


Abstract
Inflammation plays a major role in all phases of atherosclerosis. Stable plaques are characterized by a chronic inflammatory infiltrate, whereas vulnerable and ruptured plaques are characterized by an “active” inflammation involved in the thinning of the fibrous cap, predisposing the plaque to rupture. Although a single vulnerable atherosclerotic plaque rupture may cause the event, there are many other types of plaques, several of which are vulnerable. The existence of multiple types of vulnerable plaques suggests that atherosclerosis is a diffuse inflammatory process. A current challenge is to identify morphologic and molecular markers able to discriminate stable plaques from vulnerable ones, allowing the stratification of patients at high risk for acute cardiovascular and cerebrovascular events before clinical syndromes develop. With that aim in mind, this article summarizes the natural history of atherosclerotic plaques, focusing on molecular mechanisms affecting plaque progression and serum markers correlated with plaque inflammation.
Atherosclerosis has a broad spectrum of clinical presentation. Some patients are asymptomatic for life, even though they harbor atherosclerotic plaques in their vasculature. Others have ischemic symptoms, such as myocardial infarction and stroke. The first condition is usually characterized by slowly growing, silent lesions defined as “stable plaques.” In the second condition, clinical events are associated with one or more “unstable plaques.” The clinical symptoms of an atheroma typically occur in adults and usually involve thrombosis (1). The risk of major thrombotic and thromboembolic complications of atherosclerosis is related more to the instability of an atheroma than to the extent of disease (1–3). Stable angina is associated with smooth fibrous coronary artery plaques, whereas unstable angina, acute myocardial infarction (AMI), and sudden cardiac death are almost invariably associated with irregular or ruptured plaques (4). Similarly, in patients with carotid artery disease, plaque irregularity and rupture are associated with cerebral ischemic events. Patients with irregular or ulcerated plaques (as shown by carotid artery angiography) have a higher risk of ischemic stroke irrespective of the degree of stenosis of the vessel lumen (5).
Inflammation is a component of all forms of plaque (6,7). Moreover, a topographic relationship among an inflammatory infiltrate, plaque rupture, and thrombosis was proved by van der Wall et al. (8), suggesting a pathogenetic role for macrophages at the site of cap rupture in patients with fatal AMI. Further observations demonstrated the role of activated macrophages and activated T lymphocytes in plaque destabilization (7,9). The combination of macrophages and lymphocytes in vulnerable plaque is associated with the secretion of cytokines and lytic enzymes that result in thinning of the fibrous cap, predisposing a lesion to rupture (7,9).
This article summarizes the natural history of atherosclerotic plaques, focusing on molecular mechanisms affecting plaque progression and serum markers correlated with plaque inflammation.
NATURAL HISTORY OF ATHEROSCLEROTIC PLAQUES
Atherosclerotic lesions, according to the American Heart Association classification recently modified by Virmani et al. (4) and Naghavi et al. (10), are divided into 2 groups: nonatherosclerotic intimal lesions and progressive atherosclerotic lesions. A third group of lesions, healed atherosclerotic plaques, are the most prevalent lesions, particularly in the carotid arteries (Table 1). An alternative approach to characterizing atherosclerotic lesions is based on the thickness of the fibrous cap and its grade of inflammatory infiltrate. As a lesion grows from a fatty streak to an atheroma, the increasing size of the lesion is accommodated by adaptive positive remodeling of the vessel: The external elastic lamina expands to accommodate the lesion, maintaining the size of the lumen (11). This expansion continues until the lesion causes the vessel to expand to 180% of its original area. The lesion contains monocyte-derived macrophages, smooth muscle cells, and T lymphocytes. The interaction between these cell types and the connective tissue determines the development and progression of the plaque, including important complications, such as thrombosis and rupture.
Classification of Atherosclerotic Lesions
Nonatherosclerotic Intimal Lesions
Most adult human lesions originate from preexisting intimal lesions consisting of intimal thickening and fatty streaks.
Intimal Thickening.
Intimal thickening involves mainly smooth muscle cells in a proteoglycan-rich matrix (Fig. 1A). The distribution of such lesions in children correlates with the distribution of atherosclerotic lesions in adults (12). Moderate cell replication has been demonstrated in early lesions, whereas smooth muscle cells of adult lesions are usually clonal (12). There have been very few investigations of the evolution of early intimal lesions in humans, and none of these has clarified their precise pathologic mechanisms of development.

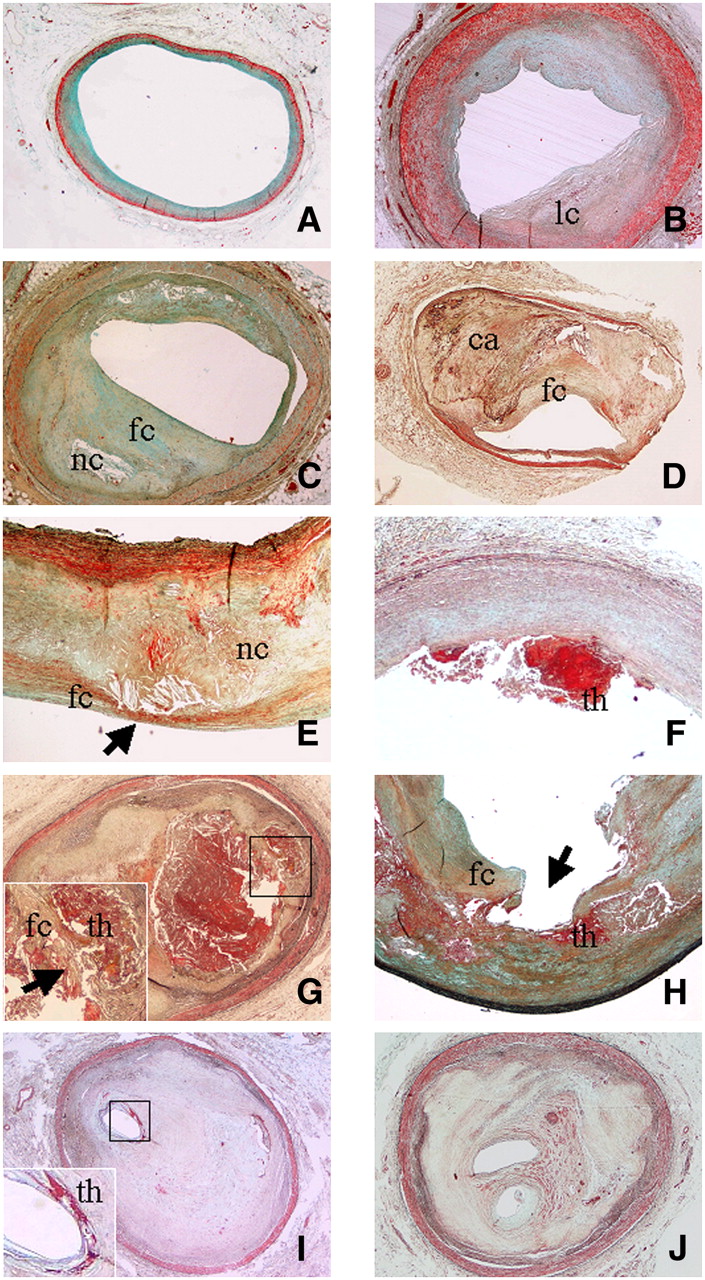
Various types of atherosclerotic lesions. (A) Diffuse intimal thickening consisting mainly of smooth muscle cells in proteoglycan-rich matrix (Movat stain; magnification, ×2). (B) Pathologic intimal thickening associated with some deep lipid core (lc) without necrosis (Movat stain; magnification, ×2). (C) Fibrous cap atheroma characterized by presence of large lipidic–necrotic core (nc) consisting of extracellular lipid, cholesterol crystals, and necrotic debris, covered by thick fibrous cap (fc), with various degrees of infiltration by macrophages and T lymphocytes (Movat stain; magnification, ×2). (D) Fibrocalcific plaque characterized by small lipid-laden necrotic core and thick fibrous cap (fc) overlying extensive accumulation of calcium (ca) in intima (Movat stain; magnification, ×2). (E) Vulnerable plaque (thin fibrous cap atheroma) characterized by large lipidic–necrotic core (nc) associated with thin inflamed fibrous cap (fc; arrow) (Movat stain; magnification, ×2). (F) Plaque erosion showing area of acute thrombosis (th) associated with superficial erosion of endothelium without fibrous cap rupture (Movat stain; magnification, ×2). (G) Fibrous cap (fc) rupture with lumen-occluding thrombus (th) (Movat stain; magnification, ×2). Inset shows site of cap rupture. Arrow indicates acute thrombosis. (H) Plaque rupture with ulceration (arrow), characterized by excavated necrotic core with discontinuation of fibrous cap (fc) (Movat stain; magnification, ×2). Acute thrombus is indicated by th. (I) Thrombotically active plaque characterized by stratified organizing thrombus (dense collagen interspersed with proteoglycan matrix) associated with area of acute thrombosis (th; inset) near residual lumen (Movat stain; magnification, ×2). (J) Healed lesion with lumen almost totally occluded, characterized by distinct layers of dense collagen interspersed with proteoglycan matrix (Movat stain; magnification, ×2).
Fatty Streaks.
Fatty streaks correspond to the intimal xanthomata of the classification of Virmani et al. (4) and are characterized by an intimal accumulation of fat-laden macrophages. These types of lesions may contain a few smooth muscle cells and T lymphocytes.
Progressive Atherosclerotic Lesions
Stable Plaques.
A plaque with pathologic intimal thickening is characterized by intimal thickening associated with lipid deposition but without evidence of necrosis (4). The area overlying the lipid is rich in smooth muscle cells and proteoglycans and may contain variable numbers of macrophages and T lymphocytes (Figs. 1B and 2A).
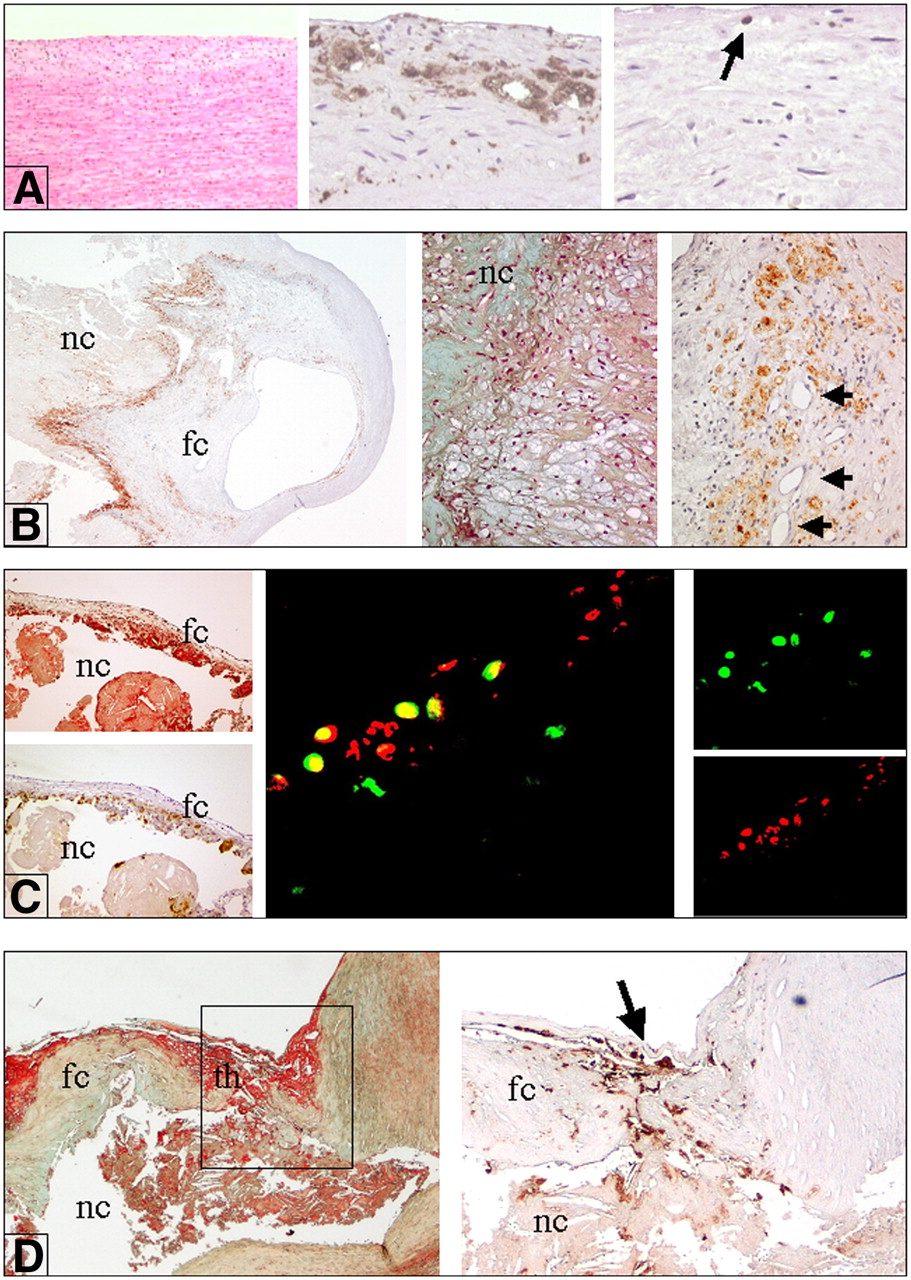
Inflammation in various types of atherosclerotic plaques. (A) Intimal thickening, characterized by smooth muscle cells (left; hematoxylin–eosin stain; magnification, ×10), a few fat-laden macrophages (foam cells) (middle; immunostaining with anti-CD68; magnification, ×10), and scattered T lymphocytes (arrow) (right; immunostaining with anti-CD3; magnification, ×10). (B) Stable plaque (fibrous cap atheroma). Immunohistochemical stain for CD68 (anti-human monocytes and macrophages) shows diffuse positive reaction near lipidic–necrotic core (nc) and large numbers of macrophage foam cells (left; anti-CD68; magnification, ×2) (middle; Movat stain; magnification, ×10). In contrast, only a few macrophages are present in fibrous cap (fc). Numerous macrophage foam cells, positive for CD68, are present near newly formed vessels (arrows) (right; anti-CD68; magnification, ×10). (C) (Left) Vulnerable plaque, characterized by large lipidic–necrotic core (nc) associated with thin fibrous cap (fc) (top; Movat stain; magnification, ×4) rich in inflammatory macrophage foam cells (bottom; immunostaining for CD68; magnification, ×4). (Middle) CXCR3 (fractalkine receptor) expression in activated T lymphocytes. Double fluorescence immunostain studied by 2-dimensional confocal analysis clearly shows diffuse positive reaction for CXCR3 in activated T lymphocytes (concordant double positivity appears as yellow stain) (magnification, ×800). (Right Top) CXCR3 reaction revealed by streptavidin–fluorescein conjugate (green stain). (Right Bottom) CD25 (IL-2 receptor antigen) antibody revealed by streptavidin–Texas Red fluorescent conjugate (red stain). (D) Unstable thrombotic plaque. nc = lipidic–necrotic core. (Left) Site of rupture of thin cap (fc) associated with acute thrombus (th) (Movat stain; magnification, ×4). (Right) Fibrous cap at site of rupture (arrow) showing many CD68-positive macrophages.
A fibrous cap atheroma has a large lipidic–necrotic core containing extracellular lipid, cholesterol crystals, and necrotic debris covered by a thick fibrous cap. The cap consists of smooth muscle cells in a collagen-proteoglycan matrix, with various degrees of infiltration by macrophages and T lymphocytes (Figs. 1C and 2B) (4). Variable numbers of inflammatory cells (macrophage foam cells and T lymphocytes) are also present in the shoulder of the plaque, near the lipidic–necrotic core. This type of lesion may progress to a highly calcified stable lesion or develop complications such as mural hemorrhage.
A plaque with a small or absent lipid-laden necrotic core and a thick fibrous cap overlying extensive accumulations of calcium in the intima close to the media is classified as fibrocalcific (Fig. 1D) (4). Biomechanical studies have shown that intimal tears often occur at the interface of calcified and adjacent noncalcified arterial tissues (13), and it is likely that calcification plays an active role in plaque rupture. A study with electron-beam CT revealed that the vast majority of patients with AMI or unstable angina have levels of coronary calcium measurable by electron-beam CT (14). Conversely, Hunt et al. demonstrated that patients with carotid artery disease and calcification of their carotid artery plaques had fewer symptoms of stroke and transient ischemic attack than those without calcification (15). Atheromas in the carotid and coronary arteries are different, because calcification in the carotid arteries is more likely to begin on the surface, resulting in the eruption of calcified nodules.
A thin fibrous cap atheroma, also called a vulnerable or high-risk plaque, is a plaque prone to rupture, releasing thrombogenic material and causing a thrombus to form. The lesion is characterized by a large necrotic core containing numerous cholesterol clefts. The overlying cap is rich in inflammatory cells, macrophages, and T lymphocytes, with a few smooth muscle cells (Figs. 1E and 2C) (4,10).
Burke et al. (16) defined a vulnerable plaque in the coronary arteries as a lesion with a cap thickness of ≤65 μm. In the carotid arteries, the cap thickness of a vulnerable lesion is ≤165 μm (A. Mauriello, unpublished data, January 2007).
Thin fibrous cap atheromas are most frequently observed in the proximal coronary arteries of patients with fatal AMI. The vessels exhibiting thin fibrous cap atheromas do not usually show severe narrowing but do show positive remodeling. In thin fibrous cap atheromas, the necrotic core length is approximately 2–17 mm (mean, 8 mm), and the underlying cross-sectional area narrowing in over 75% of cases is less than 75% (diameter of stenosis, <50%). The area of the necrotic core in at least 75% of cases is ≤3 mm2 (17).
Unstable Thrombotic Plaques.
Thrombi occur as a consequence of 1 of 3 events: plaque rupture, plaque erosion or, less frequently, a calcified nodule (Table 1). Ulceration and plaque rupture have been variably defined and alternatively used in the literature and are correlated with the presence of vulnerable plaques. The observation that most ruptured plaques are covered by a thrombus with or without luminal occlusion provides convincing evidence that these plaques are causally related to clinical events.
Plaque rupture is defined as an area of fibrous cap disruption in which the overlying thrombus is in continuity with the underlying necrotic core (Figs. 1G and 1J) (18). Ruptured lesions typically have a large necrotic core and a disrupted fibrous cap infiltrated by macrophages and lymphocytes (Fig. 2D). The smooth muscle cell content within the fibrous cap at the rupture site may be quite sparse.
Plaque erosion is identified when serial sectioning of a thrombosed arterial segment fails to reveal fibrous cap rupture (18). Typically, the endothelium is absent at the erosion site (Fig. 1F). The exposed intima consists predominantly of smooth muscle cells and proteoglycans and, surprisingly, the eroded site contains minimal inflammation (19). Unlike a rupture, an erosion can occur in an area of pathologic intimal thickening. Recent studies suggested that plaque erosion is associated with the presence of mast cells in the cap and occurs as a result of mast cell proteases (20).
Another, rare cause of a thrombotic lesion is a calcified nodule. This term refers to a lesion with fibrous cap disruption and thrombi associated with eruptive, dense, calcified nodules (4). It is unclear whether the fibrous cap wears down because of physical forces exerted by the nodules themselves, because of proteases derived from the surrounding cellular infiltrate, or both.
The cellular and molecular mechanisms responsible for thrombus formation on atherosclerotic plaques, whether ruptured, stenotic, or eroded, are still scarcely known. As indicated by Virchow's triad, the occurrence of arterial thrombosis depends on the arterial wall substrate, the local rheologic characteristic of blood flow, and systemic factors in the circulating blood.
Although a plaque-dependent thrombogenic substrate and rheologic factors appear to be implicated in thrombus formation in the carotid arteries (21), the role of systemic factors is less well known.
Current understanding of the pathophysiologic mechanisms of atherothrombosis is based on pathologic, experimental, and clinical studies of acute coronary artery syndromes (ACS). The exposure of a thrombogenic substrate, represented by lipids with tissue factor, mostly located in macrophage-rich areas, is a key factor determining the thrombogenicity of a lesion (22). The degree of stenosis caused by the ruptured plaque and the overlying mural thrombus also determines thrombogenicity, because they alter the flow rate at the lesion site. Changes in vessel geometry that increase the shear forces that are directly related to flow velocity and inversely related to the third power of the luminal diameter may result in an increase in platelet deposition at the apex of the stenosis. This process gives rise to a vicious cycle, that is, mural thrombus formation may contribute to vasoconstriction through factors released from platelets (serotonin and thromboxane A2), in turn increasing shear force–dependent platelet deposition (23).
Recently, some carotid artery plaques that remained thrombotically active for a long time after the initial clinical event, predisposing patients to a continuous release of emboli in the intracranial vascular bed, were described (5). This plaque pattern is characterized by an organizing thrombus consisting of fibrous tissue interspersed with a proteoglycan matrix containing a network of large, thin-walled vascular channels. A small area of acute thrombosis containing fibrin or platelets is always present, in association with variable numbers of macrophages and T cells (Fig. 1I) (5). Thrombotically active plaques have been detected up to 30 mo after the first acute cerebrovascular event. They were still present in 53.8% of plaques in patients undergoing surgery 24 mo after symptom onset (5).
Healed Lesions
Healed lesions often show a totally occluded lumen and contain distinct layers of dense collagen. The necrotic core is usually absent, but some lesions with healed ruptures exhibit multiple layers of lipid and necrotic core, suggesting multiple episodes of thrombosis (Fig. 1J).
Morphologic studies of coronary arteries have suggested that plaque progression beyond 50% cross-sectional–luminal narrowing usually occurs as a result of repeated ruptures, most of which are clinically silent (24). The same may be true in carotid artery disease.
MOLECULAR FACTORS ACTING ON NATURAL HISTORY OF ATHEROSCLEROSIS
Inception of Plaque
Endothelial injury has been proposed to be an early and clinically relevant pathophysiologic event in the atherosclerotic process (6). Patients with endothelial dysfunction have an increased risk for future cardiovascular events, including stroke (25). The loss of biologic activity of the endothelium reduces nitric oxide (NO) and is associated with the increased expression of prothrombotic factors, proinflammatory adhesion molecules, cytokines, and chemotactic factors. Cytokines may decrease NO bioavailability, increasing the production of reactive oxygen species (ROS). ROS reduce NO activity both directly, by reacting with endothelial cells, and indirectly, through oxidative modification of iNOS (inducible Nitric Oxide Synthase) or guanylyl cyclase (26). Low NO bioavailability upregulates vascular adhesion molecule 1 (VCAM-1) expression. VCAM-1 binds monocytes and lymphocytes to the endothelium, the first step in the invasion of the vascular wall, through the induction of nuclear factor κB expression (27). Another effect of NO is the inhibition of leukocyte adhesion (28). The reduction of NO induces the expression of monocyte chemotactic protein 1 (MCP-1), which recruits monocytes (29). NO is in a sensitive balance with endothelin 1 (ET-1), regulating vascular tone (30). Plasma ET-1 concentrations are increased in patients with advanced atherosclerosis and correlate with the severity of the disease (31). In addition to its vasoconstrictor activity, ET-1 also promotes leukocyte adhesion (32) and thrombus formation (33). Dysfunctional endothelium expresses P-selectin (stimulation by agonists such as thrombin) and E-selectin (induced by interleukin 1 [IL-1] or tumor necrosis factor-α [TNF-α]) (34). The expression both of intercellular adhesion molecule 1 (ICAM-1) by macrophages and endothelium and of VCAM-1 by endothelial cells is induced by inflammatory cytokines such as IL-1, TNF-α, and interferon-γ [IFN-γ] (35). Endothelial cells also produce MCP-1, monocyte colony-stimulating factor (M-CSF), and IL-6, which further amplify the inflammatory cascade (35). IL-6 production by smooth muscle cells represents the main stimulus for C-reactive protein (CRP) production (Fig. 3C) (36). Recent evidence suggests that CRP may contribute to the proinflammatory state of the plaque both by mediating monocyte recruitment and by stimulating monocytes to release IL-1, IL-6, and TNF-α (37). The damaged endothelium allows the passage of lipids into the subendothelial space. Fatty streaks represent the first step in the atherosclerotic process (Fig. 4).
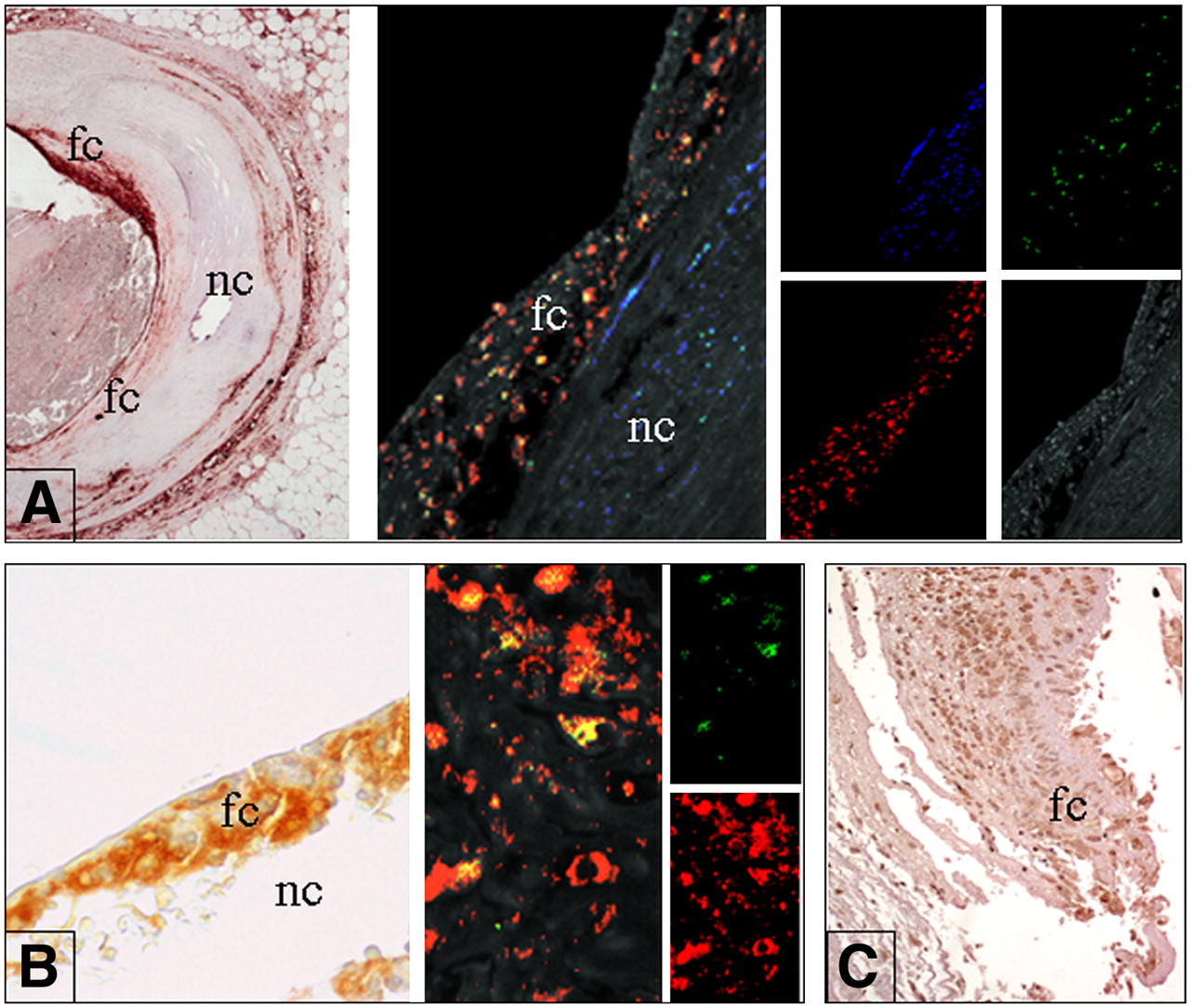
In situ expression of molecular factors. (A) In situ expression of PTX-3. (Left) Cross section of coronary artery (low-power field; magnification, ×4). Shoulder area of eroded plaque shows strong positivity for PTX-3 (conventional immunohistochemistry; 3,3′-diaminobenzidine [DAB] revealed). (Middle) Triple fluorescence immunostain studied by 2-dimensional confocal analysis demonstrates that PTX is mainly expressed by macrophages (concordant double positivity appears as yellow stain). (Right) Confocal analysis showing smooth muscle actin (smooth muscle cell antigen) reaction revealed by streptavidin–Alexa fluor 430 (Molecular Probes/Invitrogen) conjugate (blue stain), CD68 (macrophage antigen) reaction revealed by streptavidin–Texas Red fluorescent conjugate (red stain), and PTX3 reaction revealed by streptavidin–fluorescein conjugate (green stain). Plaque background is shown at bottom right (medium wave excitation UV filter). fc = fibrous cap; nc = necrotic core. (B) Expression of PAPP-A. (Left) Thin cap of ruptured plaque is rich in foam cells (fc) expressing PAPP-A at high levels and covering large necrotic core (nc) (magnification, ×40; conventional immunohistochemistry; DAB revealed). (Middle) Double fluorescent immunostain studied by 2-dimensional confocal analysis clearly shows strong and diffuse positive reaction for PAPP-A in macrophages (concordant double positivity appears as yellow stain) (magnification, ×800). (Right Top) PAPP-A reaction revealed by streptavidin–fluorescein conjugate (green stain). (Right Bottom) CD68 antibody revealed by streptavidin–Texas Red fluorescent conjugate (red stain). (C) Foam cells (fc) at site of plaque rupture strongly express IL-6 (magnification, ×20; conventional immunohistochemistry; DAB revealed).

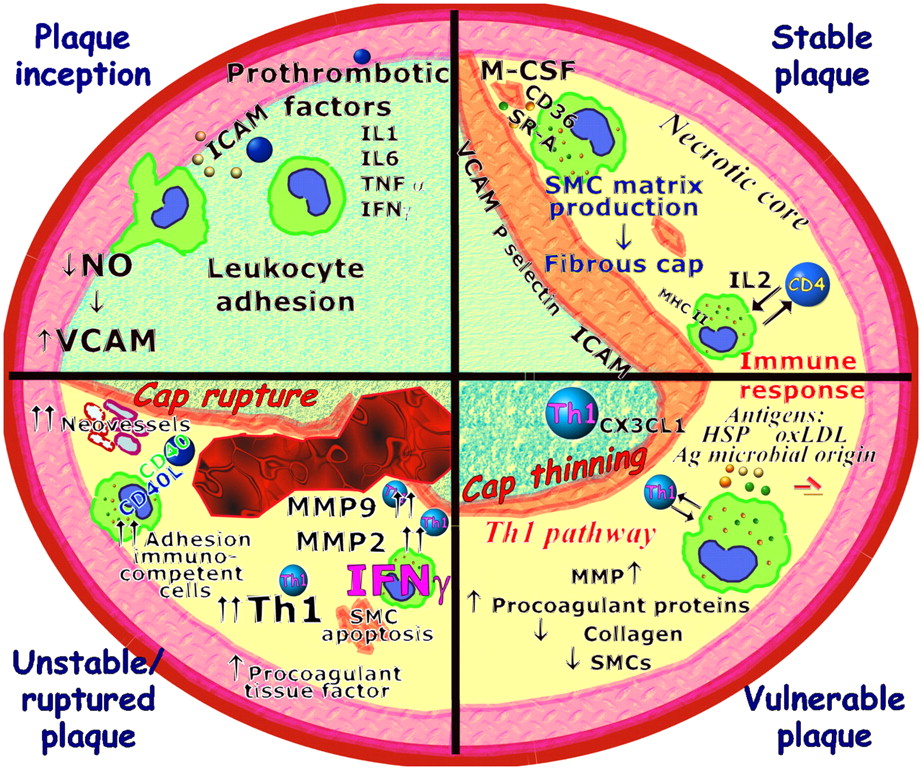
Molecular factors involved in plaque evolution. In plaque inception, activated endothelial cells increase expression of adhesion molecules and inflammatory genes. Circulating monocytes migrate into subendothelial space and differentiate into macrophages. Macrophages take up lipid deposited in intima via several receptors, including scavenger receptor A (SR-A) and CD36. Lipid-laden macrophages forming fatty streak secrete MMPs, tissue factor, and proinflammatory cytokines that amplify local inflammatory response in lesion. Repeated cycles of inflammation lead to accumulation of macrophages, some of which can die in this location, producing so-called necrotic core, and induce smooth muscle cell (SMC) proliferation and migration in lesion to form fibrous cap of advanced complicated stable atherosclerotic lesion (stable plaque). T cells may encounter antigens (Ag), such as OxLDL and heat shock proteins (HSP) of endogenous or microbial origins. Several different effector mechanisms can be elicited by immune response. Combination of IFN-γ and TNF-α upregulates expression of fractalkine (CX3CL1). This cytokine network promotes development of Th1 pathway, which is strongly proinflammatory and induces macrophage activation, superoxide production, and protease activity. Selective recruitment and activation of Th1 T cells determine potent inflammatory cascade favoring transition from stable plaque to unstable or ruptured plaque. During this transition, existence of theoretic plaque structure known as vulnerable plaque, which is very similar to unstable plaque except for plaque erosion or rupture, has been postulated.
Evolving Fibroatheromatous Plaque
The evolution of atheromas is modulated by innate and adaptive immune responses (7,36,38). The most important receptors for innate immunity in atherothrombosis are the scavenger receptors and the Toll-like receptors (39). Adaptive immunity is much more specific than innate immunity but may take several days or even weeks to be fully mobilized. It involves an organized immune response leading to the generation of T- and B-cell receptors and immunoglobulins, which can recognize foreign antigens (40).
Stable Plaque.
Macrophages phagocytose lipid deposited in the intima through several receptors, including scavenger receptor A and CD36. Deregulated uptake of modified low-density lipoprotein (LDL) through scavenger receptors leads to cholesterol accumulation and “foam cell” formation. The lipid-laden macrophages (foam cells) forming the fatty streak secrete proinflammatory cytokines that amplify the local inflammatory response in the lesion, matrix metalloproteinases (MMPs), tissue factor into the local matrix, and growth factors that stimulate the smooth muscle replication responsible for lesion growth. M-CSF acts as the main stimulator in this process, together with granulocyte-macrophage–stimulating factor and IL-2 for lymphocytes (41). Lymphocytes enter the intima by binding adhesion molecules (VCAM-1, P-selectin, ICAM-1, MCP-1 [CCL2], and IL-8 [CxCL8]) (35). Infiltrates consisting mainly of CD4+ T lymphocytes recognize antigens bound to major histocompatibility complex (MHC) class II molecules involved in antigen presentation to T lymphocytes, thus provoking an immune response (9). MHC class II molecules are expressed by endothelial cells, macrophages, and vascular smooth muscle cells in proximity to activated T lymphocytes in atherosclerotic plaque. Proinflammatory cytokines manage a central transcriptional control point mainly mediated by nuclear factor κB. Macrophage foam cells produce cytokines that activate neighboring smooth muscle cells, resulting in extracellular matrix production (9).
Repeated cycles of inflammation lead to the accumulation of macrophages, some of which die in this location, producing the so-called necrotic core, and induce smooth muscle cell proliferation and migration in the lesion to form the thick fibrous cap of advanced, complicated, stable atherosclerotic lesions (Fig. 4). These lesions are asymptomatic and often are unrecognized.
Vulnerable Plaque: Shift Toward Th1 Pattern.
T cells in plaque may encounter antigens, such as oxidized LDL (OxLDL). The number of activated T cells expressing the IL-2 receptor (CD25) is influenced by lipid-lowering treatment with statins and is correlated with 99mTc-labeled IL-2 accumulation in vulnerable carotid artery plaques (42). Moreover, a T-cell response can be triggered by heat shock proteins of endogenous or microbial origins (43).
It is still not known why the initial inflammatory response becomes a chronic inflammatory condition. However, when the plaque microenvironment triggers the selective recruitment and activation of Th1 T cells, they in turn initiate a potent inflammatory cascade.
The combination of IFN-γ and TNF-α upregulates the expression of fractalkine (CX3CL1) (44). IL-1– and TNF-α–activated endothelium also expresses fractalkine (membrane-bound form), directly mediating the capture and adhesion of CX3CR1-expressing leukocytes and causing additional leukocyte activation (45). This cytokine network promotes the development of the Th1 pathway, which is strongly proinflammatory and induces macrophage activation, superoxide production, and protease activity. In particular, Th1 T cells release IFN-γ, which plays a crucial role in atherosclerosis because it activates macrophages, promotes procoagulant protein and metalloproteinase secretion, inhibits smooth muscle proliferation, and downregulates α-actin and collagen expression (Fig. 4) (35).
PATHOBIOLOGIC DETERMINANTS OF PLAQUE RUPTURE
A plaque prone to rupture is characterized by a large lipidic–necrotic core and is separated from the adjacent vessel lumen by a thin fibrous cap containing macrophages, T lymphocytes, and other inflammatory cells. Inflammatory activity in the plaque cap has been associated with a higher incidence of preoperative ischemic neurologic and cardiovascular events (5,8,17,46,47). The major pathobiologic determinants of plaque rupture are the expression of factors that weaken the fibrous cap, physical forces acting on the fibrous cap, and newly formed microvessels (vasa vasorum).
Expression of Factors that Weaken Fibrous Cap
The fibrous cap covers the luminal side of the plaque, forming an antithrombotic wall between the highly thrombogenic lipidic–necrotic core and circulating prothrombotic factors. Its resistance to circumferential forces and shear stress depends on the presence of functioning smooth muscle cells and the related extracellular matrix that maintains the fibrous cap.
The role of the lipidic–necrotic core as a vulnerability factor is still being debated. Some authors demonstrated a larger pool of extractible lipid in symptomatic patients than in asymptomatic patients (48). Conversely, Bassiouny et al. demonstrated that in carotid artery plaque, the most important factor for plaque rupture is the distance of the lipidic–necrotic core from the fibrous cap (49). Cell migration into the lesion, proliferation of elements in the lesion, and the production and degradation of the extracellular matrix are all factors in the transition from stable plaque to vulnerable plaque. A limited number of T cells following the Th1 pathway initiate the production of a series of cytokines orchestrating the transition from stable plaque to unstable plaque (Fig. 4) (7,50).
Within the plaque, foam cells and monocyte-derived macrophages produce matrix-degrading enzymes, cytokines, and growth factors that reduce the stability of the extracellular matrix. In particular, IFN-γ suppresses the synthesis of collagen, a major component of the fibrous cap (35), whereas the infiltration of mononuclear cells results in the release of proteases, which also cause plaque disruption (51).
ROS produced within the plaque have important implications for its structural integrity (26). Deregulated oxidant production promotes the activation of matrix-degrading enzymes in the fibrous cap of the plaque. Moreover, impaired NO function, coupled with oxidative excess, may activate MMPs (MMP-2 and MMP-9), which weaken the fibrous cap (52). Another mechanism responsible for thinning of the fibrous cap is the apoptosis of smooth muscle cells. There is, in fact, evidence for extensive apoptosis of smooth muscle cells in the fibrous cap in advanced atherosclerosis as well as in cultured plaques (53).
Physical Forces Acting on Fibrous Cap
Circumstantial evidence, mostly derived from mathematic models, indicates that circumferential force, shear stress, and vasospasm may trigger the sudden rupture of a plaque that has already been modified by the factors described earlier.
The shear force is directly related to the flow rate and inversely related to the third power of the luminal diameter and contributes to determining plaque rupture and thrombus growth (54). This force acts tangentially on the interface of plaque components with different degrees of compliance, thus favoring the sliding of the fibrous cap over the lipidic–necrotic core (55).
Another triggering action may be vasospasm, which forces the plaque content through a weakened plaque cap, producing an effect like a volcanic eruption (56).
According to Laplace's law, the circumferential stress induced by blood pressure on the plaque is the result of the endoluminal pressure times the luminal radius. Hence, theoretically, moderately or slightly stenotic plaques, if covered by a thin cap, should be at greater risk for rupture than those with severe stenosis.
POTENTIAL FACTORS CONTRIBUTING TO PLAQUE INSTABILITY
Adventitia Inflammation
The tunica adventitia is involved in the inflammatory process of atherosclerosis. This information, obtained mainly in the aorta, suggests an active role of an adventitial lesion in generating an immune response (57–59). Houtkamp et al. demonstrated the presence of follicular aggregates composed of B and T cells, reticulodendritic cells (CD21+), and macrophages in the aortic adventitia (59). These infiltrates resemble mucosa-associated lymphoid tissue and could play an active role in the humoral immune response of advanced atherosclerosis.
In the abdominal aorta, the rate of inflammation was found to be higher in the tunica media and adventitia underneath ruptured plaques than in the media and adventitia under fatty streaks or fibrous plaques (60).
Few studies have been conducted for the coronary arteries. Kohchi et al. (57) and Stratford et al. (61) observed a significant increase in the rate of adventitial inflammation in patients with fatal AMI. Neither group correlated the adventitial infiltrate with the plaque type. More recently, Higuchi et al. demonstrated significantly more lymphocytes and microvessels in coronary culprit lesions than in stable lesions in patients with fatal AMI (62).
Maseri et al. (23) hypothesized a role for an adventitial inflammatory infiltrate in coronary vasospasm.
In the outer layer of the adventitia of infarct-related coronary arteries in patients with myocardial infarction, besides lymphocytes and macrophages, numerous mast cells were found in contact with sensory nerve fibers (63). Neurogenic stimulation of mast cells in the adventitia of coronary arteries may favor the release of vasoactive compounds (i.e., histamine and leukotrienes) that can contribute to the complex neurohormonal response leading to abnormal coronary vasoconstriction.
Neoangiogenesis
In the shoulder of the plaque, a plexus of newly formed small and large vessels is often seen. Microarray gene chip analysis (64) revealed that the newly formed vessels are associated with increased angiogenic gene expression (i.e., angiopoietin 2, angiogenic inducer 61, and neuropilin 1). These vessels are weak and therefore could be responsible for intraplaque hemorrhage. Intraplaque hemorrhage causes a sudden increase in plaque volume and pressure, leading to plaque instability.
Moreover, the inflamed endothelium expresses high levels of E-selectin, ICAM-1, and VCAM-1 (64). Therefore, these activated endothelial cells might be the local source of leukocytes recruited into an atherosclerotic lesion (65). Microvessels in lipid-rich plaques also express increased levels of ICAM-1, VCAM-1, E-selectin, and CD40 (66). The expression of CD40 is prominent in processes associated with angiogenesis and inflammation. CD40 and its counterpart, CD40 ligand (CD40L, also called CD154), may play a central role in both experimental and human atherosclerotic plaque progression and destabilization. Interactions between CD40 and CD40L stimulate endothelial cells to express adhesion molecules and produce several proinflammatory cytokines and chemokines (67). Moreover, binding of CD40 results in the production of metalloproteinases (68), fibroblast growth factor (69), and vascular endothelial growth factor and promotes vascular endothelial growth factor–dependent angiogenesis (Fig. 4) (68).
The new blood channels formed in the plaque are associated with mononuclear infiltrates (66). Neovascularization and expression of adhesion molecules by microvessels at sites of vulnerable plaques may sustain the influx of inflammatory cells and hence may contribute to plaque destabilization (66). Moreover, infiltration of mononuclear cells stimulates the release of proteases (MMPs), which cause plaque disruption (64).
Plaque Hemorrhage
Intraplaque hemorrhage facilitates a more rapid progression and rupture of the plaque. The origin of plaque hemorrhage is still uncertain. It has been suggested that hemorrhage into a plaque occurs from cracks or fissures originating from the luminal surface (70). The fissuring of the fibrous cap occurs at its thinnest portion, typically at the shoulder region, thereby allowing the entry of blood into the necrotic core. Alternatively, intraplaque hemorrhage has been considered to be secondary to the rupture of vasa vasorum (71), a common feature of advanced lesions showing plaque rupture and luminal thrombosis.
In neoangiogenesis, the superficial and deep newly formed vessels show a characteristic angiomatous aspect, with relatively thinner walls. These small and fragile vessels may represent the first cause of morphologic changes leading to an intramural hemorrhage. Microvessel density was shown to be increased in lesions with severe macrophage infiltration at the fibrous cap and at the shoulder of the plaque (72).
Intraplaque hemorrhage is common in advanced coronary atherosclerotic lesions. Intraplaque hemorrhage contributes to the growth of the lipid necrotic core because extravasated red cell membranes contribute cholesterol to the lesion. In fact, Kolodgie et al. (73) detected glycophorin A, an erythrocyte protein, in early lesions, such as pathologic intimal thickening or fibrous cap atheromas. Fibroatheromas with late-stage core necrosis or thin caps showed a marked increase in glycophorin A expression, closely related to cholesterol clefts and associated with greater macrophage infiltration.
In the carotid arteries, as in the coronary arteries, the presence of intraplaque hemorrhage seems to stimulate plaque progression, as recently demonstrated by Takaya et al. (74) and Saam et al. (75) by means of MRI studies.
DIFFUSE INFLAMMATION AND VULNERABILITY
Despite the predominant hypothesis focusing on the responsibility of a specific vulnerable atherosclerotic plaque rupture (1) for ACS, some physiopathologic, clinical, and angiographic observations suggest that the principal cause of coronary instability is not to be found in the vulnerability of a single atherosclerotic plaque but in the presence of multiple vulnerable plaques in the entire coronary tree, correlated with the presence of a diffuse inflammatory process (46,47,76,77). Recent angiographic studies demonstrated multiple vulnerable plaques in patients with unstable angina (78) and in those with transmural myocardial infarction (77). Flow cytometry studies demonstrated the presence of an active multicentric inflammatory infiltrate in the coronary vessels of patients with fatal AMI (47). Support for the multicentric hypothesis was demonstrated by Buffon et al., on the basis of neutrophil myeloperoxidase (MPO) activity in the coronary vessels of individuals with unstable angina (76). In addition, we performed a morphologic study and demonstrated an inflammatory infiltrate of activated macrophages and T lymphocytes in the entire coronary tree (including stable plaques) in individuals with fatal AMI. These plaques showed a 2- to 4-fold-higher level of inflammatory infiltrate than samples from age-matched individuals with chronic stable angina or without a clinical history of cardiac disease and with noncardiac causes of death (46). Furthermore, in that study, histopathologic examination revealed an average of 6.7 vulnerable coronary plaques per patient with fatal AMI, in addition to plaques with endoluminal thrombosis, compared with 0.8 and 1.4 vulnerable lesions per patient in the individuals with chronic stable angina and in the individuals without a clinical history of cardiac disease, respectively (46).
Moreover, we recently demonstrated that activated T lymphocytes infiltrate the myocardium both in the periinfarct area and in remote, unaffected myocardial regions in patients with a first AMI (79). The simultaneous occurrence of diffuse coronary inflammation and myocardial inflammation in these patients further supports the concept that both coronary and myocardial vulnerabilities concur in the pathogenesis of fatal AMI. AMI is therefore likely to be the consequence of a diffuse “active” chronic inflammatory process that determines the destabilization of lesions in the entire coronary tree and not just the culprit lesion. Little is known about the causes of the diffuse inflammation associated with myocardial infarction. The presence of activated T lymphocytes suggests the in situ presence of an antigenic stimulus that triggers adaptive immunity.
SERUM MARKERS CORRELATED WITH PLAQUE INFLAMMATION
In recent years, several studies have correlated different serologic biomarkers with cardiovascular disease, leading to a rapid increase in the number of available biomarkers (Table 2). These biomarkers are useful in that they can identify a population at risk of an acute ischemic event and detect the presence of so-called vulnerable plaques or vulnerable patients. Ideally, a biomarker must have certain characteristics to predict the incidence of vascular disease. Measurements have to be reproducible in multiple independent samples, the method for determination should be standardized, variability should be controlled, and sensitivity and specificity should be high. In addition, the biomarker should add information to that provided by other, established risk markers and should reflect the underlying biologic process associated with plaque burden and progression.
Serologic Markers of Vulnerable Plaques and Patients
Traditional biomarkers for cardiovascular risk include LDL cholesterol and glucose. However, 50% of heart attacks and strokes occur in individuals who have normal LDL cholesterol levels, and 20% of major adverse events occur in patients with no accepted risk factors (80). Therefore, in light of changing atherosclerosis models, vulnerable blood may be better described as blood that has an increased level of activity of plasma determinants of plaque progression and rupture.
In this context, proposed biomarkers fall into 9 general categories: inflammatory markers, markers of plaque erosion, markers of thrombosis, lipid-associated markers, markers of endothelial dysfunction, oxidative stress, metabolic markers, markers of neovascularization, and genetic markers. As mentioned earlier, some of these markers may indeed reflect the natural history of atherosclerotic plaque growth and may not be directly related to an increased risk of cardiovascular events. On the other hand, markers related to complex plaque morphologic features may reflect an active process within the plaque which, in turn, relates to the onset of local complications and acute clinical events.
The best outcomes may be achieved by use of a panel of markers that will capture all of the different processes involved in plaque progression and plaque rupture and that will enable clinicians to quantify an individual patient's true risk for cardiovascular events. In all likelihood, a combination of genetic markers (representing heredity) and serum markers (representing the net interaction between heredity and environment) will ultimately be used in primary prevention. Finally, different noninvasive and invasive imaging techniques may be coupled with biomarker detection to increase the specificity, sensitivity, and overall predictive value of each potential diagnostic technique.
Markers of Inflammation
Markers of inflammation include CRP, pentraxin 3 (PTX-3), inflammatory cytokine soluble CD40L (sCD40L), soluble vascular adhesion molecules, and TNF. All of these are variably expressed in situ in vulnerable and unstable plaques (Fig. 3).
CRP is a circulating pentraxin that plays a major role in the human innate immune response (81) and provides a stable plasma biomarker for low-grade systemic inflammation. CRP is produced predominantly in the liver as part of the acute-phase response. However, CRP is also expressed in smooth muscle cells within diseased atherosclerotic arteries and has been implicated in multiple aspects of atherogenesis and plaque vulnerability, including the expression of adhesion molecules, the induction of NO, altered complement function, and inhibition of intrinsic fibrinolysis (82). CRP is considered to be an independent predictor of unfavorable cardiovascular events in patients with atherosclerotic disease. Beyond the ability of CRP to predict risk for both primary and secondary prevention purposes, interest in it has increased with the recognition that a statin-induced reduction of CRP levels is associated with a lower incidence of adverse cardiovascular events, independent of the lipid-associated changes (83). The efficacy of statin therapy may be related to the underlying level of vascular inflammation, as detected by high sensitive CRP (hs-CRP). Among patients with stable angina and established coronary artery disease (CAD), plasma levels of hs-CRP have consistently been associated with the risk of recurrent cardiovascular events (84). Similarly, in the presence of acute coronary ischemia, levels of hs-CRP are predictive of a high risk of vascular events even if troponin levels are not detectable, suggesting that inflammation is associated with plaque vulnerability even in the absence of detectable myocardial necrosis (85). Despite these data, the most relevant use of hs-CRP remains in the setting of primary prevention. To date, over 2 dozen large-scale prospective studies have shown baseline levels of hs-CRP to independently predict future myocardial infarction, stroke, death from cardiovascular disease, and peripheral arterial disease (86). Moreover, 8 major prospective studies have had adequate power to evaluate hs-CRP after adjustment for all Framingham Heart Study covariates, and all have confirmed the independence of hs-CRP (87). Despite the evidence described earlier, it is important to recognize that there are currently no firm data showing that lowering CRP levels per se will lower the risk of vascular events. Further, as with other biomarkers of inflammation, it remains controversial whether CRP plays a direct causal role in atherogenesis (88), and ongoing work with targeted CRP-lowering agents will be required to fully test this hypothesis. However, the clinical utility of hs-CRP has been well established, and on the basis of data available through 2002, the Centers for Disease Control and Prevention and the American Heart Association endorsed the use of hs-CRP as an adjunct to global risk prediction, particularly among individuals with intermediate risk (89). Data available since 2002 strongly reinforce these recommendations and suggest expansion to lower-risk groups as well as individuals taking statin therapy. Perhaps most importantly, data for hs-CRP provide evidence that biomarkers beyond those traditionally used for vascular event risk detection and monitoring can play important clinical roles in prevention and treatment.
Cellular adhesion molecules can be considered potential markers of vulnerability because such molecules are activated by inflammatory cytokines and then released by the endothelium. These molecules represent the sole available markers for assessing endothelial activation and vascular inflammation. The Physicians' Health Study evaluated more than 14,000 healthy subjects, demonstrated a positive correlation of ICAM-1 expression with the risk of cardiovascular events, and showed that subjects in the higher quartile of ICAM-1 expression had 1.8-times-higher risk than subjects in the lower quartile (90). Furthermore, soluble ICAM-1 and VCAM-1 levels showed a positive correlation with atherosclerosis disease burden (91). IL-6 is expressed during the early phases of inflammation and is the principle stimulus for liver CRP production (Fig. 3C). In addition, CD40L, a molecule expressed on the cellular membrane, is a TNF-α homolog that stimulates activated macrophage proteolytic substance production (92).
CD40 and CD40L have been found on platelets and several other cell types in functionally bound and soluble (sCD40L) forms. Although many platelet-derived factors have been identified, recent evidence suggests that CD40L is actively involved in the pathogenesis of ACS. CD40L drives the inflammatory response through the interaction between CD40L on activated platelets and the CD40 receptor on endothelial cells. This interaction facilitates the increased expression of adhesion molecules on the surface of endothelial cells and the release of various stimulatory chemokines. These events, in turn, facilitate the activation of circulating monocytes as a trigger of atherosclerosis. Beyond the known proinflammatory and thrombotic properties of CD40L, experimental evidence suggests that CD40L-induced platelet activation leads to the production of reactive oxygen and nitrogen species, which are able to prevent endothelial cell migration and angiogenesis (93). As a consequence of the inhibition of endothelial cell recovery, the risk of subsequent coronary events may be higher. Clinical studies have supported the involvement of CD40L in ACS and the prognostic value of CD40L in individuals with ACS. Levels of sCD40L have been shown to be an independent predictor of adverse cardiovascular events after ACS (94), with increased levels portending a worse prognosis (95). Importantly, specific therapeutic strategies have shown to be beneficial in reducing risk associated with sCD40L (96).
IL-18 is a proinflammatory cytokine that is mainly produced by monocytes and macrophages, and it acts synergistically with IL-12 (21). Both of these interleukins are expressed in the atherosclerotic plaque and stimulate the induction of IFN-γ which, in turn, inhibits collagen synthesis, preventing thick fibrous cap formation and facilitating plaque destabilization. Mallat et al. (97) examined 40 stable and unstable atherosclerotic plaques obtained from patients undergoing carotid endarterectomy; they highlighted how IL-18 expression was higher in macrophages and endothelial cells extracted from unstable lesions than in those extracted from stable lesions and was correlated with clinical (symptomatic plaques) and pathologic signs of vulnerability. Pregnancy-associated plasma protein A (PAPP-A) is a high-molecular-weight, zinc-binding metalloproteinase that is typically measured in blood during pregnancy and that is later found in macrophages and smooth muscle cells in unstable coronary atherosclerotic plaques. This protease cleaves the bond between insulinlike growth factor 1 (IGF-1) and its specific inhibitors (IGFBP-4 and IGFBP-5 [IGFBP is insulinlike growth factor binding protein]), increasing free IGF-1 levels (98). IGF-1 is important for monocyte and macrophage chemotaxis and activation in atherosclerotic lesions, with consequent proinflammatory cytokine and proteolytic enzyme release, and stimulates endothelial cell migration and organizational behavior, with consequent neoangiogenesis. Hence, IGF-1 represents one of the most important mediators in the transformation of a stable lesion into an unstable one. Bayes-Genis et al. (99) demonstrated that PAPP-A is expressed at higher levels in the serum of patients with ACS (unstable angina, myocardial infarction) than in the serum of patients with stable angina. In particular, serum PAPP-A levels of greater than 10 mIU/L recognize patient vulnerability with a specificity of 78% and a sensitivity of 89%. Recently, we demonstrated that the level of PAPP-A histologic expression is higher in complex, vulnerable or ruptured carotid artery plaques than in stable lesions (Fig. 3B) (100). Because serum PAPP-A levels can be easily measured by means of an enzyme-linked immunosorbent assay, this protease could represent an easily quantifiable marker of vulnerability, with a reproducible method, allowing the identification of a subgroup of patients at high risk for cerebrovascular events before the manifestation of a clinical event.
Jaffer et al. recently published a detailed review of different techniques, based on several biomarkers that have been implemented in recent years, for the detection of vulnerable plaque (101). In this context, plaques with active inflammation may be identified directly by extensive macrophage accumulation. Possible intravascular diagnostic techniques based on the determination of inflammatory infiltration within the plaque include thermography (102), contrast-enhanced MRI (103), 18F-FDG PET (104), and immunoscintigraphy (42). In addition, noninvasive techniques include MRI with superparamagnetic iron oxide (105) and gadolinium fluorine compounds (106).
Metabolic Markers
Insulin and glucose are classic metabolic markers of insulin resistance. Recent research has focused on adipokines that may be involved in atherogenesis, including leptin and resistin, and inflammatory cytokines released by adipose tissue (i.e., TNF) or in response to their release (i.e., CRP) (107). Adiponectin, an adipose tissue–derived cytokine that appears to be vasoprotective, may be a prognostic marker of a good cardiovascular outcome.
Lipid Markers
Lipid markers, in addition to the classic LDL and high-density lipoprotein (HDL) cholesterol, include OxLDL cholesterol, small dense LDL cholesterol, lipoprotein (a) [Lp(a)], and lipoprotein-associated phospholipase A2 (Lp-PLA2). Oxidation of cholesterol typically occurs in the diseased vessel wall and plays a major role in foam cell formation. There are different forms of OxLDL cholesterol, depending on which component—apolipoprotein or lipid—is oxidized. Human carotid and coronary arteries are significantly enriched in OxLDL (108) and, importantly, unstable plaques appear to be preferentially enriched in OxLDL (109). In the last 5 y, an increasing number of studies have evaluated the role of OxLDL in preclinical atherosclerosis, endothelial dysfunction, stable CAD, ACS, percutaneous coronary intervention, and response to statins. Increased OxLDL cholesterol levels are associated with increased carotid artery intima–media thickness in asymptomatic subjects and impaired flow-mediated vasodilatation. Plasma OxLDL levels correlate with the presence of CAD (110). Toshima et al. (111) showed that plasma OxLDL-DLH3 levels were higher in patients with CAD than in healthy control subjects and reported that the receiver operating characteristic curves showed that the area under the curve was higher for OxLDL-DLH3 levels than for total cholesterol, apolipoprotein B, HDL-C, and triglyceride levels. Similarly, Holvoet et al. (112) showed that OxLDL-4E6 levels were higher in older (mean age, 74 y) patients with CAD, risk equivalent to the risk of CAD, and metabolic syndrome. Three recent studies assessed the prognostic utility of OxLDL measures. In a cross-sectional study, Holvoet et al. (113) showed that OxLDL-4E6 levels did not predict overall CAD but did predict myocardial infarction in an older cohort. In a prospective study, Shimada et al. (114) observed 238 patients with CAD for a mean of 52 mo and showed that baseline levels of OxLDL-DLH3 were significantly higher in patients with the subsequent development of cardiac death, nonfatal myocardial infarction, and unstable angina. In a prospective study, Wallenfeldt et al. (115) showed that baseline OxLDL-4E6 levels predicted the progression of carotid artery intima–media thickness in asymptomatic and presumably healthy 58-y-old Swedish men, independent of other cardiovascular risk factors. In summary, OxLDL may serve as an attractive biomarker because it may provide a link between lipoprotein disorders and inflammation.
Lp(a) is a unique lipoprotein, similar to LDL cholesterol except for additional apoprotein (a), which is homologous to plasminogen. The association of Lp(a) with CAD and its ability to act as a biomarker of risk appear to be strongest in patients with hypercholesterolemia and, in particular, in young patients with premature atherosclerosis. In this regard, increased levels of Lp(a) (>30 mg/dL) in plasma independently predict the presence of symptomatic and angiographically determined CAD, particularly in patients with elevated LDL cholesterol levels (116).
Lp-PLA2 is a 50-kDa, Ca2+-independent enzyme associated with LDL. Small dense LDL particles are very atherogenic and readily undergo oxidative modification (117). The enzyme is a subtype of a growing family of A2 phospholipases and is secreted mainly by macrophages, monocytes, mast cells, and T lymphocytes. The enzyme has proinflammatory properties because it hydrolyzes oxidized phospholipids to lysophosphatidylcholine and free oxidized fatty acids and thus is the enzyme responsible for most of the increased lysophosphatidylcholine content of OxLDL particles. The atherogenic potential of OxLDL has been attributed to this high lysophosphatidylcholine content. Several prospective epidemiological studies have reported that Lp-PLA2 is a predictor of CAD (117), although controversy persists as to its independence from LDL cholesterol. The relationship of Lp-PLA2 to LDL cholesterol is also supported by several studies showing equivalent decreases in Lp-PLA2 and LDL cholesterol levels in response to several different classes of lipid-lowering agents (118). Further, in contrast to the situation for CRP, the levels of which are reduced by statin therapy in a manner independent of the effects on LDL cholesterol, there is little evidence that statins lower Lp-PLA2 levels once LDL cholesterol level reduction is accounted for (119). Nonetheless, in a nested case-control study of hyperlipidemic patients in the West of Scotland Coronary Prevention Study, elevated baseline Lp-PLA2 levels were found to be independent predictors of death, myocardial infarction, and revascularization in men, although the odds ratio was only ∼1.2 (120).
Markers of Plaque Neovascularization and Thrombosis
Several experimental and clinical studies have suggested that plaque neovascularization plays a role in plaque growth and progression (121). Different angiogenic cytokines, including placental growth factor and stroma-derived factor 1, may be potential biomarkers for these processes (122). Nicotine is an angiogenic agent that also plays a role in plaque progression. In an apolipoprotein E-deficient, hypercholesterolemic mouse model, nicotine increased plaque growth, with increased neovascularization (123). Tissue factor, a thrombogenic protein secreted by macrophages, plays a dominant role in thrombosis subsequent to plaque rupture. Tissue factor is highly concentrated in the lipid core, and levels of tissue factor in plasma are elevated in patients bearing multiple cardiovascular risk factors (21).
Markers of Endothelial Dysfunction
Compromise of endothelial integrity is believed to be fundamental not only to the initiation and progression of atherosclerotic disease but also to the onset of ACS. Leukocytes are believed to contribute to direct endothelial damage in this setting. Irrespective of the underlying contributor, endothelial damage and dysfunction remain integral to atherogenesis and the development of ACS. Numerous studies have confirmed that endothelial vasodilator dysfunction is independently predictive of cardiovascular events (124). Acetylcholine releases NO, prostacyclin, and other vasodilators from the endothelium. The results of studies of acetylcholine-induced vasoreactivity in patients undergoing catheterization showed that patients with endothelial vasodilator dysfunction exhibited vasoconstriction, rather than vasodilatation, in response to acetylcholine. Such patients also had a worse prognosis than patients who responded normally (125).
Potential markers of endothelial dysfunction include NO, asymmetric dimethylarginine (ADMA), soluble vascular adhesion molecules, von Willebrand factor, and endothelial progenitor cells. NO, which is a vasodilator, is also a vasoprotective molecule that inhibits muscle cell proliferation, leukocyte adhesion, and platelet adherence and aggregation. In the circulatory system, ADMA, an arginine analog, competes with arginine and inhibits the production of NO. Several studies have shown that ADMA levels are elevated in individuals with cardiovascular risk factors and that ADMA may itself predispose individuals to cardiovascular events (126). The levels of soluble vascular adhesion molecules and von Willebrand factor are known to be increased with endothelial dysfunction. Endothelial progenitor cells are bone marrow–derived stem cells for the endothelium and for the vascular smooth muscle cells that can resurface injured endothelium or participate in angiogenesis (127). Recent studies indicated that the number of progenitor cells is inversely correlated with ADMA and major cardiovascular events and is directly correlated with the endothelial vasodilator response (128). Because it is difficult to measure endothelial progenitor cells directly, potential biomarkers of circulating endothelial progenitor cells include sKit ligand and stroma-derived factor, which are present at increased levels and participate in mobilizing endothelial progenitor cells from the bone marrow (129).
Oxidative Stress Markers
Oxidative stress plays a very important role in atherogenesis (26). Evidence shows that the activation of vascular oxidative enzymes leads to lipid oxidation, foam cell formation, expression of vascular adhesion molecules and chemokines, and ultimately atherogenesis. MPO is a heme peroxidase that is present in and secreted by activated phagocytes at sites of inflammation. MPO can generate several reactive, oxidation-derived intermediates, all mediated through a reaction with hydrogen peroxide, to induce oxidative damage to cells and tissues (130). Oxidation products from MPO are found at significantly increased levels (up to 100-fold higher than circulating LDL) on LDL isolated from atherosclerotic lesions (131) and lead to accelerated foam cell formation through nitrated apolipoprotein B-100 on LDL and uptake by scavenger receptors (132). Accumulating evidence suggests that MPO may play a causal role in plaque vulnerability (133). Sugiyama et al. (134) showed that advanced ruptured human atherosclerotic plaques derived from patients with sudden cardiac death strongly expressed MPO at sites of plaque rupture, in superficial erosions, and in the lipid core, whereas fatty streaks exhibited little MPO expression. In addition, MPO macrophage expression and HOCl were highly colocalized immunochemically in culprit lesions in these patients. Several inflammatory triggers, such as cholesterol crystals and CD40L, induce the release of MPO and HOCl production from MPO-positive macrophages in vitro (134). Consistent with the potential role of MPO in the atherosclerotic process, genetic polymorphisms resulting in MPO deficiency or diminished activity are associated with lower cardiovascular risk, although the generalizability of these findings is uncertain (135). In parallel with the effects of MPO on NO, LDL oxidation by MPO, and the presence of MPO within ruptured plaques, several recent clinical studies have suggested that MPO levels may provide diagnostic and prognostic data regarding endothelial function, angiographically determined CAD, and ACS. In a case-control study of 175 patients with angiographically determined CAD, Zhang et al. (136) showed that the highest quartiles of both blood and leukocyte MPO levels were associated with odds ratios of 11.9 and 20.4, respectively, for the presence of CAD, in comparison with the lowest quartiles. Brennan et al. (135) obtained MPO levels in the emergency department in 604 patients presenting with chest pain and found no initial evidence of myocardial infarction but showed that MPO levels predicted the in-hospital development of myocardial infarction, independent of other markers of inflammation, such as CRP. In addition, they showed that MPO levels were strong predictors of death, myocardial infarction, and revascularization 6 mo after the initial event. The current data suggest that MPO may serve as both a marker of disease, providing independent information on diagnosis and prognosis for patients with chest pain, and a potential marker for the assessment of plaque progression and destabilization at the time of acute ischemia.
FUTURE CHALLENGES IN TREATMENT OF VULNERABLE PLAQUES
With the concept of vulnerable plaque not nearly as straightforward as once thought, there are challenges to creating a therapeutic strategy for assessing the risk of rupture of vulnerable plaques in asymptomatic patients.
First, there must be an ability to identify the vulnerable plaque with noninvasive or invasive techniques. It has been demonstrated that coronary artery plaque composition can be predicted by invasive and noninvasive imaging techniques, allowing real-time analysis and in vivo plaque characterization, but clear identification of thin fibrous cap fibroatheroma is not possible yet; moreover, the severity of inflammatory infiltration of the cap, which certainly plays a major role in plaque disruption, cannot be evaluated yet. Moreover, dynamic plaque changes, such as abrupt intraplaque hemorrhages from the vasa vasorum, which may be fundamental in predicting the potential for a plaque to rupture, will be extremely difficult to identify with real-time imaging techniques.
A second challenge is that a lesion-specific approach requires that the number of vulnerable plaques in each patient needs to be known and the number of such lesions needs to be limited. That is not the case, however. Several pathologic studies have indicated the presence of multiple lipid-rich vulnerable plaques in patients with fatal ACS or with sudden coronary death (46,77). Further complicating the issue is that coronary artery occlusion and myocardial infarction usually evolve from mild to moderate stenosis 68% of the time, according to an analysis of data from different studies.
The third and fourth challenges are that the natural history of the vulnerable plaque (with respect to the incidence of acute events) has to be documented in patients treated with patient-specific systemic therapy, and the approach has to be proven to significantly reduce the incidence of future events relative to the natural history. At this time, neither is documented or proved.
Fifth, we believe that at the present time, it is not possible to know which vulnerable plaques will never rupture. Although we suspect that it is the vast majority of them, our focus may have to shift to a more appropriate therapeutic target. In addition, targeting not only the vulnerable plaque but also the vulnerable blood (prone to thrombosis) or vulnerable myocardium (prone to life-threatening arrhythmia) may be important in reducing the risk of fatal events.
CONCLUSION
Because atherosclerosis is now recognized as a diffuse, multisystemic, and chronic inflammatory disorder involving the vascular, metabolic, and immune systems, with various local and systemic manifestations, it is essential to assess total patient vulnerability and not just search for a single, unstable plaque. A composite vulnerability index score comprising the total burden of atherosclerosis and vulnerable plaques in the aorta and coronary, carotid, and femoral arteries and the blood vulnerability factors should be the method of risk stratification. Obviously, such an index is difficult to achieve with currently available tools. The challenge is to identify patients at high risk of acute vascular events before clinical syndromes develop. At present, aside from imaging modalities, such as ultrasonography and MRI, and local temperature probes that could help to identify vulnerable plaques, highly sensitive inflammatory circulating markers, such as hs-CRP, cytokines, PAPP-A, and pentraxin 3 are currently the best candidates for diffuse active plaque detection. In order to achieve this aim, a coordinated effort is needed to promote the application of the most promising tools and to develop new screening and diagnostic techniques for identifying vulnerable patients.
Footnotes
-
↵* NOTE: FOR CE CREDIT, YOU CAN ACCESS THIS ACTIVITY THROUGH THE SNM WEB SITE (http://www.snm.org/ce_online) THROUGH November 2008.
-
No potential conflict of interest relevant to this article was reported.
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- 1.↵
- 2.
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- Received for publication March 12, 2007.
- Accepted for publication August 8, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- NATURAL HISTORY OF ATHEROSCLEROTIC PLAQUES
- MOLECULAR FACTORS ACTING ON NATURAL HISTORY OF ATHEROSCLEROSIS
- PATHOBIOLOGIC DETERMINANTS OF PLAQUE RUPTURE
- POTENTIAL FACTORS CONTRIBUTING TO PLAQUE INSTABILITY
- DIFFUSE INFLAMMATION AND VULNERABILITY
- SERUM MARKERS CORRELATED WITH PLAQUE INFLAMMATION
- FUTURE CHALLENGES IN TREATMENT OF VULNERABLE PLAQUES
- CONCLUSION
- Footnotes
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- Association between endostatin and mortality in patients with acute dyspnoea, with or without congestive heart failure: a single-centre, prospective, observational study
- A Prediction Tool for Plaque Progression Based on Patient-specific Multi-Physical Modeling
- High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1
- Supraclavicular Brown Adipose Tissue 18F-FDG Uptake and Cardiovascular Disease
- Age- and Hypertension-Associated Protein Aggregates in Mouse Heart Have Similar Proteomic Profiles
- Relationship Between Measures of Adiposity, Arterial Inflammation, and Subsequent Cardiovascular Events
- Myocardin Regulates Vascular Smooth Muscle Cell Inflammatory Activation and Disease
- In Vivo Evaluation of Atherosclerotic Plaque Inflammation and of Anti-Inflammatory Effects of Statins by 18F-Fluorodeoxyglucose Positron Emission Tomography
- Interleukin-1 Mediates Neuroinflammatory Changes Associated With Diet-Induced Atherosclerosis
- Carotid Plaque Inflammation Is Associated With Cerebral Microembolism in Patients With Recent Transient Ischemic Attack or Stroke: A Pilot Study
- Watershed Infarcts in Transient Ischemic Attack/Minor Stroke With >=50% Carotid Stenosis: Hemodynamic or Embolic?
- Multimodality Cardiovascular Molecular Imaging Technology
- Insufficient Deactivation of the Protein Tyrosine Kinase Lck Amplifies T-Cell Responsiveness in Acute Coronary Syndrome
- One Step Closer to Imaging Vulnerable Plaque in the Coronary Arteries