavor by reporting on the development of an 18F-labeled carboplatin derivative (18F-FCP) that has the potential to image drug uptake and retention, including intratumoral distribution, by PET. Methods: Fluorinated carboplatin (19F-FCP) was synthesized using 19F-labeled 2-(5-fluoro-pentyl)-2-methyl malonic acid (19F-FPMA) as the labeling agent to coordinate with the cisplatin–aqua complex. It was then used to treat cell lines and compared with cisplatin and carboplatin at different concentrations. Manual radiosynthesis and characterization of 18F-FCP were performed using 18F-FPMA for coordination with the cisplatin–aqua complex. Automated radiosynthesis of 18F-FCP was optimized on the basis of manual synthesis procedures. The stability of 18F-FCP was verified using high-performance liquid chromatography. 18F-FCP was evaluated using ex vivo biodistribution and in vivo PET imaging in non–tumor-bearing animals as well as in KB-3-1 and COLO-205 tumor xenograft–bearing nude mice. Results: In vitro cytotoxicity studies demonstrated that 19F-FCP has an antitumor activity profile similar to that of the parent drug carboplatin. In vivo plasma and urine stability analysis showed intact 18F-FCP at 24 h after injection. PET imaging and biodistribution studies showed fast clearance from blood and major accumulation in the kidneys, indicating substantial renal clearance of 18F-FCP. Using 18F-FCP PET, we could image and identify the intratumor drug profile. Conclusion: Our results demonstrated that 19F-FCP, like carboplatin, retains antitumor activity in various cell lines. 18F-FCP could be a useful imaging tool for measuring the intratumor drug distribution. This strategy of using a new therapeutic carboplatin derivative to quantify and track platinum drugs in tumors using PET has the potential to translate into a clinically useful imaging tool for individual patients.
Since the discovery of cisplatin by Rosenberg et al. (1), platinum drugs have been used as a major chemotherapy treatment for cancer (2–5). In fact, platinum drugs are administered to nearly half of all cancer patients receiving chemotherapy (6). Despite some treatment efficacy of cisplatin and carboplatin chemotherapy, drug resistance, toxic side effects, and tumor recurrence are still critical barriers that need to be addressed. Over the last several years, a resurgence of platinum-based cancer chemotherapy has been driven by the development of new derivatives and by the circumvention of mechanisms of resistance, thereby broadening the clinical use of these agents. Overcoming the limitations of platinum drugs has stimulated further research toward understanding and improving platinum-based chemotherapy (6).
Recent developments have also addressed novel targeting strategies and better drug delivery approaches (7). Several mechanisms have been implicated in tumor resistance to platinum drugs (8,9). In addition, studies have revealed that reduced drug accumulation is most consistently associated with resistance, emphasizing the need to directly measure the intratumor drug concentration (10). Contributing to this endeavor, we report on the development of an 18F-labeled carboplatin derivative (18F-FCP) for imaging drug distribution in tumors by PET. Such a noninvasive method may allow the optimization of an individual’s drug exposure at the tumor site as well as provide a screening measure for determining efficacy in individual patients.
Previous strategies for synthesizing radiolabeled platinum drugs, such as cisplatin and carboplatin, relied on the incorporation of a platinum isotope (e.g., 191Pt, 195mPt) as its chloride salt early in drug synthesis (11–14). These studies provided valuable in vivo data, but this approach was not clinically translatable because of limited platinum radioisotope availability, long radiosynthesis, and logistic cost. Another strategy used 13N-cisplatin to assess pharmacokinetics in brain tumors (15,16). However, this method was limited by the ultrashort half-life (10 min) of 13N.
In the present work, we sought to develop an alternative strategy by synthesizing 18F-FCP. The rationale for the radiolabeling strategy was based on the coordination of platinum by malonic acid derivatives (17). We previously developed a malonic acid precursor to produce an 18F-malonic acid derivative (18) that has been proposed as a potential PET apoptosis imaging agent (19). In this coordination-based labeling strategy, the fluorine isotope (19F or 18F) is introduced as a malonic acid derivative attached to the carboxylate group, which is subsequently replaced with water to form the active positively charged species. The widely accepted mechanism of action of platinum agents considers the Pt(NH3)2 moiety in cisplatin or carboplatin to be the carrier group that binds to DNA on eventual substitution of the chloride (cisplatin) or carboxylate (carboplatin) moiety. However, because the aquation rate for carboplatin (7 × 10−7 s−1) is much slower than that for cisplatin (8 × 10−5 s−1), the rate of binding to DNA is also much slower for carboplatin (20); therefore, binding to DNA is expected to be slow but compatible with the short half-life of 18F. In the present work, we report on the development of 19F-FCP and its 18F-FCP analog.
MATERIALS AND METHODS
Materials
All reagents and solvents were purchased from Sigma–Aldrich and used without further purification. Nuclear magnetic resonance (NMR) spectra (1H-NMR, 13C-NMR, 19F-NMR, and 195Pt-NMR) were obtained using Varian Mercury 300-MHz and Varian Inova 400-MHz spectrometers. Tetramethylsilane was used as an internal standard. Radio–high-performance liquid chromatography (HPLC) analysis was carried out using an HPLC pump (Waters model 1525) equipped with an ultraviolet detector (Waters model 2489) and a radiation detector (BioScan model B-FC-3300). Radioactivity was measured with a dose calibrator (Capintec CRC-15 PET), and γ-counting was done using an LKB Wallac 1282 counter.
Reference Compound (19F-FCP)
The synthesis of intermediates and 19F-labeled 2-(5-fluoro-pentyl)-2-methyl malonic acid (19F-FPMA) was performed as previously described (18). Cisplatin (0.2 mmol) and silver nitrate (0.3 mmol) were dissolved in Nanopure Water (Thermo Fisher Scientific) (2.5 mL), and the mixture was stirred at room temperature overnight in the dark. After filtration of the precipitated silver chloride, 1 equivalent (0.2 mmol) of 19F-FPMA (compound 4) was added, and the mixture was stirred at room temperature for 2 d. The product was purified by solid-phase extraction using a quaternary methyl ammonium cartridge and water as the eluent, and the fractions were lyophilized to give 19F-FCP (compound 5) in a 90% yield. 1H-NMR (300 MHz) (D2O) δ 1.32–1.47 (m, 4H), 1.49 (s, 3H), 1.65–1.78 (m, 2H), 1.90–1.94 (m, 2H), 4.50 (t, J = 6.05 Hz, 1H), 4.66 (t, J = 6.05 Hz, 1H); 19F-NMR (300 MHz) (D2O) δ −218.88 (m); and 195Pt-NMR (300 MHz) (D2O) δ −1,596 (broad singlet). The ESI-MS [M + Na]+ m/z values for C9H19FN2O4Pt + Na were 456.33 (calculated) and 456.33 (found).
Radiosynthesis and Characterization of 18F-FCP
The synthesis of 18F-FPMA was performed as previously described (18). The final 18F-FPMA (3.3 × 104 MBq) was redissolved in water (0.25 mL), an aqua–platinum complex (0.5 mL) was added, and the vial was heated at 75°C for 30 min. In the reaction, about 50% of the unreacted starting compound (18F-FPMA) was observed by HPLC analysis. The final 18F-FCP was purified by solid-phase extraction using a quaternary methyl ammonium Sep-Pak ion-exchange cartridge (Waters), water as the mobile phase, and elution in 0.25-mL fractions. A total of 7 fractions were collected, 18F-FCP was eluted in fractions 2–4, and most of the 18F-FCP radioactivity was eluted in fractions 3 and 4. 18F-FPMA was not detected in any of the water-eluted fractions by HPLC analysis. The remaining radioactivity detected in the cartridge was eluted with 0.5 mL of ethanol, and HPLC analysis of this sample showed an 18F-FPMA peak.
The automated synthesis of 18F-FCP was conducted using an AllinOne module (Trasis SA). The AllinOne synthesizer was driven by interactive software. The synthesis of 18F-FCP was programmed using in-house reaction sequences that were derived from the manual synthesis to automate the steps that can be viewed in real time. The automated experiments were conducted using a range of activities (1.9 × 104–2.8 × 104 MBq of 18F). The 18F fluorination reaction was optimized using the same parameters as those used in the manual synthesis. After the final samples were collected, the radiochemical purity was analyzed using HPLC. For in vivo studies, the required amount of activity was brought up to a 200-μL volume with phosphate-buffered saline for intravenous injection.
Cell Lines and Culture Conditions
All cell lines were maintained in a humidified incubator at 37°C and 5% CO2. The human cervical epidermoid carcinoma (KB-3-1) cell line was a generous gift from Michael Gottesman (NCI, National Institutes of Health). The human lung carcinoma (A549), ovarian carcinoma (SK-OV-3), colon adenocarcinoma (COLO-205), renal carcinoma (A498), prostate carcinoma (LNCaP), and normal prostate (RWPE-1) cell lines were purchased from the American Type Culture Collection. The head and neck carcinoma (FaDu) cell line was a gift from Andrei Pugachev (Massey Cancer Center, Virginia Commonwealth University). KB-3-1 cells were grown in Dulbecco modified Eagle medium containing a high glucose concentration and supplemented with 10% fetal bovine serum, 5 mM l-glutamine, penicillin (100 U/mL), streptomycin (100 μg/mL), and amphotericin B (0.25 μg/mL). SK-OV-3, COLO-205, FaDu, A549, A498, LNCaP, and RWPE-1 cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin G (100 U/mL), streptomycin (100 μg/mL), and amphotericin B (0.25 μg/mL). The cells were grown to 70%–80% confluence.
In Vitro Cytotoxicity of 19F FCP
The cytotoxicity of 19F-FCP for various cell lines was compared with that of cisplatin and carboplatin. The viability of COLO-205, SK-OV-3, FaDu, A549, A498, LNCaP, RWPE-1, and KB-3-1 cells treated with cisplatin, carboplatin, or 19F-FCP was evaluated. Cells plated in triplicate and grown in well plates were treated with 25 μL of drug at final concentrations of 0.001–100 μM for 72 or 96 h. At the end of the incubation period, cell viability was assessed using a CellTiter-Glo assay kit (Promega) and a plate reader (Beckman Coulter). The luminescence detected in untreated cells was used as a control (100% viability) to calculate the percentage of viability in treatment groups. The percentage of viability of each cell line was plotted and used to calculate the 50% inhibitory concentration (IC50) for each drug. Statistical analysis was done using the Student t test, and the P value was calculated on the basis of a 2-tailed test using Microsoft Excel software.
Animals
Four- to 6-wk-old female nude mice from Harlan Laboratories were used for experimentation. Animal experiments were approved and performed according to the policies and guidelines of the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
Animal Tumor Xenografts
KB-3-1 tumor cells (2.5 × 106/100 μL) and COLO-205 tumor cells (2.5 × 106/100 μL) in medium without serum were injected subcutaneously into the right flank (KB-3-1) and left shoulder (COLO-205) of athymic nude mice (4–6 wk old; weight, 18–25 g). After subcutaneous implantation, digital caliper measurements of tumor size were obtained when the tumor was palpable at the injection site (approximately 5 d). The tumor volume was calculated using the formula (length × width squared)/2 and was assessed twice per week.
Biodistribution and in Vivo Stability Analysis
The biodistribution of 18F-FCP was measured in adult female nude mice. Under 2% isoflurane, 3 mice in each of 4 groups received an intravenous injection (tail vein) of 18F-FCP (2.6–5.2 MBq in 200 μL of phosphate-buffered saline). Under anesthesia, mice were sacrificed at 5, 30, 60, and 90 min after injection. Whole blood was collected, other major organs were harvested and weighed, and the radioactivity in the tissues was counted in a γ-counter (LKB Wallac 1282). The blood activity was used to calculate the blood half-life. Decay-corrected radiotracer uptake in each tissue at various time points was then calculated as the percentage injected dose per gram of tissue (%ID/g).
The in vivo stability of 18F-FCP in plasma and urine was determined after radiotracer administration (2.6–5.2 MBq in 200 μL of phosphate-buffered saline) in female nude mice under 2% isoflurane anesthesia. Samples were collected at 5, 30, 60, and 90 min after intravenous injection of 18F-FCP. The collected samples of plasma were centrifuged for 10 min at 10,000g. Supernatants from these samples were filtered through 0.2-μm filters, and 200 μL of filtrate was injected into a reversed-phase HPLC column (Waters Nova-Pak 4-μm C18 150 × 3.9 mm column, ACN:H2O [25:75], containing 0.1% trifluoroacetic acid, at a flow rate of 1 mL/min). Urine samples were also filtered and analyzed in the same HPLC system.
PET Imaging
Small-animal PET/CT imaging studies were performed using a multimodality preclinical system (Siemens Medical Solutions Inc.). At 10 min before imaging, animals were anesthetized with 2% isoflurane in oxygen. After sedation, the animal was placed onto the imaging bed, and anesthesia was maintained for the duration of the imaging. A 90-min dynamic scan was initiated, and 18F-FCP (5.5–6 MBq in 200 μL of phosphate-buffered saline) was injected via the tail vein. micro-CT imaging was acquired after PET imaging at 80 kV and 500 μA with a focal spot of 58 μm.
PET images were reconstructed using Fourier rebinning and an ordered-subsets expectation maximization 3-dimensional algorithm with a dynamic framing sequence. Reconstructed PET/CT images were fused and analyzed using Inveon Research Workplace software (IAW 1.6; Siemens). For quantitation, regions of interest were drawn around areas of high radiotracer activity in each organ. The resulting quantitative data were expressed as %ID/g.
RESULTS
Synthesis of Cold Reference Standard Compound (19F-FCP)
Figure 1 describes the synthesis of the tosylated precursor and reference standard compound (19F-FCP; compound 5). Fluorination of compound 5 was confirmed by 19F-NMR, which showed the expected multiplet pattern and a chemical shift in the expected region (218.88 for terminal CH2F). The chemical purity of compound 5 was determined to be greater than 98% by analytic HPLC.
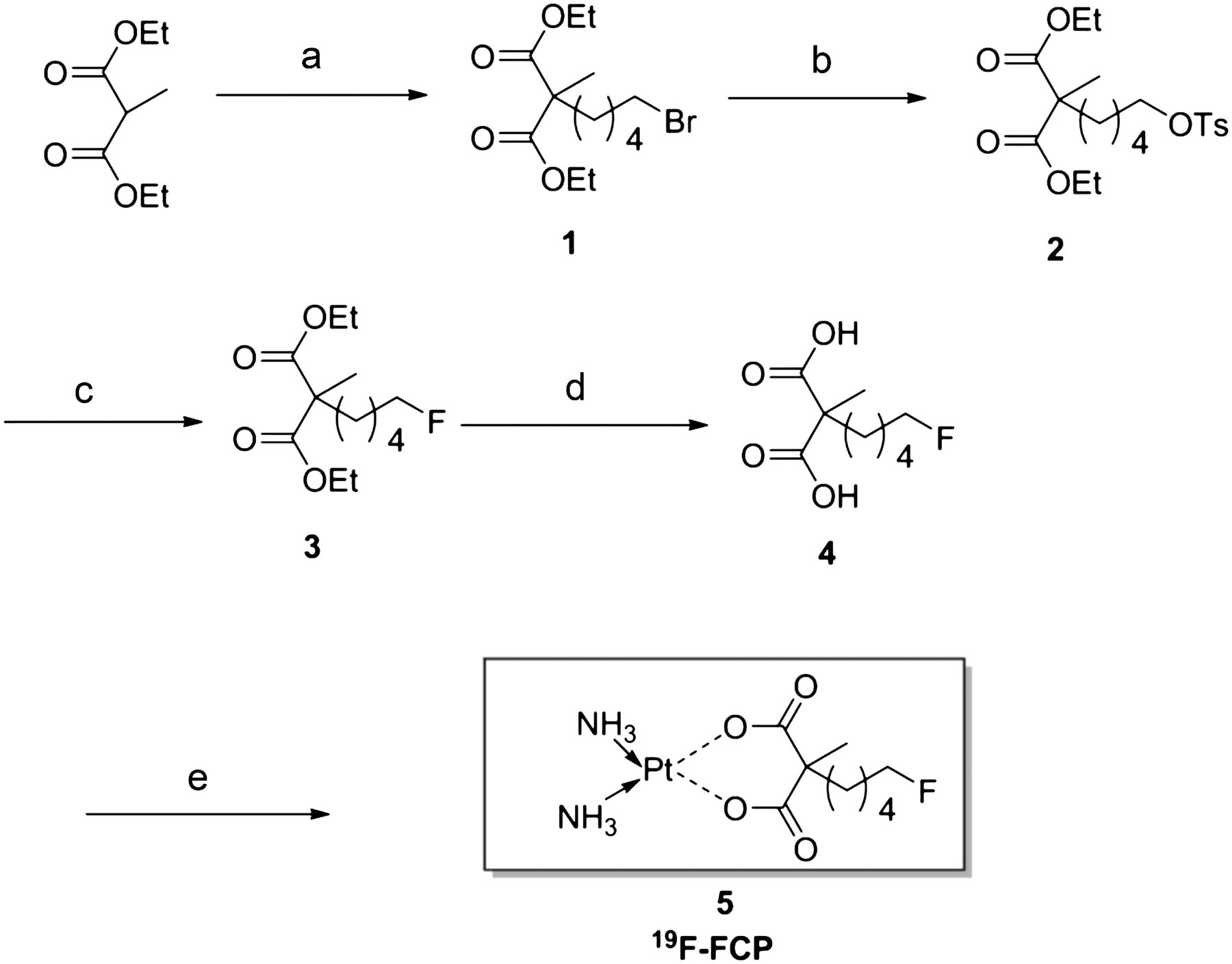
Reagents and conditions: 1,5-dibromopentane, NaH, DMF, 50°C, 12 h, 72% (a); silver tosylate, ACN, reflux, 12 h, 96% (b); TBAF, THF, 60°C, 3 h, 84% (c); LiOH/H2O THF/MeOH/H2O (6:3:1), rt, 24 h, 86% (d); and platinum aqua complex, rt, 2 d (e).
Cytotoxicity of 19F-FCP
On the basis of the cell viability data (Table 1), the IC50 values for the various platinum compounds showed that cisplatin reduced cell viability to a greater extent than did carboplatin and 19F-FCP; these results are consistent with the greater toxicity of cisplatin than of carboplatin (P < 0.05). The relative cytotoxicities were similar across cell lines, but 19F-FCP was slightly more potent than carboplatin. The difference was significant at 72 h in the A549, KB-3-1, and RWPE-1 cell lines (P < 0.005) but less so at 96 h in all cell lines (P < 0.05). The cytotoxic effect for all 3 compounds was time dependent. Increasing the incubation time from 72 h to 96 h reduced the IC50 values for all 3 compounds, indicating an increase in cytotoxicity over time.
IC50 Values for Cisplatin, Carboplatin, and 19F-FCP in Cancer Cell Lines
Radiosynthesis of 18F-FCP
Figure 2 represents the radiosynthesis of 18F-FCP (compound 8) in 3 steps: Kryptofix 222 (Merck)–mediated direct nucleophilic fluorination, NaOH base hydrolysis, and coordination with the platinum–aqua complex. The decay-corrected yield of the final product 180 min after the introduction of 18F-fluoride activity (isolated RCY of 18F-FCP) was 14.3% ± 3.8% (mean ± SD) (n = 3), and the radiochemical purity was more than 98%. Automation reduced the synthesis time by only 30 min; this result is plausible because the reaction steps were replicated from the manual procedures. The decay-corrected yield of the final product 150 min after the introduction of 18F-fluoride (isolated RCY of 18F-FCP) was 16.2 ± 0.95 (n = 4), and the radiochemical purity was more than 98%, as confirmed by HPLC. The retention time of 18F-FCP was 1.43 min (Fig. 3), which was distinguishable from that of free fluoride (retention time, 0.7–0.8 min) under these conditions. It was previously reported (21) that the retention of free fluoride is variable and depends on column characteristics and mobile phase constituents.

Reagents and conditions: K18F/K222, K2CO3, ACN, 110°C, 10 min (a); 3M NaOH, DCM/MeOH (9:1), 45°C, 30 min (b); 3M HCl (c); and platinum aqua complex, 75°C, 30 min (d).

HPLC analysis of radiosynthesis steps and purified final product. Sample was run on Waters 4-μm C18 150 × 3.9 mm column (25:75 acetonitrile:0.1% trifluoroacetic acid, at flow rate of 1 mL/min). (A) 18F-FPMA (retention time = 3.38 min). (B) Sample taken from platinum coordination reaction before purification. (C) Sample taken from 18F-FCP product (retention time = 1.43 min).
Biodistribution and in Vivo Stability Analysis
Figure 4 shows the biodistribution of 18F-FCP in female nude mice. The radiotracer demonstrated rapid blood clearance, with less than 2% of the radioactivity remaining in the circulation after 1 h. The blood half-life was calculated to be 16 min. Most organs showed low accumulation of the radiotracer, and radioactivity was predominantly cleared through the kidneys, with greater than 95% being cleared 1 h after injection.
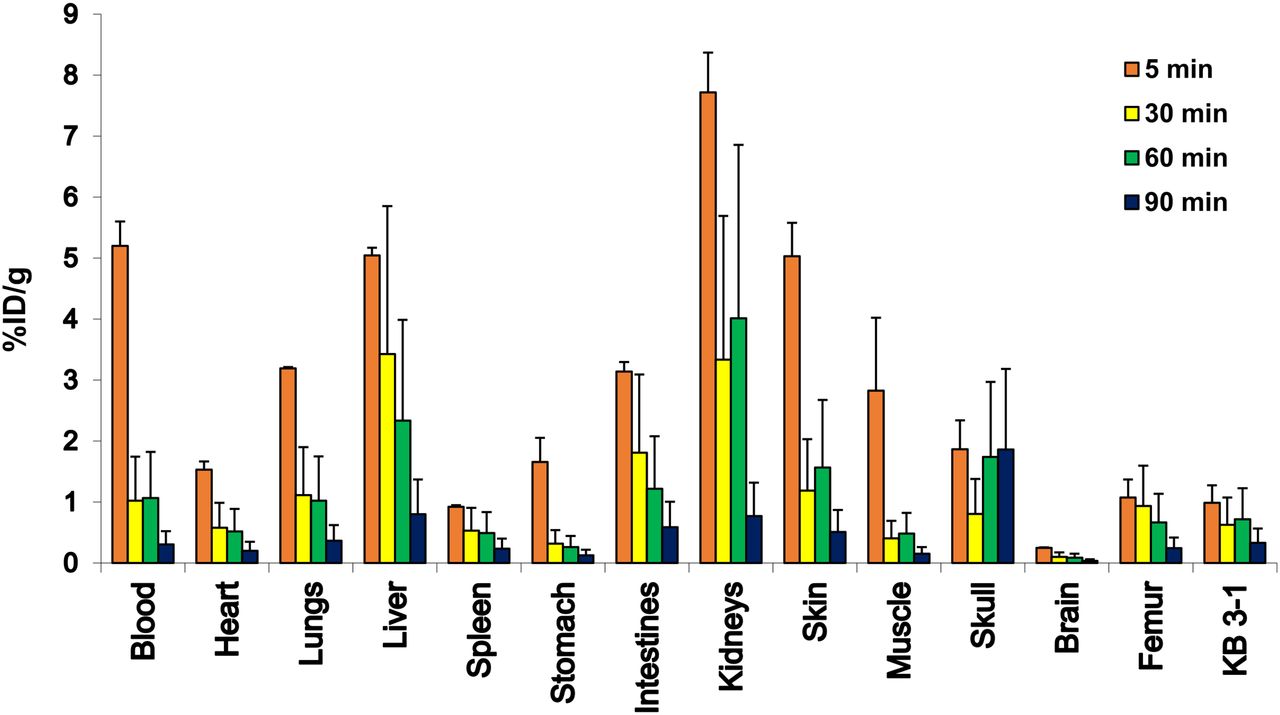
Biodistribution of 18F-FCP in KB-3-1 cervical tumor xenograft–bearing adult female nude mice (n = 3/time point) at 5, 30, 60, and 90 min after intravenous injection. Radiotracer uptake in %ID/g was determined by γ-counting.
The in vivo stability of 18F-FCP in plasma and urine was assessed during the first 60 min after intravenous administration in normal nude mice. The stability of 18F-FCP was determined by HPLC at 5, 30, 60, and 90 min after administration. The HPLC peak of the injected plasma samples appeared at the same retention time as the initial injected radiotracer (Fig. 5), demonstrating intact 18F-FCP 60 min after injection. No radioactivity was detected in 90-min samples; this result could be attributed to the fast physiologic clearance of the radiotracer from blood. The HPLC peak of the injected urine samples also appeared at the same retention time as the initial injected radiotracer, demonstrating intact 18F-FCP (Supplemental Fig. 1 at http://jnm.snmjournals.org).
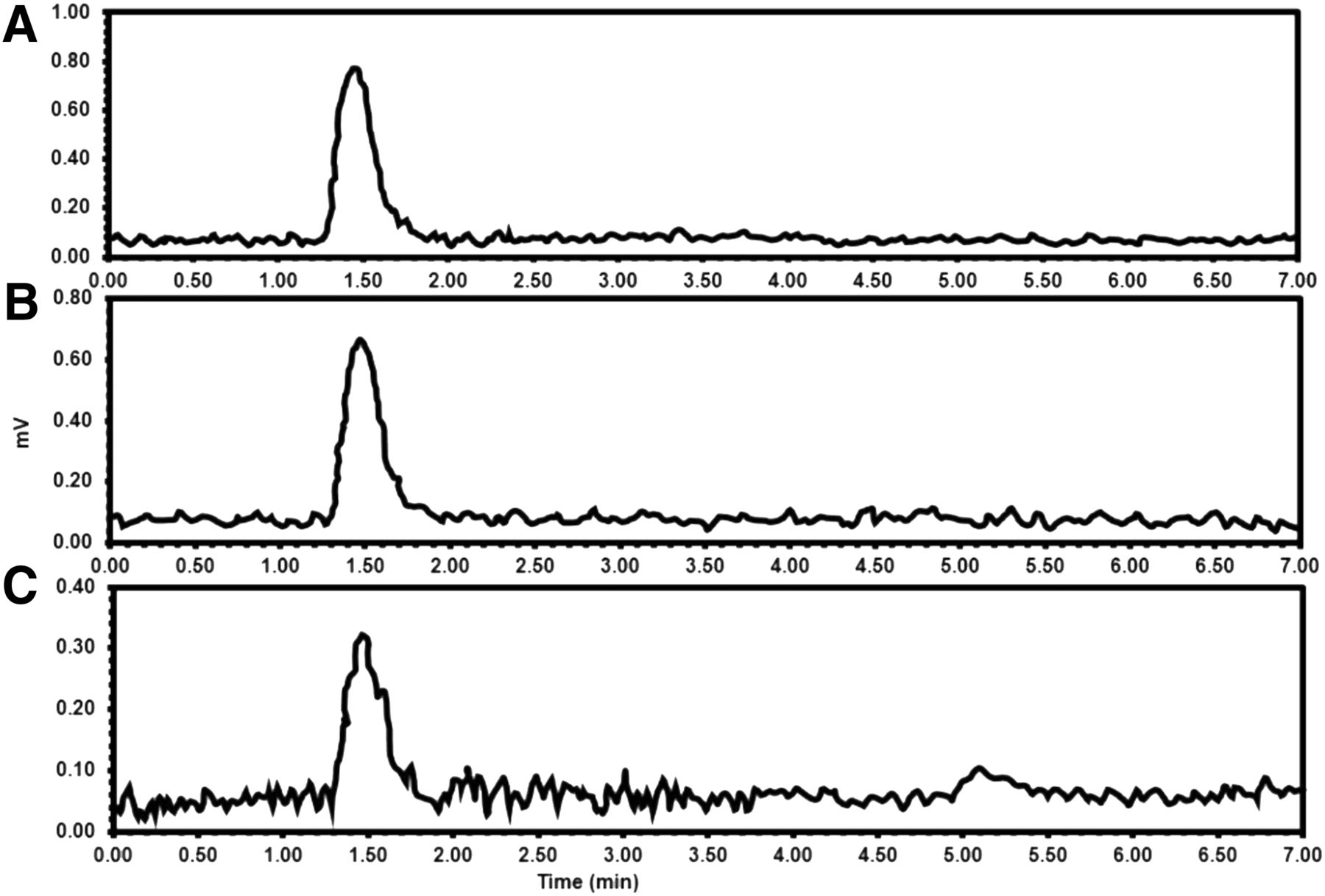
HPLC analysis of plasma samples run on Waters 4-μm C18 150 × 3.9 mm column (25:75 acetonitrile:0.1% trifluoroacetic acid, at flow rate of 1 mL/min) at different time points after intravenous injection of 18F-FCP. (A) 5 min. (B) 30 min. (C) 60 min.
PET Imaging
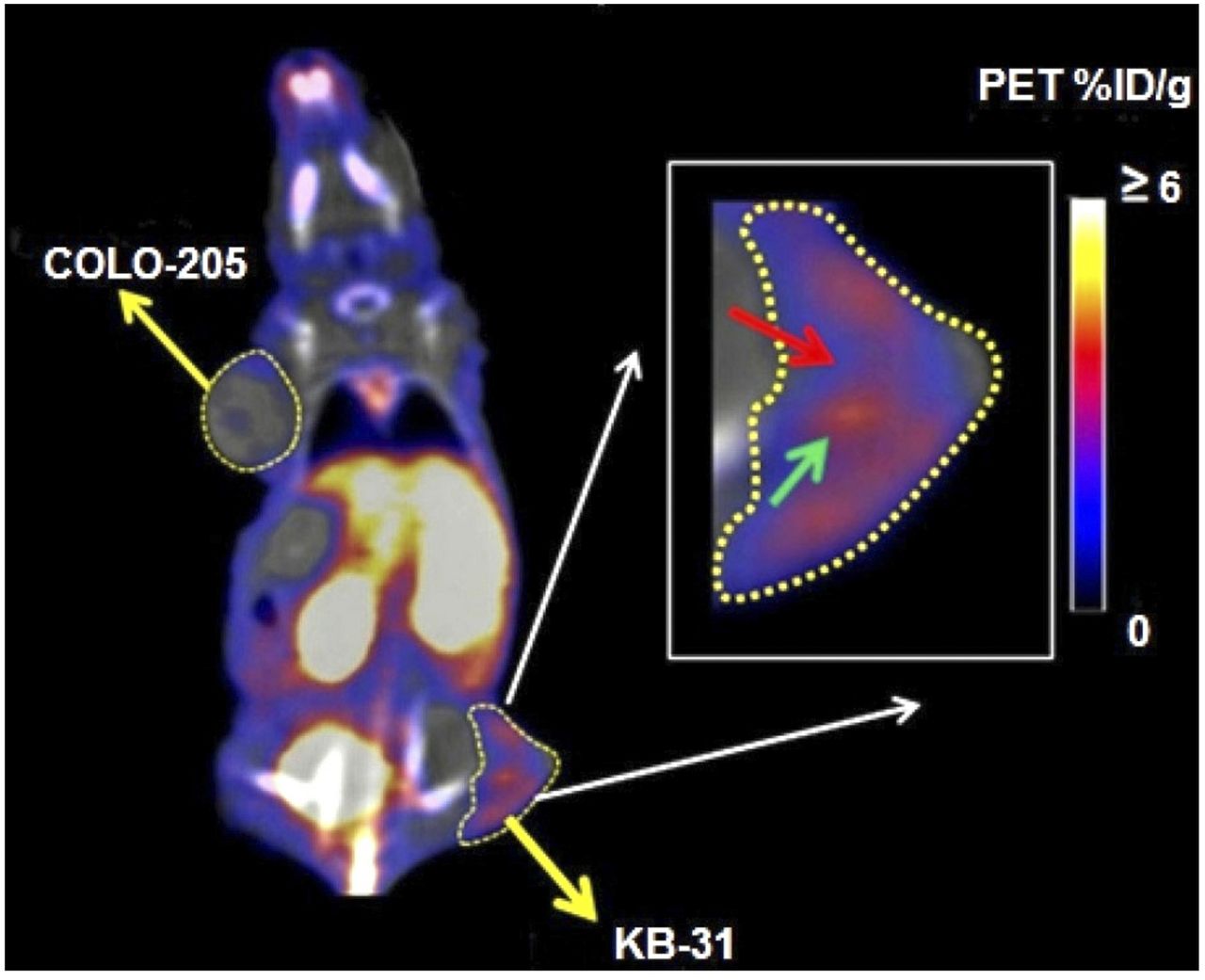
Dynamic images acquired for 90 min illustrated the full-body distribution of 18F-FCP (Fig. 6). 18F-FCP had the highest uptake in the kidneys. The accumulation of the radiotracer was also seen in the lungs and liver at early time points. High uptake in the kidneys seemed to reflect urinary excretion of the tracer, which could be attributed to the radioactivity present in the urine during the scan. Images showed the subsequent clearance of the radiotracer over time. Static images acquired 90 min after radiotracer administration illustrated the full-body distribution of 18F-FCP in tumor-bearing mice (Fig. 7). Major uptake was observed in the liver and abdominal region. Region-of-interest analysis showed that KB-3-1 tumors had higher uptake (2.6 %ID/g) than COLO-205 tumors (0.8 %ID/g). The inset image in Figure 7 also showed clear heterogeneous uptake in a KB-3-1 tumor.

Female nude mouse was injected intravenously with 5.5–6 MBq of 18F-FCP and imaged using Inveon PET/CT scanner (Siemens). (A) Selected slices of dynamic image data that were acquired for up to 90 min. (B) Time–activity curve data generated from dynamic image dataset showing time-dependent kinetics and tissue clearance.

18F-FCP (5.5–6 MBq) was injected intravenously via tail vein into nude mouse bearing KB-3-1 and COLO-205 xenografts. Imaging was performed 90 min after injection using Inveon PET/CT scanner, and static image was reconstructed and analyzed. Images showed obvious differences in uptake of 18F-FCP between KB-3-1 (2.6 %ID/g) and COLO-205 (0.8 %ID/g) tumors. Furthermore, heterogeneity of uptake was evident in KB-3-1 tumor (inset), showing regions of high (cyan arrow) and low (red arrow) 18F-FCP uptake. This observation is now being further investigated in our laboratory using carboplatin-sensitive (A2780) and carboplatin-resistant (A2780-CP20) tumor models.
DISCUSSION
Reduced intracellular drug accumulation has been identified as a major factor associated with drug resistance (10). A direct in vivo measure of a drug in tumors is therefore an important means of addressing this issue. PET imaging with a radiolabeled drug provides a noninvasive direct measure of intratumor drug distribution. This measure can then be related to the detection of any proteome alterations that may have contributed to the degree of regional drug uptake, retention, or efflux culminating in drug resistance.
Radiolabeled platinum drugs have been synthesized with platinum radioisotopes as radioactive drugs for imaging and therapy studies. Areberg et al. (11) studied the antitumor effect of radioactive cisplatin (191Pt) in tumor-bearing nude mice and demonstrated that 191Pt-cisplatin was more effective than unlabeled cisplatin in retarding tumor growth. Patient studies by Areberg et al. (22,23) demonstrated the use of cisplatin radiolabeled with 191Pt, 193mPt, or 195mPt to visualize the uptake of platinum in tumor and tissues noninvasively after cisplatin treatment. Dowell et al. (13) used 195mPt-labeled cisplatin and carboplatin to estimate, through imaging, the amount of drug at the tumor site and in selected organs. However, the production and supply of platinum radionuclides were limited and costly because of the need to use enriched platinum isotopes as target material and the logistics of radiosynthesis (13). These factors did not facilitate wide applications of platinum-radiolabeled drugs. Positron-emitting radionuclide–labeled drugs—specifically, 18F-labeled drugs—represent a feasible strategy for developing PET imaging that can be clinically applied to evaluate the biodistribution of drugs.
In this work, we developed FCP as a drug analog of carboplatin and 18F-FCP as a companion imaging diagnostic agent. Such development represents an advance over previous work using platinum or nitrogen radionuclides for labeling and imaging. We demonstrated that the nonradioactive FCP analog was at least as potent as the parent drug in various cancer cell lines in vitro. It remains to be seen whether the in vivo antitumor effects of the analog are as good as or better than those of carboplatin. Therapeutic studies to address this notion are under way, and the findings will be reported in a separate article.
Using PET imaging, we showed that the 18F-FCP biodistribution profile was like the previously reported overall pharmacokinetic data for carboplatin (24–26). The radiotracer was stable in plasma and urine at 60 min after administration (Fig. 5). A small peak at about 5.3 min in the plasma HPLC chromatogram could be attributed to a metabolite, but the presence or absence of this metabolite needs to be confirmed in future studies. 18F-FCP exhibited distribution throughout the body, and the blood clearance pattern followed monoexponential blood kinetics. The blood half-life of 18F-FCP was calculated to be 16 min, similar to the reported value for carboplatin (26).
After intravenous injection of 18F-FCP in normal nude mice, quantification of the dynamic images up to 90 min after injection showed the highest activity in the kidneys, bladder, and urine. The accumulation of 18F-FCP was also recorded in the liver and lungs at early time points, with subsequent clearance over time; these results are consistent with the findings of Ginos et al. (15) for 13N-labeled cisplatin. Ex vivo biodistribution data corresponded to the imaging results, with a major accumulation of the radiotracer in the kidneys and liver (Fig. 4).
The data generated here showed that 18F-FCP PET/CT can be used to noninvasively assess the drug distribution profile in different tumor types. 18F-FCP uptake in KB-3-1 tumors was much higher than that in COLO-205 tumors. Furthermore, PET imaging revealed that the uptake in KB-3-1 tumors was heterogeneous, with significant differences in uptake in different tumor regions. In this work, we demonstrated that 18F-FCP PET/CT is a unique tool that can measure heterogeneous intratumor drug distribution. When combined with proteomic analysis, this information may provide a better understanding of the relationships among intratumor drug distribution, proteome alterations in distinct tumor regions, drug–tumor interactions, mechanisms of resistance, and tumor recurrence. 18F-FCP can also depict the kinetics of drug efflux, which is related to drug resistance.
Although many anticancer drugs are widely used in clinics, many questions about how individual patient tumors interact with drugs and how much actual drug is retained in different tumors remain unanswered—leading to the question of why patients with the same type of tumor respond differently to the same drug treatment. PET using radiolabeled drugs is the only imaging method that may further understanding of the clinical behavior of drug therapy and contribute to better guidance of treatment planning in individual patients.
CONCLUSION
The synthesis of a fluorinated analog of carboplatin was developed. The compound retained the antitumor activity of the parent drug. The feasibility of producing 18F-FCP for PET imaging was demonstrated. The pharmacokinetics of 18F-FCP mirrored those of carboplatin, and the profile showed major accumulation in the kidneys and liver. This PET tool enabled the imaging of intratumor drug distribution and the detection of heterogeneous retention within the tumor. 18F-FCP PET is a novel tool that can be used to guide platinum therapy with the aim of enhancing the overall efficacy of platinum-based chemotherapy on an individual-patient basis.
DISCLOSURE
This work was supported by Virginia Commonwealth University School of Medicine. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Bob Ylimaki, IBA Molecular, for 18F-fluoride production and Michael D’Cona for technical assistance.
Footnotes
↵* Contributed equally to this work.
Published online Jul. 20, 2017.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication February 27, 2017.
- Accepted for publication June 27, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.