


Abstract
Lung inflammatory diseases contribute significantly to the socioeconomic burden of disease. Yet very few new, effective therapies for respiratory disease have been approved for use. A major contributing factor is the lack of biomarkers that can accurately quantify the lung inflammatory burden and can be used to understand the contribution of lung inflammation to loss in lung function. Molecular imaging approaches can detect and quantify the recruitment and activation of specific immune cells in lung inflammation. We review the clinical techniques used to image lung inflammation, provide an overview of clinical and emerging PET techniques for quantifying lung inflammation, and discuss potential clinical applications.
Inflammatory lung diseases, with their high morbidity and mortality, pose a significant socioeconomic burden on society. Chronic lower respiratory diseases, which include chronic obstructive pulmonary disease (COPD) and asthma, are the third leading cause of mortality in the United States and contribute significantly to societal health-care costs (1,2). Despite this, the number of new first-in-class drugs being approved for pulmonary indications is disappointingly low, with these drugs incurring the highest research and development costs (3,4). Pulmonary function testing is the mainstay of determining lung disease severity and is the most frequently used primary endpoint for clinical trials in lung disease. However, pulmonary function tests do not directly measure the cause of lung dysfunction and often take at least a year or more to change. Thus, the long periods of observation needed to detect a drug effect increase the costs associated with respiratory drug development.
The lack of quantitative biomarkers that accurately reflect lung disease severity has contributed in part to these higher drug-development costs. Extensive efforts have focused on developing blood- or lung tissue–based molecular biomarkers obtained by induced sputum or bronchoscopy (5–7). However, invasive bronchoscopic procedures carry higher complication risks for patients, and peripheral blood samples do not necessarily reflect lung-specific processes. Induced sputum, although noninvasive, samples only the central airways and not the distal alveolar spaces. Therefore, noninvasive, quantitative imaging approaches that can directly measure molecular processes related to lung disease severity could overcome some of these limitations. Although all diagnostic imaging modalities can detect the presence of inflammation, these signals are frequently not specific for inflammatory processes. Therefore, molecular imaging techniques that provide inflammation-specific measures could complement the information from existing diagnostic imaging modalities and potentially facilitate basic investigations that can aid in the drug-development process.
This review focuses on the molecular and nuclear medicine imaging approaches that have been used to image lung parenchymal inflammation in humans and provides a brief evaluation of the clinical manifestations of lung inflammation on CT, the clinical gold standard method for characterizing lung disease. Although many novel, targeted inflammation tracers in preclinical studies have been reported that hold great promise, these are not reviewed here. Disease-specific data are presented for COPD, asthma, acute respiratory distress syndrome (ARDS), and interstitial lung diseases (ILDs). In addition, data describing the use of PET with 18F-FDG for measuring lung inflammation in cystic fibrosis have been summarized recently and are, therefore, not reviewed here (8).
METHODS FOR IMAGING LUNG INFLAMMATION
CT Imaging of Lung Inflammation
Pulmonary inflammation can have varying manifestations depending on the affected compartment, including the airways, vasculature, or interstitium. Although the density changes in the lungs themselves are not specific for inflammation, characteristic patterns can be helpful in distinguishing inflammation from other processes. Nemec et al. recently described common CT findings and imaging clues for noninfectious inflammatory lung disease, summarized in part below (9).
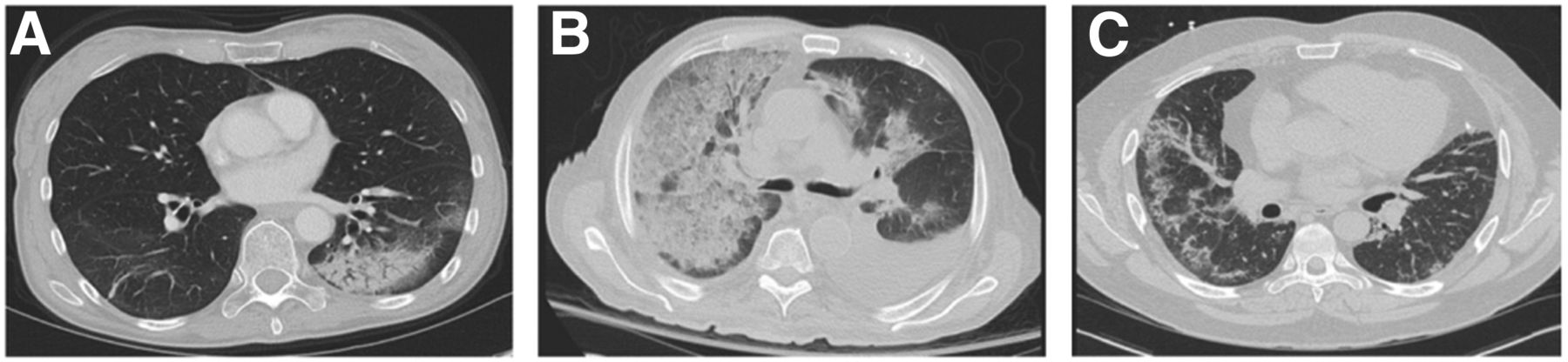
Airway-predominant diseases frequently involve infiltration of the alveoli and alveolar ducts with inflammatory cells, debris, or fluid, as seen in bacterial and organizing pneumonia (Fig. 1A), as well as aspiration of secretions or food debris. These filled airways cause airspace opacities that can become more confluent and appear as consolidation, with patent airways within these regions appearing as air bronchograms. Characteristic involvement of the small respiratory bronchioles in diseases such as Langerhans cell histiocytosis or smoking-related lung diseases, with pigmented macrophages in respiratory bronchiolitis-associated ILD and desquamative interstitial pneumonia, result in centrilobular and peribronchiolar ground-glass opacities and nodules with bronchial wall thickening. The alveolar spaces can also fill with fluid through leaky capillaries such as in ARDS, which is the clinical manifestation of diffuse alveolar damage caused by direct or indirect lung injury. Such leakage can mimic pulmonary edema or pulmonary hemorrhage (Fig. 1B).
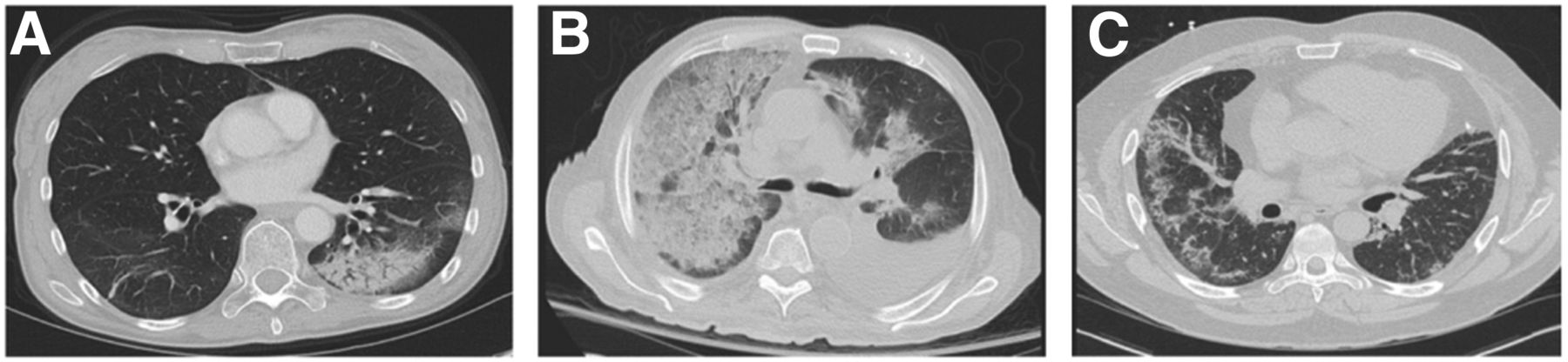
CT manifestations of inflammatory lung disease. (A) Organizing pneumonia is characterized by left lower lobe focal consolidation with peripheral ground-glass opacity and air bronchograms. Chronic eosinophilic pneumonia and adenocarcinoma in situ can appear similar. (B) Diffuse, confluent ground-glass opacity and interlobular septal thickening with a “crazy paving” pattern in right lung, with lesser involvement of left lung can reflect multifocal pneumonia, atypical edema pattern, or pulmonary parenchymal hemorrhage. Pleural effusions are also present bilaterally. (C) Nonspecific interstitial pneumonia exhibits predominantly peripheral ground-glass opacity with fine reticulation and subpleural sparring in lower lobes, right greater than left. Absence of honeycombing makes usual interstitial pneumonia unlikely.
ILDs are characterized by inflammation of the interstitial spaces in addition to fibrotic changes with varying degrees of airway involvement. In pulmonary fibrosis, such as usual interstitial pneumonia, interstitial inflammation—with or without concomitant fibrosis—results in temporally and spatially heterogeneous reticular subpleural opacities, bronchial wall thickening and dilatation, architectural distortion, and peripheral cystic change or honeycombing, leading to lung volume loss and a restrictive physiology. Nonspecific interstitial pneumonia (Fig. 1C) is characterized more by ground-glass opacity related to the symmetric subpleural interstitial inflammation that is more homogeneous in lung distribution. Interestingly, a fibrotic form of nonspecific interstitial pneumonia can demonstrate several features similar to usual interstitial pneumonia. A recent review of the radiographic manifestations of the spectrum of ILDs is provided by Mueller-Mang et al. (10).
In summary, lung inflammation manifests itself in recognizable and sometimes predictable ways. These methods can be used to differentiate various causes of lung inflammation, but as indicated above, few manifestations are specific for inflammation.
18F-FDG
Although 18F-FDG uptake itself is not a specific indicator of inflammation, inflammatory processes clearly demonstrate increased 18F-FDG uptake, as frequently observed in oncology staging 18F-FDG PET scans. Initial studies using 18F-FDG to image lung inflammation focused on measuring neutrophilic recruitment (11,12). However, 18F-FDG also accumulates in macrophages (13), lymphocytes (14,15), and eosinophils (16,17). The increased 18F-FDG uptake in these activated cell types supports various functions related to their specific immune responses, including cytokine-induced activation of neutrophils and eosinophils (18,19), antigen receptor–mediated activation of lymphocytes (15,20), and polarization state changes in macrophages (21). Structural cells within the lungs also increase glucose utilization during inflammation (22,23). Therefore, increased lung 18F-FDG uptake most likely represents a measure of the lungs’ integrated inflammatory response in the context of lung inflammatory diseases.
67Ga-Citrate and 68Ga-Citrate
67Ga is a group IIIb transition metal similar to ferric ion that binds to multiple iron-binding molecules, including transferrin, lactoferrin, ferritin, and siderophores, and has most frequently been used to detect infections. However, 67Ga localizes in inflammatory processes as well, most likely from binding to lactoferrin within neutrophils (24), though 67Ga localization to sites of infection and inflammation has been observed in patients with no circulating leukocytes. 68Ga-citrate, the positron-emitting isotope, has characteristics similar to 67Ga-citrate for imaging infection, with the added advantages inherent to PET imaging, including improved spatial resolution and the ability to quantify uptake (25). Several 68Ga-labeled peptides are being evaluated for imaging inflammation in preclinical models that have recently been reviewed (26).
Radiolabeled Leukocytes
Leukocytes labeled in vitro with 111In-oxine or 99mTc-exametazime are commonly used in clinical practice to identify infection. 18F-FDG–labeled leukocytes and 111In-tropolonate in neutrophils and eosinophils have been used for human research investigations (27,28). With all of these approaches, the leukocytes are handled in a manner that avoids activating the leukocytes, thus preserving their ability to respond to inflammatory signals in the body for localization. Recently published data on radiolabeled neutrophils have shown that unprimed neutrophils and eosinophils transit quickly through the pulmonary circulation without significant first-pass retention, whereas primed neutrophils are retained significantly in the lungs of healthy volunteers (27,29). For radiolabeled eosinophils, the intravascular residence time is 25.2 h, with margination into the liver, spleen, and bone marrow.
Somatostatin Receptor (SSTR) Imaging
SSTR imaging, most commonly used for neuroendocrine tumor imaging, is being increasingly used to study inflammatory lung disease. 111In-pentetreotide, which has been used for decades for imaging SSTRs in neuroendocrine tumors, also images granulomatous inflammatory lesions (30). 68Ga-DOTANOC, another SSTR imaging agent, has a high affinity for SSTRs 2, 3, and 5, whereas 68Ga-DOTATATE has the highest affinity for SSTRs 2, 4, and 5 (31). Variable expression of all 5 SSTR isoforms has been found on human lymphocytes, monocytes, and macrophages but not on neutrophils (32,33), explaining why inflammatory lesions are frequently seen on SSTR scintigraphy (32,33). Fibroblasts and endothelial cells from tissue samples from idiopathic pulmonary fibrosis (IPF) patients also express SSTRs, which appear to promote fibroblast activity (34). Thus, SSTR expression in lung disease likely reflects a combination of these processes.
Translocator Protein Imaging
The 18-kDa translocator protein, previously known as the peripheral benzodiazepine receptor, is upregulated in activated microglia, a macrophage lineage cell in the brain (35). The radiolabeled isoquinoline 11C-PK11195 binds specifically to translocator protein, and its binding correlates with macrophage recruitment in rabbit lungs after silica particle challenge (36). Despite this potential specificity for macrophages, translocator protein expression can be found on resting peripheral blood monocytes and neutrophils (37) that increases with endotoxin-induced inflammation, leading to increased 11C-PK11195 uptake in the lungs (38).
DISEASE-SPECIFIC INVESTIGATIONS
COPD and Asthma
COPD is projected to become one of the leading causes of mortality worldwide (3). COPD is a heterogeneous disease that is defined by progressive, irreversible airflow limitation. The lungs’ inflammatory response to the inhalation of particles and gases, particularly from cigarette smoke, is thought to cause lung tissue injury and destruction, thus leading to the development of COPD (39,40). Evidence suggest that macrophages drive the development of COPD, leading to increased neutrophil recruitment in the airways (41,42). Neutrophilia in COPD can persist even after smoking cessation, is associated with areas of greater air trapping and airflow limitation, and correlates with disease severity (43–47). Given the hypothesized link between inflammation and lung tissue destruction, imaging markers that can quantify the lung inflammatory burden in COPD could be more useful in predicting treatment responses to antiinflammatory interventions than changes in forced expiratory volume 1, which is often the primary endpoint for COPD drug trials.
Asthma affects 300 million people worldwide, causing significant morbidity and leading to high socioeconomic costs (48). Eosinophils are the most common and numerous cell type present in the airways of patients with mild asthma; therefore, asthma has long been regarded an allergic disease caused by T2 lymphocytes. Large cohort analyses, however, have revealed additional clinical and functional phenotypes beyond the classic phenotype of allergic, eosinophilic, steroid-sensitive asthma (49). As many COPD patients also have evidence of asthma, asthma–COPD overlap syndrome is becoming increasingly recognized as an independent entity (50). Because the inflammatory phenotype in both COPD and asthma may help guide therapy choices, noninvasive molecular imaging approaches that can differentiate such phenotypes could aid in selecting patients for targeted therapies.
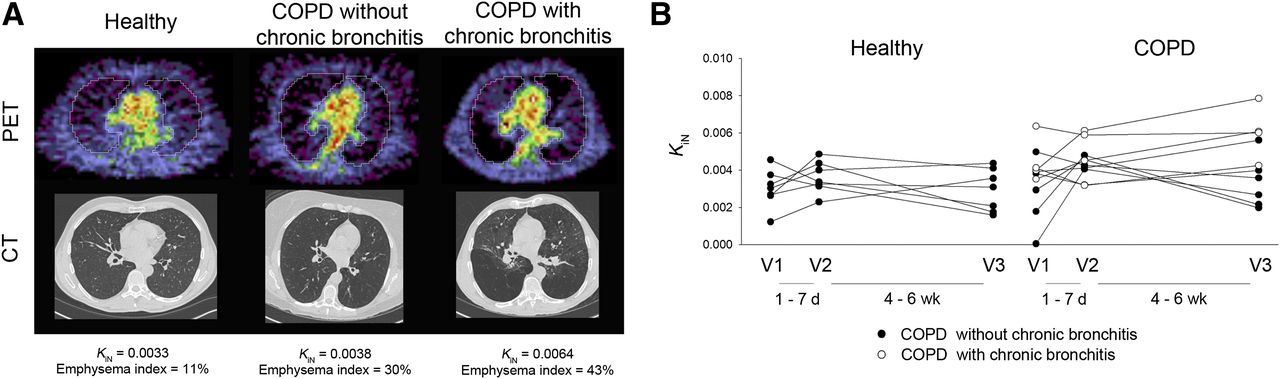
Several small studies have investigated the use of 18F-FDG for monitoring inflammation in both COPD and asthma. COPD studies have focused on quantifying the whole-lung inflammatory burden in COPD participants and healthy volunteers with 18F-FDG (51–53). The uptake was quantified using the intercept-normalized Patlak influx constant, Ki, to account for differences in lung density as originally described by Jones et al. (54). These studies, with a combined 26 COPD participants and 24 age-matched healthy volunteers, demonstrated a range of increased intercept-normalized Ki values in the COPD group when compared with that of the healthy volunteers across all studies. Subramanian et al. demonstrated in an additional 10 participants with α1-antitrypsin deficiency that the intercept-normalized Ki was unexpectedly not increased in this group relative to healthy volunteers (53). Preliminary results from our own institution, from a protocol approved by our Institutional Review Board and with written consent from participants, further suggest that patients with chronic bronchitis symptomatology have increased 18F-FDG uptake compared with those without such symptoms (Fig. 2; data published in abstract form from Chen et al. (52)), in line with the known increased lung inflammation that occurs with chronic bronchitis (55). Together, the available data suggest that 18F-FDG uptake may be useful in distinguishing inflammatory phenotypes within COPD.
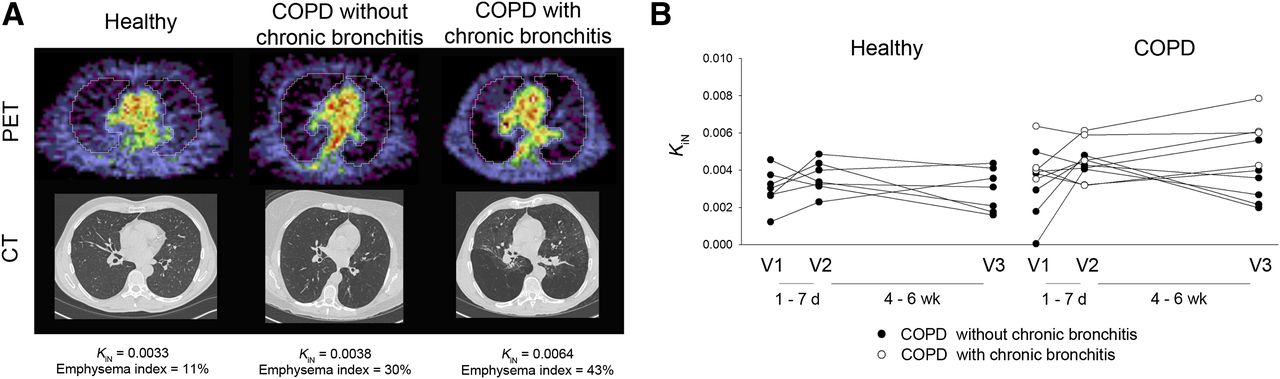
PET/CT images in COPD and age-matched healthy volunteers in pilot study assessing reproducibility of 18F-FDG uptake in COPD lungs. (A) Representative PET and CT images from healthy volunteer and volunteers with COPD without or with chronic bronchitis symptoms. PET images shown are sum of last 5 min of data from 60-min dynamic acquisition. COPD participant without chronic bronchitis had emphysema diffusely throughout both lungs on CT. COPD participant with chronic bronchitis had more heterogeneously distributed emphysema. Units of intercept-normalized Ki (KiN) = min−1. (B) All data points in healthy volunteers (n = 7) and COPD participants without (n = 6) and with chronic bronchitis symptoms (n = 4). (Data reported in abstract form in Chen et al. (52)).
In asthma, 18F-FDG has been used to image the effects of allergen challenges or exacerbations. Two studies have shown that 18F-FDG uptake, quantified as the Patlak Ki, increased with eosinophilic inflammation induced by segmental allergen challenge in patients with atopic asthma (16,17) but not with nebulized allergen (16). Viral-induced asthma exacerbations also increase pulmonary 18F-FDG uptake, though whether this specifically is related to eosinophilic inflammation is unclear (56, published in abstract form). Taken together, these data support the potential for using 18F-FDG to assess for changes in the inflammatory burden in asthma.
The translocator protein–targeted tracer 11C-PK11195 has also been used to image the presence of macrophages in asthma and COPD and compared with 18F-FDG (51). In this study, 11C-PK11195 was increased in both patients with COPD and patients with asthma compared with healthy, age-matched controls (51). This increase in both groups was in contrast to the 18F-FDG signal, which was increased only in the COPD group. Although the presence of increased macrophage numbers has long been known in COPD, emerging evidence points to a larger role for macrophages in asthma than originally appreciated (57). Therefore, the data from this pilot study suggest that 11C-PK11195 may be useful in imaging the macrophage burden in the lungs.
ARDS
ARDS continues to cause significant morbidity and mortality once it develops (58). Although its pathogenesis remains ill-defined, neutrophilic accumulation and activation are prominent and universal features of ARDS and acute lung injury (59). ARDS can be caused by a variety of pathologic conditions, including sepsis, trauma, transfusion of blood products, and ventilator-induced lung injury (60). The common proposed mechanism for ARDS is the persistence of activated neutrophils that release cytokines that destroy lung tissue (61). However, the exact mechanisms by which ARDS develops have yet to be elucidated.
18F-FDG has been used extensively to study the kinetics of neutrophil activation and recruitment in ARDS in preclinical models (12,62). Several small studies have further evaluated 18F-FDG PET in patients with ARDS or at risk of developing ARDS. 18F-FDG uptake is increased not only in areas of infiltrate or consolidation in patients with ARDS but also in areas of normal-appearing lung (63). This uptake most likely reflects areas at risk of being further injured by ventilation as these more compliant areas are subject to increased stretch, leading to neutrophil recruitment and subsequent lung injury (64). This finding may also explain why low tidal volume ventilation strategies lead to a significant reduction in ARDS mortality because such strategies will reduce the distention of the more normal areas of the lungs (65). Radiolabeled primed and unprimed neutrophils (primed neutrophils being in an enhanced state for responding to inflammatory stimuli) have also been used to demonstrate that normal lungs in healthy volunteers can deprime neutrophils, allowing them to leave the pulmonary circulation (29). In patients with ARDS, on the other hand, this depriming step fails to occur, leading to increased retention of neutrophils that likely contributes to the development of ARDS. These studies demonstrate the potential for using molecular and targeted cell imaging to better understand the contribution of neutrophilic inflammation to ARDS.
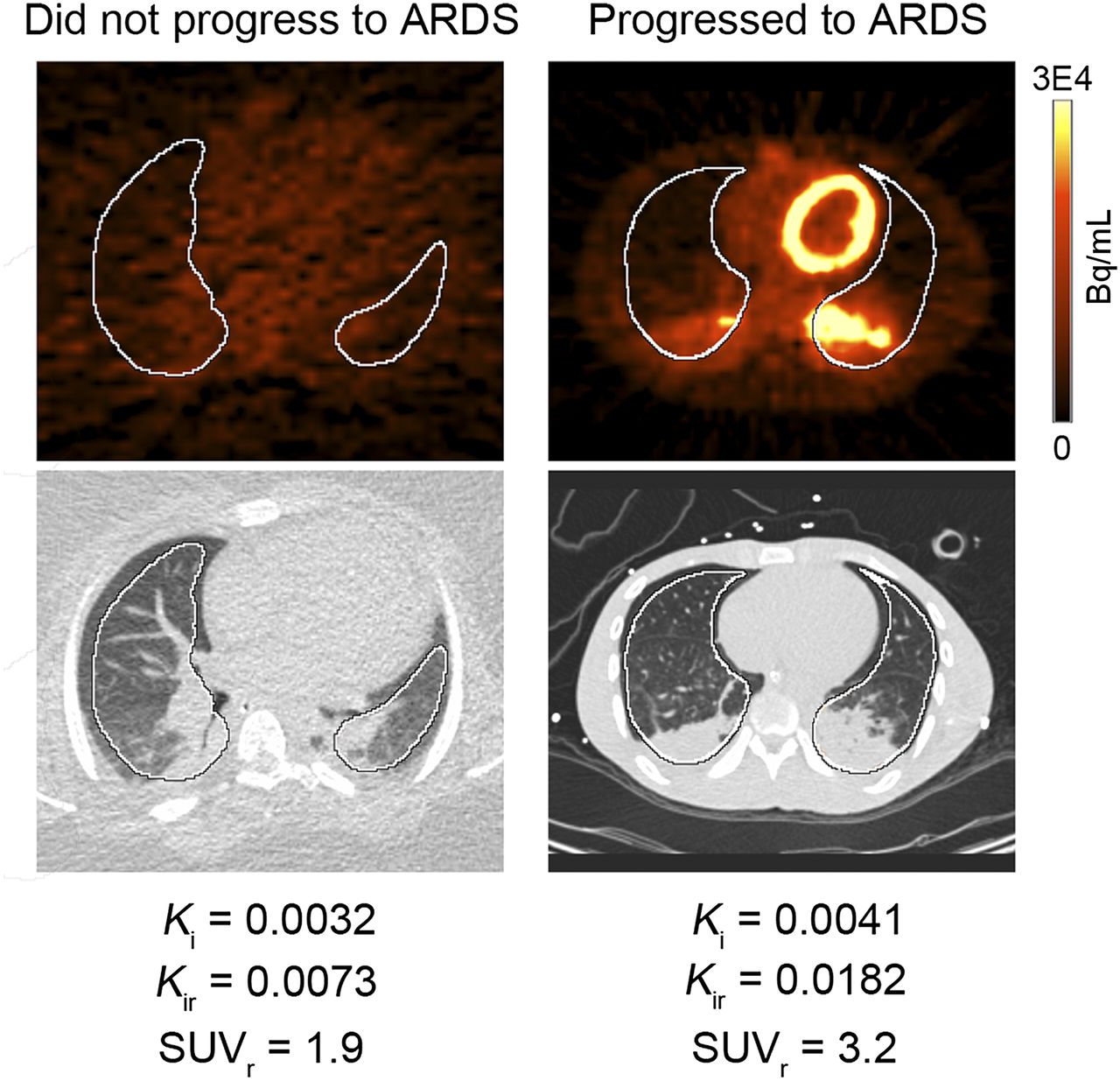
Predicting which ventilated patients will progress to develop ARDS remains challenging. Another small study demonstrated that increased 18F-FDG uptake precedes the development of clinically defined ARDS and therefore may be a useful method for assessing ARDS risk (66). Our own pilot data corroborated the findings from this pilot study, collected under a protocol approved by our Institutional Review Board and with written consent from legal authorized representatives for the patient. In this study, 1 patient with a high lung injury predictive score, a validated measure for predicting acute lung injury risk (67), had increased 18F-FDG uptake, and went on to develop clinical ARDS (Fig. 3). Four others with low lung injury predictive scores had no increased lung 18F-FDG and were successfully weaned off of ventilator support without further incident. Newer tracers such as 18F(+/−)NOS, which targets the inducible nitric oxide synthase (iNOS) and appears specific for iNOS expression in endotoxin-induced lung inflammation in healthy volunteers (68), may also be helpful in identifying early lung inflammation that increases the risk of progression to ARDS. Prospective studies will be needed to validate these initial results and assess the contribution of new, inflammation-targeted tracers for predicting ARDS development.

PET and CT images from 2 patients obtained within 24 h of being placed on ventilator. Images on left demonstrate no abnormal 18F-FDG uptake within lungs. Images on right demonstrate visibly increased 18F-FDG uptake in both lungs. SUVs of whole lung were not different between patients, demonstrating relative insensitivity of SUV for quantifying whole-lung inflammation compared with whole-lung Ki, which is higher in patient on right. Kir and SUVr are Ki and SUV in 50% of pixels with highest activity within each region of interest (in white). Images obtained in collaboration with Brian Fuller, MD, Washington University School of Medicine.
ILDs
The ILDs, or idiopathic interstitial pneumonias, comprise a heterogeneous group of diffuse parenchymal diseases characterized by chronic inflammation and fibrosis. IPF is the most common form. ILDs can also develop from multiple other causes, such as connective tissue disorders, sarcoidosis, and certain drug exposures. Chronic inflammation had long been thought to cause the fibrotic changes and structural destruction in ILDs. Therefore, 67Ga-citrate scintigraphy was used in the past to assess the inflammatory activity in these diseases and is still used for this purpose in some centers. Increased 67Ga-citrate lung uptake is frequently seen in patients with ILDs and has been used to measure the antiinflammatory effect of steroids. However, reduced 67Ga uptake in response to steroid therapy does not correlate with clinical improvement in patients with IPF (69). These results are in line with multiple studies demonstrating the ineffectiveness of steroids and immunosuppression for mitigating ILD progression (70). Therefore, inflammation is less likely to be a causative factor in ILDs.
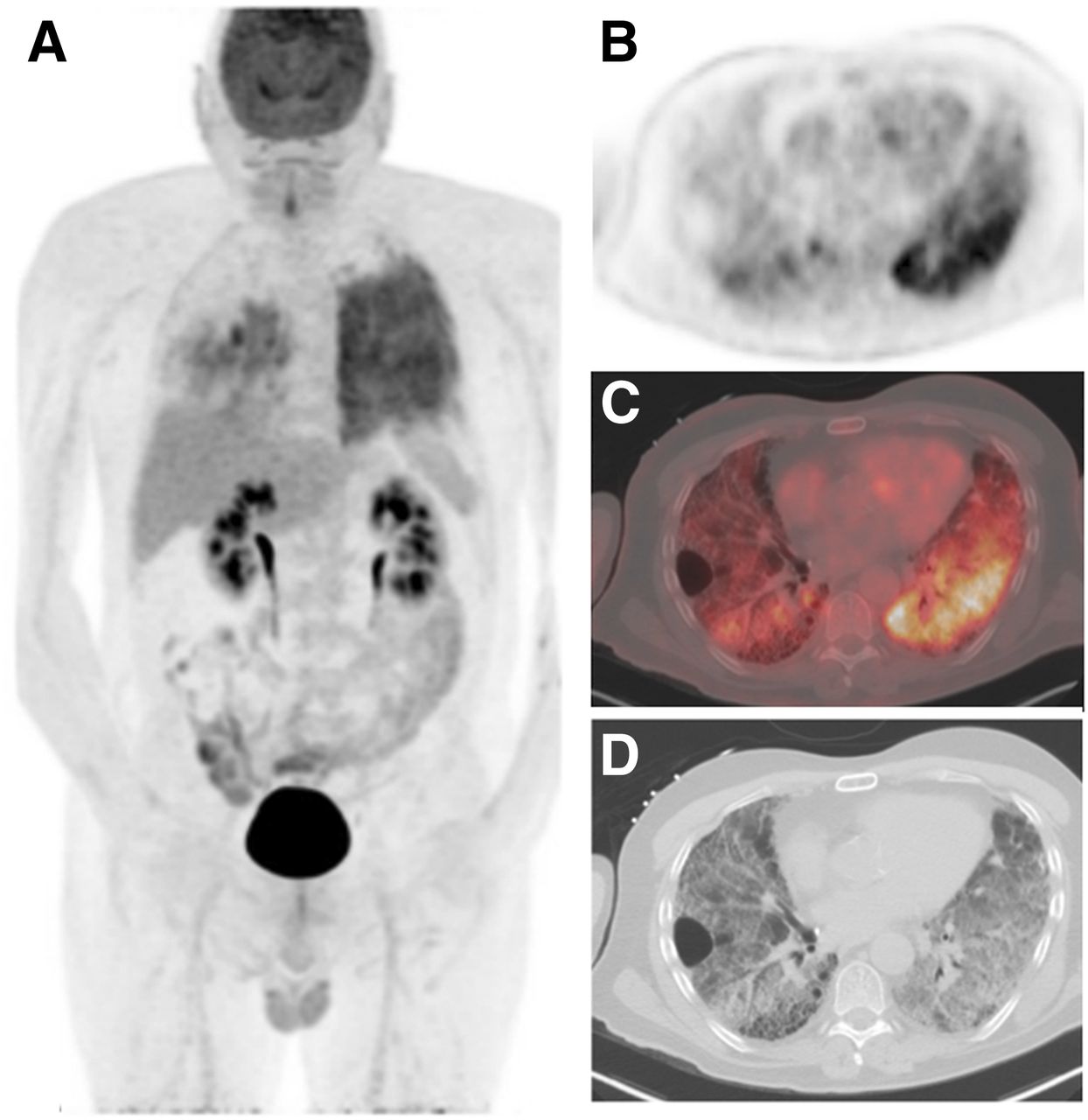
Growing evidence suggests that the cause of fibrosis is an initial epithelial injury coupled with an inadequate wound-repair response that leads to fibroblast proliferation and parenchymal destruction in ILDs (71). 18F-FDG uptake is frequently seen in ILDs (Figs. 4 and 5). The more intense 18F-FDG uptake in areas of honeycombing, in light of data from IPF lung tissue samples showing increased expression of glucose metabolism genes, suggests that increased 18F-FDG uptake in IPF may reflect an active fibrotic remodeling process (72,73). Although the pattern and intensity of 18F-FDG uptake cannot distinguish between ILD subtypes (74), differences in 18F-FDG retention using dual-time-point imaging predicted poor survival in a small study of 50 IPF patients (75). When accounting for air and blood in regional lung 18F-FDG uptake, however, fibrotic areas had lower uptake than more normal-appearing areas (76). Whether these quantification corrections more accurately represent the underlying biology is unclear in the absence of direct regional validation. Despite this, the data available suggest that 18F-FDG uptake may reflect fibroblast activity in addition to inflammatory cell activity and thus serve as a marker of treatment response to antifibrotic treatments. Further prospective studies will be needed to determine the clinical utility of 18F-FDG PET imaging in ILDs.

High-resolution CT and 18F-FDG PET/CT in usual interstitial pneumonia. (A) Axial high-resolution CT image of chest demonstrates honeycombing, fibrosis, and traction bronchiectasis, most evident in lingula. There is also emphysema. (B and C) Axial attenuation-corrected PET and fused PET/CT images demonstrate mild, diffusely increased 18F-FDG uptake throughout lung parenchy3ma, with more focally increased 18F-FDG uptake in area of honeycombing and fibrosis in lingula.

High-resolution CT and 18F-FDG PET/CT in ILD. (A) Coronal maximum-intensity-projection PET image demonstrates diffusely increased 18F-FDG uptake in left lung, with lesser involvement in right lung. (B and C) Axial PET and PET/CT fused images shows heterogeneously increased 18F-FDG uptake, suggesting active inflammation or possible active fibrosis. (D) CT images demonstrate regions of diffuse ground-glass opacity with honeycombing, interlobular septal thickening, and bronchiectasis, more pronounced on right, in areas of increased 18F-FDG uptake. Although usual interstitial pneumonia was suspected due to presence of honeycombing, left lung biopsies demonstrated emphysematous changes, subpleural and interstitial fibrosis, and focal organizing pneumonitis, with considerations including hypersensitivity pneumonitis or smoking-related ILD.
Studies with 111In-pentetreotide and 68Ga-DOTANOC have shown SSTR expression in IPF (77,78). 111In-pentetreotide uptake, expressed as a target-to-background ratio, correlates with altered lung function and the intensity of alveolitis, suggesting a disease-related functional role of SSTRs (78). 68Ga-DOTANOC is increased in peripheral and subpleural high-resolution CT abnormalities in IPF patients, with a strong linear correlation between the SUVmax and disease extent on high-resolution CT (77). Lung 18F-FDG and 68Ga-DOTATATE uptake in diffuse parenchymal lung diseases appear to be similar, suggesting that these 2 tracers image different aspects of the ongoing inflammatory and fibroblastic processes in ILDs (79). Given that a pilot open-label study evaluating long-acting octreotide in patients with IPF showed some improvement in pulmonary function when compared with historical controls (80), SSTR imaging may be a useful method for guiding such treatment decisions.
CONCLUSION
Molecular imaging approaches may serve as useful tools for better understanding the factors that contribute to lung disease progression and selecting patients for targeted interventions or therapies. Although firm conclusions about the clinical utility of the imaging approaches described here cannot be drawn from the multiple small studies that have been published to date, the data support evaluating these imaging approaches as biomarkers of lung disease activity or severity. As new targeted tracers are developed, molecular imaging will likely advance basic investigations in patients with pulmonary disease that could lead to improved outcomes.
Footnotes
Published online Sep. 1, 2016.
Learning Objectives: On successful completion of this activity, participants should be able to (1) describe imaging approaches that have been used to measure lung inflammation; (2) discuss potential applications of 18F-FDG PET for quantifying lung inflammation; and (3) gain familiarity with new imaging approaches being used to study lung inflammation.
Financial Disclosure: The NIH (R01 HL121218) funded Dr. Chen’s effort in part. The COPD data were obtained with funding from Pfizer, Inc. The ARDS data were obtained with funding from the Washington University School of Medicine Institute of Clinical and Translational Sciences and Mallinckrodt Institute of Radiology. No other potential conflict of interest relevant to this article was reported.
CME Credit: SNMMI is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing education for physicians. SNMMI designates each JNM continuing education article for a maximum of 2.0 AMA PRA Category 1 Credits. Physicians should claim only credit commensurate with the extent of their participation in the activity. For CE credit, SAM, and other credit types, participants can access this activity through the SNMMI website (http://www.snmmilearningcenter.org) through November 2019.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.
- 45.
- 46.
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- Received for publication June 20, 2016.
- Accepted for publication August 22, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Association of radiological severity with inflammatory biomarkers for prognostic prediction in patients with COVID-19
- PET Imaging of Neutrophil Elastase with 11C-GW457427 in Acute Respiratory Distress Syndrome in Pigs
- Consensus Recommendations on the Use of 18F-FDG PET/CT in Lung Disease
- Quantification of Lung PET Images: Challenges and Opportunities