


Abstract
A novel synthetic approach to 6-18F-fluoro-3,4-dihydroxy-l-phenylalanine (18F-DOPA), involving the nucleophilic substitution of a diaryliodonium salt precursor with non-carrier-added 18F-fluoride, yielded a product with a specific activity that was 3 orders of magnitude higher than the product of the conventional synthesis method, involving an electrophilic substitution of a trialkylstannane precursor with 18F2. We performed a direct comparison of high- and low-specific-activity 18F-DOPA in a neuroendocrine tumor model to determine whether this difference in specific activity has implications for the biologic behavior and imaging properties of 18F-DOPA. Methods: 18F-DOPA was produced via the novel synthesis method, yielding 18F-DOPA-H with a high specific activity (35,050 ± 4,000 GBq/mmol). This product was compared in several experiments with conventional 18F-DOPA-L with a low specific activity (11 ± 2 GBq/mmol). In vitro accumulation experiments with the human pancreatic neuroendocrine tumor cell line BON-1 were performed at both 0°C and 37°C and at 37°C in the presence of pharmacologic inhibitors of proteins involved in the uptake mechanism of 18F-DOPA. Small-animal PET experiments were performed in athymic nude mice bearing a BON-1 tumor xenograft. Results: At 37°C, the uptake of both 18F-DOPA-H and 18F-DOPA-L did not differ significantly during a 60-min accumulation experiment in BON-1 cells. At 0°C, the uptake of 18F-DOPA-L was significantly decreased, whereas the lower temperature did not alter the uptake of 18F-DOPA-H. The pharmacologic inhibitors carbidopa and tetrabenazine also revealed differential effects between the 2 types of 18F-DOPA in the 60-min accumulation experiment. The small-animal PET experiments did not show any significant differences in distribution and metabolism of 18F-DOPA-H and 18F-DOPA-L in carbidopa-pretreated mice. Conclusion: The advantages of the novel synthesis of 18F-DOPA, which relies on nucleophilic fluorination of a diaryliodonium salt precursor, lie in the simplicity of the synthesis method, compared with the conventional, electrophilic approach and in the reduced mass of administered, pharmacologically active 19F-DOPA. 18F-DOPA-H demonstrated comparable imaging properties in an in vivo model for neuroendocrine tumors, despite the fact that the injected mass of material was 3 orders of magnitude less than 18F-DOPA-L.
The PET tracer 6-18F-fluoro-3,4-dihydroxy-l-phenylalanine (18F-DOPA) is abundantly used for visualizing neuroendocrine tumors and neurologic imaging. This amino acid tracer visualizes those cells that possess the capability to take up the aromatic amino acid precursor and are involved in the biosynthesis of dopamine. 18F-DOPA has been shown to be a good tracer for well-differentiated neuroendocrine tumors originating from the gastrointestinal tract (1–3). In addition, 18F-DOPA has also been shown to be a good tracer for other types of neuroendocrine tumors, such as neuroblastoma (4), medullary thyroid carcinoma, pheochromocytoma, and paraganglioma (1), and for malignancies of the central nervous system (5). Finally, imaging dopamine metabolism in the central nervous system with 18F-DOPA is an effective means to determine the extent of degeneration of presynaptic dopaminergic neurons in Parkinson disease (6). Despite the general applicability of 18F-DOPA imaging in nuclear medicine, 18F-DOPA is available in only a few countries worldwide. The major drawback limiting a more widespread use is its labeling method.
The conventional synthesis of 18F-DOPA involves an electrophilic substitution of a trialkylstannane precursor with 18F gas (7,8). The difficulty of generating and handling 18F2 and the large amount of carrier 19F-DOPA that is generated during the electrophilic process are major disadvantages, which have led to the development of various nucleophilic substitution protocols for the production of 18F-DOPA using no-carrier-added fluoride (9–13). DiMagno has recently developed a fully elaborated diaryliodonium salt precursor, which allows for the facile production of no-carrier-added 18F-DOPA via radiofluorination, followed by deprotection (14).
We applied this method to synthesize 18F-DOPA (Fig. 1) and tested whether the high-specific-activity product possesses discernibly different biologic behavior and imaging properties in comparison to conventional 18F-DOPA. This article presents the first comparison of 18F-DOPA with high specific activity, produced from a diaryliodonium salt precursor (18F-DOPA-H), with conventional, low-specific-activity 18F-DOPA (18F-DOPA-L) in a biologic model for neuroendocrine tumors.

Two-step synthesis of 18F-DOPA from precursor ALPDOPA, ((S)-methyl-3-(4,5-bis(ethoxymethoxy)2-iodophenyl)-2-(di-(tert-butoxycarbonyl))amino)propanoate)(4-methoxyphenyl)-λ3-iodane trifluoromethanesulfonate. Radiochemical yield, 14% ± 4% EOB; enantiomeric excess > 99%.
MATERIALS AND METHODS
Chemicals
All chemicals were obtained from Sigma-Aldrich, unless stated otherwise. Matrigel was obtained from BD Biosciences. The culture medium DMEM (Dulbecco modified Eagle medium) and Ham F-12 were obtained from Invitrogen, Life Technologies. The fetal calf serum was obtained from Bodinco. The phosphate-buffered saline (PBS) used in the experiments was obtained from the Department of Hospital Pharmacy, University Medical Center Groningen. The PBS contained 140 mM NaCl, 9 mM Na2HPO4, and 1.3 mM NaH2PO4, and pH was 7.4. For incubation during in vitro experiments GMC (5.6 mM d-glucose, 0.49 mM MgCl2, 0.68 mM CaCl2) was added to the PBS. The diaryliodonium precursors for the synthesis of 18F-DOPA and 6-18F-fluorodopamine were provided by Ground Fluor Pharmaceuticals. The synthesis and characterization of the diaryliodonium precursor to 18F-DOPA, ((S)-methyl-3-(4,5-bis(ethoxymethoxy)2-iodophenyl)-2-(di-(tert-butoxycarbonyl))amino)propanoate)(4-methoxyphenyl)-λ3-iodane trifluoromethanesulfonate (ALPDOPA), are provided in the supplemental materials (available at http://jnm.snmjournals.org).
Radiosynthesis
Experiments were performed with either 18F-DOPA-L, obtained from the conventional synthesis procedure (electrophilic substitution) as described by de Vries et al. (8), resulting in a product with a specific activity of 11 ± 2 GBq/mmol and a decay-corrected radiochemical yield of 33% ± 4% at the end of bombardment (EOB), or with 18F-DOPA-H, synthesized via the novel method starting from diaryliodonium salt precursor, resulting in a product with a specific activity of 35,050 ± 4,000 GBq/mmol and a decay-corrected radiochemical yield of 14% ± 4% EOB. The radiosynthesis of 18F-DOPA-H was based on the instructions of the supplier of the precursor, as detailed in the supplemental information. 18F-DOPA-H was produced in 117 ± 4 min at the EOB. Both products were produced with a radiochemical purity higher than 95% and a high enantiomeric purity (enantiomeric excess > 99%).
Cell Culture
In vitro experiments were performed with the human pancreatic neuroendocrine tumor cell line BON-1. The cells were cultured in 25 cm2 culture flasks containing 5 mL of culture medium (DMEM:Ham F-12:fetal calf serum, 9/9/2). Cells were grown in a humidified atmosphere containing 5% CO2 and subcultured twice a week.
Cells were harvested by trypsin treatment, resuspended, and plated at a density of 500,000 cells per well in culture medium on 12-well plates at the day before the cell experiment (1 mL/well). The viability and the number of cells were determined with the trypan blue exclusion technique.
Accumulation Experiments in BON-1 Cells
On the day of the experiment, cells were washed with PBS (2 × 1 mL), and 1 mL of PBS-GMC buffer, optionally spiked with a pharmacologic inhibitor, was added per well. The pharmacologic inhibitors were applied in the following concentrations: 1 mM 2-aminobicyclo[2.2.1]heptane-2-carboxylic acid (BCH), 80 μM carbidopa, 100 μM pargyline, or 10 μM tetrabenazine. The cells were placed in an incubator for 1 h at 37°C to allow depletion of residual nutrients from the culture medium.
After the 1-h depletion period, 200 kBq of 18F-DOPA-L or 18F-DOPA-H, dissolved in a volume of 50 μL of the tracer formulation, were added to each well. The time of addition of the tracer (0, 15, 30, 45, 50, and 55 min after the 1-h depletion period) corresponded with the incubation times of 60, 45, 30, 15, 10, and 5 min, respectively. Afterward, the cells were placed back in the incubator at 37°C. At 60 min, all wells were washed with ice-chilled PBS (2 × 1 mL). To each well, 0.2 mL of 0.05% (w/v) trypsin in PBS was added, and the cells were incubated at 37°C for 5 min. Then 1 mL of culture medium was added. The contents of the wells were resuspended and transferred to 5-mL counting tubes. The radioactivity was measured with a γ counter (1282CS; LKB Wallac Compugamma). Then the viability and the number of cells were determined with the trypan blue exclusion technique. The viability was greater than 90% within 2 h after the experiment. The radioactivity in the samples was expressed as the percentage of the added dose per 100,000 cells.
Animals
Male BALB/cOlaHsdFoxn1nu/nu athymic nude mice (body weight, 18–24 g) were obtained from a breeding facility (Harlan). The animals were housed in temperature- and humidity-controlled rooms with 12-h day and 12-h night cycles. They were provided with forage and water ad libitum. Animals were housed in individually ventilated cages with HEPA filters in the research facility of the University Medical Center Groningen. All animal experiments were done by licensed investigators in compliance with the Law on Animal Experiments of The Netherlands. The study protocol was approved by the Committee on Animal Ethics of the University of Groningen.
Small-Animal PET Experiments in BON-1 Tumor–Xenografted Mice
The nude mice were injected subcutaneously on the right shoulder with an ice-cold tumor cell suspension, consisting of 1 million BON-1 cells in 100 μL of culture medium and 100 μL of Matrigel. Three weeks after inoculation of the tumor, the animals were scanned (small-animal PET). The tumor growth and the welfare and body weight of the animal were checked regularly in this period (at least 3 times a week).
The 3 experimental groups each consisted of 4 mice. At least 2 h before the start of the small-animal PET scan, the mice were acclimatized to the laboratory conditions. Animals from 2 of 3 experimental groups were pretreated with an intraperitoneal injection of 20 μg of carbidopa in 100 μL of PBS 60 min before the start of the small-animal PET scan. The mice were anesthetized with isoflurane (5% in medical air for induction and 1.5% in medical air for maintenance of anesthesia) before an injection in the penile vein with 1 MBq of either 18F-DOPA-L (228.3 ± 5.7 pmol/g, 1 experimental group) or novel 18F-DOPA-H (0.49 ± 0.09 pmol/g, 2 experimental groups). The mice were scanned during 90 min with a Concorde microPET Focus 220 system equipped with microPET Manager software for data acquisition. The dynamic PET scan was followed by a 10-min transmission scan using a 51Co source. The animals were sacrificed by application of 5% isoflurane in medical air for 15 min, which was applied from the start of the transmission scan. Quantitative analysis was performed using ordered-subset expectation maximization. The PET acquisition data were fully corrected for random coincidences, attenuation, and scatter. The reconstructed PET images were analyzed using Siemens Inveon Research Workplace software. Organs and tissues, including the tumor xenograft, were dissected and measured with a γ counter.
In Vivo Stability
After measurement of the radioactivity with the γ camera, tissue samples from the brain and the tumor were cut into small pieces and submerged in 0.5 mL of water and 1 mL of acetonitrile in a plastic test tube. After homogenizing the tissue samples with a rotor-stator mixer, the resulting suspension was spun down at 6,000 rpm, and a sample of the supernatant was spotted on a silica plate. The protein content was precipitated by adding 2 volumes of acetonitrile to 1 volume of plasma. Also, samples of the deproteinated plasma and of urine were spotted on a silica plate. The plate was developed with n-BuOH:CH3COOH:H2O:EtOH 4/1/1.6/0.5 (v/v/v/v). The thin-layer chromatography (TLC) plate was dried and placed on a phosphor storage screen, which was exposed overnight. The intensity of the spots on the phosphor storage screen was measured with the Cyclone (Packard Instuments).
Data Analysis
Data represent mean ± SEM, from n separate experiments. The statistical significance of differences between means was calculated using the Student t test. Differences were considered to be statistically significant when the P value was less than 0.05.
RESULTS
Accumulation Experiments in BON-1 Cells
At 37°C, the uptake of 18F-DOPA-H did not differ significantly from the uptake of 18F-DOPA-L. A differential response was revealed when the accumulation experiment was performed at 0°C. The uptake of 18F-DOPA-L was significantly reduced at 0°C, compared with 37°C at all time points, whereas the uptake of 18F-DOPA-H was only slightly reduced by changing the temperature at 10 and 15 min (Fig. 2A).

(A) Influence of temperature on uptake of 18F-DOPA by BON-1 cells (n = 5–6 for 0°C and 10–11 for 37°C). *P ≤ 0.05, 0°C: 18F-DOPA-H vs. 18F-DOPA-L; #P ≤ 0.05, 18F-DOPA-H: 0°C vs. 37°C; ##P ≤ 0.05, 18F-DOPA-L: 0°C vs. 37°C. (B) Influence of inhibition of LAT in BON-1 cells by 1 mM BCH (n = 10–11 for control and 4 for BCH-treated). #P ≤ 0.05, 18F-DOPA-H: control vs. BCH; ##P ≤ 0.05, 18F-DOPA-L; control vs. BCH. (C) Influence of inhibition of AADC in BON-1 cells by 0.08 mM carbidopa (n = 10–11 for control and 5 for carbidopa-treated). *P ≤ 0.05, carbidopa: 18F-DOPA-H vs. 18F-DOPA-L; #P ≤ 0.05, 18F-DOPA-L; control vs. carbidopa. (D) Influence of inhibition of MAO in BON-1 cells by 0.1 mM pargyline (n = 10–11 for control and 4–5 for pargyline-treated). *P ≤ 0.05, pargyline: 18F-DOPA-H vs. 18F-DOPA-L; #P ≤ 0.05, 18F-DOPA-L: control vs. pargyline. (E) Influence of inhibition of VMAT in BON-1 cells by 0.01 mM tetrabenazine (TBZ; n = 10–11 for control and 3–4 for TBZ-treated). *P ≤ 0.05, TBZ: 18F-DOPA-H vs. 18F-DOPA-L; #P ≤ 0.05, 18F-DOPA-H: control vs. TBZ; ##P ≤ 0.05, 18F-DOPA-L: control vs. TBZ.
The selective inhibitor of the L-type amino acid transporter (LAT) BCH lowered the uptake of both 18F-DOPA-L and 18F-DOPA-H in a similar fashion (Fig. 2B).
Carbidopa, an inhibitor of aromatic L-amino acid decarboxylase (AADC), was applied to determine whether inhibition of metabolic conversion to 6-18F-fluorodopamine affected tracer uptake. Carbidopa significantly decreased uptake of 18F-DOPA-L at late time points, compared with control. The uptake of 18F-DOPA-H was not significantly affected by carbidopa (Fig. 2C).
Pargyline, at the applied concentration a nonselective inhibitor of monoamine oxidase (MAO), did not significantly alter the uptake of 18F-DOPA-H or 18F-DOPA-L (Fig. 2D).
Tetrabenazine, an inhibitor of vesicular monoamine transporter type 2 (VMAT2), led to differential effects between the 2 variants of 18F-DOPA. It significantly increased the uptake of 18F-DOPA-H, whereas the uptake of 18F-DOPA-L was significantly reduced (Fig. 2E).
Small-Animal PET Experiments in BON-1 Tumor–Xenografted Mice
18F-DOPA-H was also compared with 18F-DOPA-L in in vivo experiments in mice bearing a tumor xenograft, which was created from a BON-1 tumor cell suspension. The experimental variables and other characteristics of the mice belonging to the 3 experimental groups are listed in Table 1.
Characteristics of Experimental Animals
On a representative PET image (Fig. 3A), it is clearly visible that carbidopa increased the uptake of 18F-DOPA-H in the BON-1 tumor xenograft. The uptake of 18F-DOPA-L and 18F-DOPA-H was also high in the pancreas and bladder.
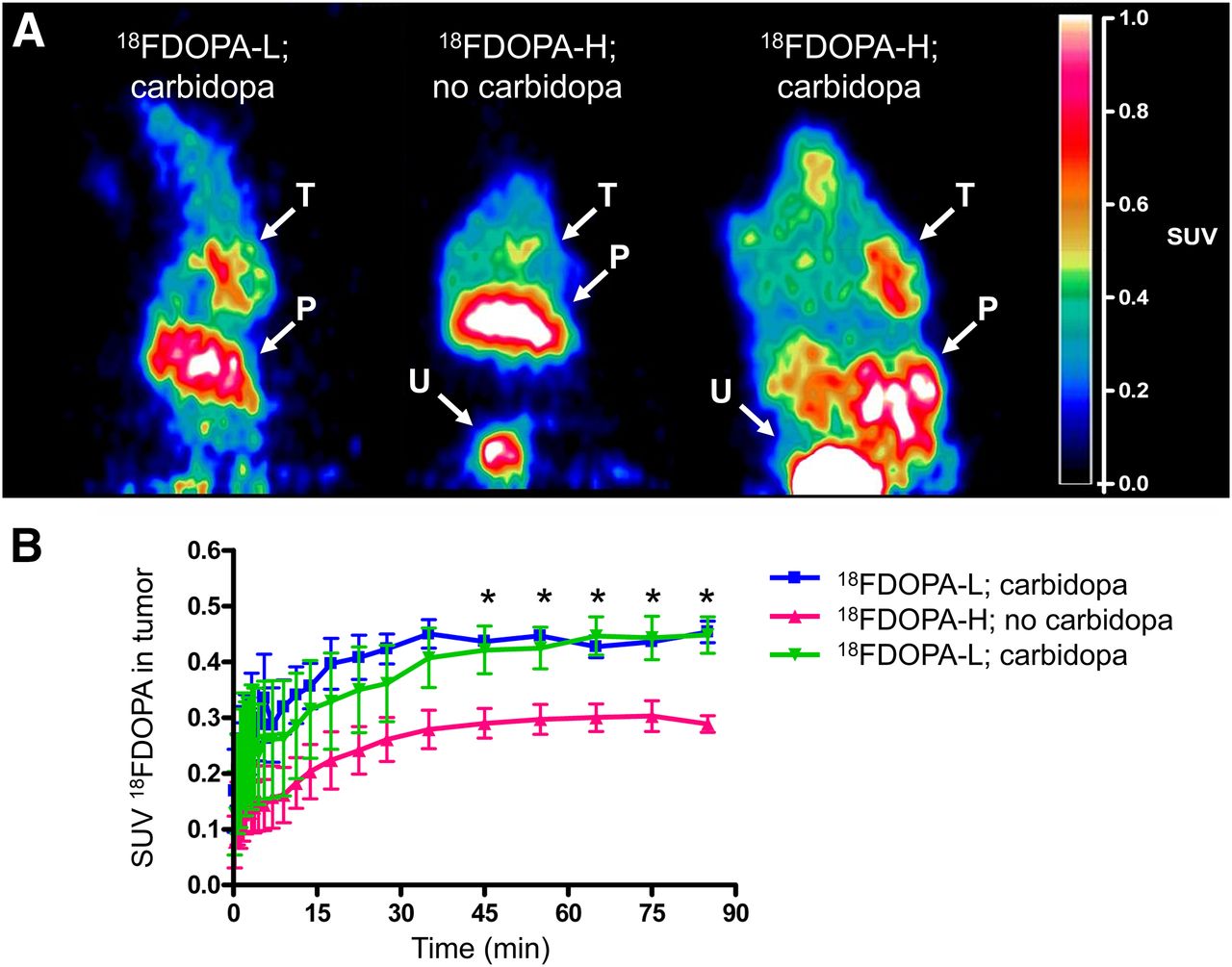
(A) Coronal sections, reconstructed from summed time frames of 30–90 min, of athymic nude mice. Intensity of signal reflects standardized uptake values (SUVs) as indicated on bar on right. Locations with relatively high uptake are indicated by white arrows: tumor xenograft on right shoulder (T), pancreas (P), and urinary bladder (U). (B) Time–activity curves of 18F-DOPA with mean SUVs determined from regions of interest drawn at location of BON-1 tumor xenograft in PET images. *P ≤ 0.05, 18F-DOPA-H; no carbidopa vs. carbidopa.
The time–activity curves (Fig. 3B) reveal that from 45 min onward, carbidopa significantly enhanced the uptake of 18F-DOPA-H in the tumor. In animals that received carbidopa pretreatment, there was no significant difference in mean standardized uptake value between those that received 18F-DOPA-H and those that received 18F-DOPA-L.
The biodistribution data revealed that there were no statistically significant differences between the uptake of 18F-DOPA-H and the uptake of 18F-DOPA-L in relevant organs. In tumor and brain, carbidopa pretreatment led to a significant increase in uptake of 18F-DOPA-H (Fig. 4A), which also translated into an increased uptake ratio of tumor versus plasma after carbidopa pretreatment (Fig. 4B).
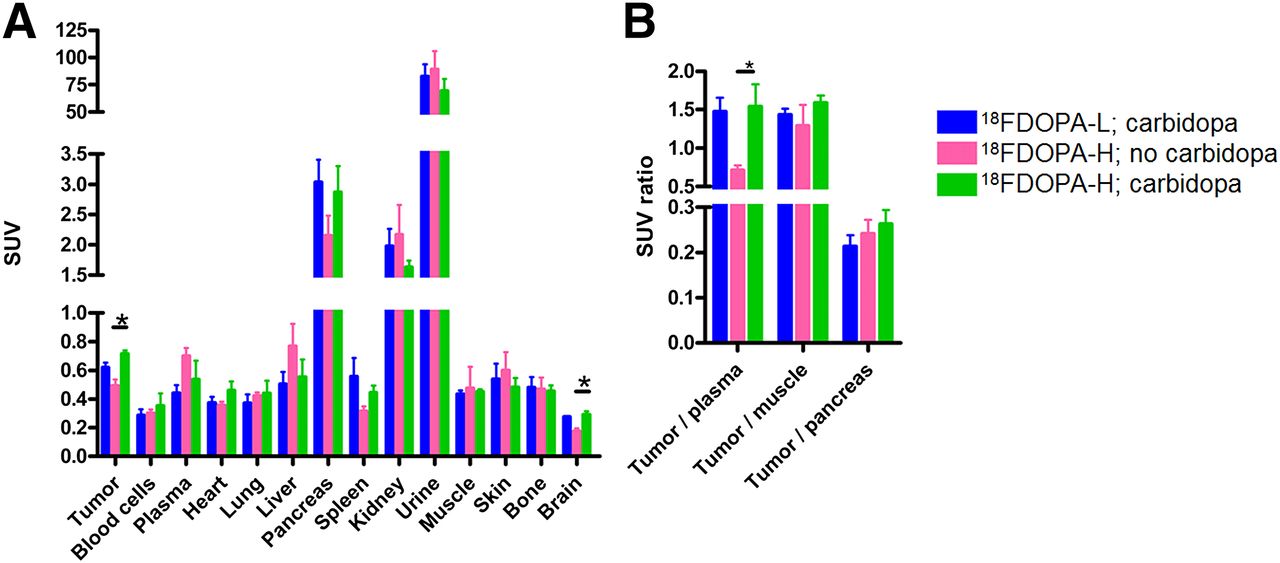
(A) Biodistribution of 18F-DOPA in mice bearing BON-1 tumor xenograft at 105 min after tracer injection. *P ≤ 0.05, 18F-DOPA-H: no carbidopa vs. carbidopa. (B) Ratio of standardized uptake values (SUVs) of tumor vs. several reference tissues, based on SUV determined by biodistribution experiment. *P ≤ 0.05, 18F-DOPA-H: no carbidopa vs. carbidopa.
In Vivo Stability
6-18F-fluorodopamine was synthesized in the same manner as 18F-DOPA (Fig. 1) to check its retention factor on radio-TLC. The Rf of 18F-DOPA was 0.43, and the Rf of the metabolite 6-18F-fluorodopamine was 0.54. Tissue samples from the biodistribution experiment were investigated by radio-TLC. The fraction of the administered 18F-DOPA found in plasma and homogenates of tumor and brain in the form of the metabolite 6-18F-fluorodopamine was significantly increased after pretreatment with carbidopa in animals that received 18F-DOPA-H (Fig. 5). The fraction of 6-18F-fluorodopamine did not differ significantly between carbidopa-pretreated animals that received 18F-DOPA-H and those that received conventional 18F-DOPA-L.

Fraction of radiometabolite 6-18F-fluorodopamine contributing to whole amount of radioactivity of applied radio-TLC sample. *P ≤ 0.05, 18F-DOPA-H: no carbidopa vs. carbidopa.
DISCUSSION
The availability of a novel synthesis method to produce 18F-DOPA from no-carrier-added 18F-fluoride and a diaryliodonium salt precursor greatly simplifies access to this tracer, because 18F2 does not need to be generated or handled. The product, 18F-DOPA-H, has a considerably higher specific activity (3 orders of magnitude) than the 18F-DOPA-L, which is produced from electrophilic fluorination. We investigated whether the increase of the specific activity had implications for the biologic behavior of the tracer due to saturation of 1 or more proteins involved in the molecular uptake mechanism of 18F-DOPA.
The uptake of 18F-DOPA in accumulation experiments in BON-1 cells under control conditions did not differ significantly between 18F-DOPA-H and 18F-DOPA-L at the time points within 60 min after addition of the tracer. The accumulation of 18F-DOPA-L was significantly reduced by lowering the temperature from 37°C to 0°C because of partial inactivation of 1 or more proteins that are involved in the uptake mechanism, whereas the accumulation of 18F-DOPA-H was unaffected. The extent of cold-induced inhibition was not large enough to reduce the capacity to sequester the 18F-DOPA-H. To further elucidate the observed cold-induced inhibition, accumulation experiments were performed in the presence of pharmacologic inhibitors of proteins involved in the molecular uptake mechanism of 18F-DOPA.
Figure 6 provides an overview of the cellular uptake mechanism of 18F-DOPA. The LAT, which is responsible for entry of a variety of amino acids, is a high-capacity transporter. Furthermore, this transporter is upregulated in a wide range of malignant cells (15). LAT1 expression is associated with poor prognosis (16–18) as well as metastases and angiogenesis (19). Because of its high expression in neuroendocrine malignancies, LAT1 is unlikely to be satiated, which is in accordance with the results of the accumulation experiment of 18F-DOPA after treatment with the specific inhibitor of LAT: BCH led to a decrease of the same extent in both 18F-DOPA-H and 18F-DOPA-L.
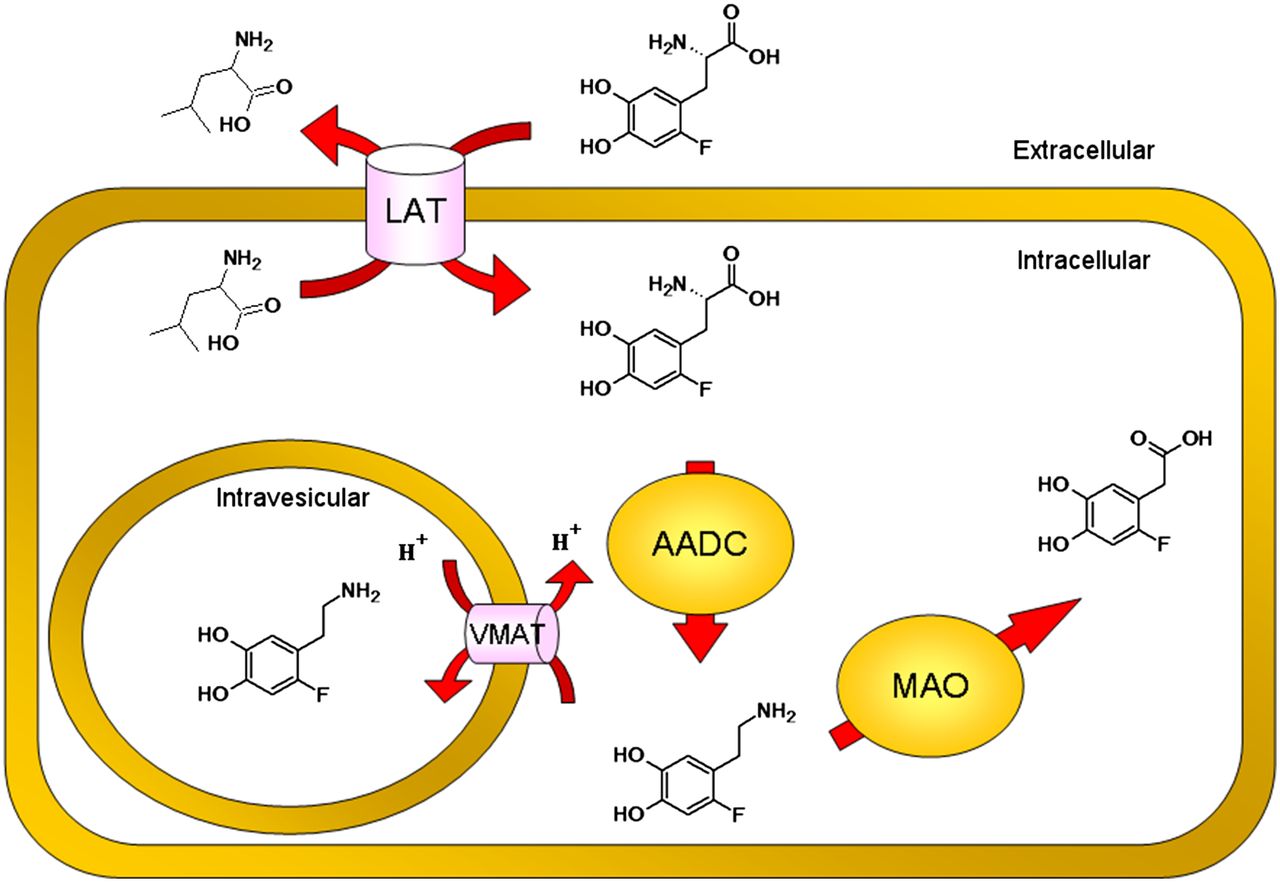
Overview cellular uptake mechanism of 18F-DOPA. 18F-DOPA is taken up by LAT in exchange for other neutral amino acids (e.g., leucine). Enzyme AADC metabolizes 18F-DOPA to 6-18F-fluorodopamine, which in turn can be either sequestered in secretory vesicles by VMAT or degraded by other enzymes, such as MAO, to break down products that are rapidly cleared from the cell.
The expression of AADC is increased in most tumors of neuroendocrine origin as well (4,20,21). The application of carbidopa (inhibitor of AADC) showed a modest decline in uptake for 18F-DOPA-L, compared with 18F-DOPA-H, at early time points (5–30 min). In addition, 18F-DOPA-L is significantly decreased at later time points (30–60 min) after carbidopa pretreatment. The difference in uptake after treatment with carbidopa between 18F-DOPA-H and 18F-DOPA-L is limited to the early time points, and therefore the differential response to lowering the temperature from 37°C to 0°C cannot be fully attributed to cold-induced inhibition of AADC.
The expression of vesicular monoamine transporter (VMAT) is inversely correlated with poor prognosis (22,23), possibly implying a potential protective function of the VMAT. However, it more likely reflects the extent of differentiation of the tumor as tumor dedifferentiation is accompanied with a poor disease outcome. The application of tetrabenazine (an inhibitor of VMAT2) led to a differential response between 18F-DOPA-H and 18F-DOPA-L. The uptake of 18F-DOPA-L was significantly decreased at late time points (45–60 min) as one would expect from the reduced activity of VMAT2, resulting in a lower sequestration of tracer in the secretory vesicles. The uptake of 18F-DOPA-H was significantly increased at time points 30–60 min. This finding may be due to the filling degree of the vesicles. Because there is probably some residual VMAT2 activity and because 18F-DOPA-H is present at a concentration lower than the concentration of endogenous substrates, it does not suffer in the same extent from direct inhibition of VMAT2 transporter function as 18F-DOPA-L and endogenous substrates. Because the vesicles are therefore less filled with endogenous substrates due to partial inhibition of VMAT2, the negative feedback loop, resulting in downregulation of VMAT2, has not been activated (24). This indirect effect of tetrabenazine treatment may have increased the uptake of 18F-DOPA-H.
The predominant form of MAO in the gastrointestinal tract and associated tumors is MAO-A. The expression of MAO-A is increased and associated with dedifferentiation and metastatic potential in prostate cancer (25), whereas MAO-A expression is downregulated in pheochromocytomas resulting in the formation of a large quantity of catecholamines (26). The expression of MAO-A in gastroenteropancreatic neuroendocrine tumors is generally high, which allows for MAO-A imaging using PET (27). Pharmacologic inhibition of MAO by pargyline did not result in any significant differences in the uptake of 18F-DOPA-H and 18F-DOPA-L. Pargyline is an inhibitor with about a 30-fold selectivity toward MAO-B over MAO-A, but at the high concentrations applied in the in vitro experiments it acts as a nonselective inhibitor of both subtypes of MAO.
To discover whether the saturation effects discussed above have implications on the distribution and imaging properties of the PET tracer in vivo, 18F-DOPA-H and 18F-DOPA-L were compared in an animal model for neuroendocrine tumors. In mice that were pretreated with carbidopa, there were no statistically significant differences found between 18F-DOPA-H and 18F-DOPA-L with respect to the tracer uptake, the time–activity curve, or the fraction converted to 6-18F-fluorodopamine at the end of the scan. The saturation effects as observed in the in vitro experiments had no implications for the imaging properties of the novel 18F-DOPA-H in comparison to the conventional 18F-DOPA-L.
Several explanations for the different findings between the in vitro and the in vivo experiments are possible. The most logical explanation is that the saturation effects in the in vitro experiments were seen only under specific experimental conditions, for which either thermal or pharmacologic inhibition was applied. Although carbidopa has been administered to the mice, carbidopa mainly inhibited the peripheral metabolism.
Furthermore, the total load of administered 18F-DOPA in the in vivo model (±50 MBq/kg) is lower than the load of 18F-DOPA in the incubation solution containing the BON-1 cells in an in vitro experiment (200 kBq in a solution of 1 mL, 200 MBq/kg). This difference in administration alone makes the observation of saturation effects in in vivo models less probable, even for 18F-DOPA-L, which is a comforting message, because scaling up from an in vivo model in small models to clinical application of this PET tracer for the imaging of neuroendocrine tumors will further decrease the load of 18F-DOPA (±3 MBq/kg). The absence of saturation effects in vivo means that the novel 18F-DOPA-H is biologically equivalent to conventional 18F-DOPA-L for imaging neuroendocrine tumors in mouse models.
The main advantages of 18F-DOPA-H are its facile and easier-to-implement production method and the dramatic reduction in the administered mass of pharmacologically active 19F-DOPA. The latter might prevent adverse reactions such as induction of a carcinoid crisis. In the event in which administration of 18F-DOPA led to a carcinoid crisis, 5.63 mg of compound were administered to the patient (28). Whether the properties of 18F-DOPA-H with a 3-orders-of-magnitude higher specific activity differ from those of 18F-DOPA-L in imaging of the dopamine biosynthesis pathway in other diseases, such as Parkinson disease, remains to be investigated. As several proteins involved in the uptake mechanism of 18F-DOPA are downregulated in Parkinson disease (29–31), changing from 18F-DOPA-L to 18F-DOPA-H might affect neurologic imaging.
CONCLUSION
The in vitro accumulation experiments showed that changing from conventionally produced 18F-DOPA-L to the novel 18F-DOPA-H has implications for the accumulation of the tracer under specific experimental conditions. However, the uptake levels were similar under control conditions, in which proteins involved in the uptake mechanism of 18F-DOPA were neither thermally nor pharmacologically inhibited. There were no differences observed between the uptake of 18F-DOPA-H and the uptake of 18F-DOPA-L in an in vivo model for neuroendocrine tumors. We conclude that for the application of 18F-DOPA in imaging neuroendocrine tumors, both 18F-DOPA-H and 18F-DOPA-L offer identical imaging quality. Thus, 18F-DOPA-H offers the potential for high-quality imaging with a dramatic reduction in the risk of pharmacologic effects due to the coadministration of 19F-DOPA. Studies in patients are needed to establish whether these findings also hold true in humans.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This study was supported by a grant from the Dutch Cancer Society, 2009-4338. One the authors of the manuscript, Kiel D. Neumann, is an employee of Ground Fluor Pharmaceuticals, Inc., Lincoln, Nebraska, which gave nonfinancial support by providing diaryliodonium salt precursor materials for the nucleophilic synthesis of both 18F-DOPA and 6-18F-fluorodopamine. Another author, Stephen G. DiMagno, holds a patent for the nucleophilic fluorination of aromatic ring systems, which includes the nucleophilic synthesis of 18F-DOPA via a diaryliodonium salt precursor (US Patent 8,604,213 B2, Dec. 10, 2013). No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Dec. 11, 2014.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication July 23, 2014.
- Accepted for publication November 13, 2014.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Production of diverse PET probes with limited resources: 24 18F-labeled compounds prepared with a single radiosynthesizer
- Norepinephrine Transporter as a Target for Imaging and Therapy
- Effect of Carbidopa on 18F-FDOPA Uptake in Insulinoma: From Cell Culture to Small-Animal PET Imaging
- Molecular Imaging of Gastroenteropancreatic Neuroendocrine Tumors: Current Status and Future Directions