


Visual Abstract

Abstract
α-Particle emitters targeting the prostate-specific membrane antigen (PSMA) proved effective in treating patients with prostate cancer who were unresponsive to the corresponding β-particle therapy. 211At is an α-emitter that may engender less toxicity than other α-emitting agents. We synthesized a new 211At-labeled radiotracer targeting PSMA that resulted from the search for a pharmacokinetically optimized agent. Methods: A small series of 125I-labeled compounds was synthesized from tin precursors to evaluate the effect of the location of the radiohalogen within the molecule and the presence of lutetium in the chelate on biodistribution. On that basis, 211At-3-Lu was selected and evaluated in cell uptake and internalization studies, and biodistribution and PSMA-expressing (PSMA+) PC3 PIP tumor growth control were evaluated in experimental flank and metastatic (PC3-ML-Luc) models. A long-term (13-mo) toxicity study was performed for 211At-3-Lu, including tissue chemistries and histopathology. Results: The radiochemical yield of 211At-3-Lu was 17.8% ± 8.2%. Lead compound 211At-3-Lu demonstrated total uptake within PSMA+ PC3 PIP cells of 13.4 ± 0.5% of the input dose after 4 h of incubation, with little uptake in control cells. In SCID mice, 211At-3-Lu provided uptake that was 30.6 ± 4.8 percentage injected dose per gram (%ID/g) in PSMA+ PC3 PIP tumor at 1 h after injection, and this uptake decreased to 9.46 ± 0.96 %ID/g by 24 h. Tumor–to–salivary gland and tumor-to-kidney ratios were 129 ± 99 at 4 h and 130 ± 113 at 24 h, respectively. Deastatination was not significant (stomach, 0.34 ± 0.20 %ID/g at 4 h). Dose-dependent survival was demonstrated at higher doses (>1.48 MBq) in both flank and metastatic models. There was little off-target toxicity, as demonstrated by hematopoietic stability, unchanged tissue chemistries, weight gain rather than loss throughout treatment, and favorable histopathologic findings. Conclusion: Compound 211At-3-Lu or close analogs may provide limited and acceptable toxicity while retaining efficacy in management of prostate cancer.
Radiopharmaceutical therapy targeting prostate-specific membrane antigen (PSMA) using low-molecular-weight agents is becoming viable for metastatic prostate cancer (1–3). Such treatments have used β-particle emitters, including 131I and 177Lu, or α-particle emitters such as 213Bi, 212Pb, 227Th, and 225Ac (4–10). To date, most clinical trials have used 177Lu. In one such trial, PSA levels decreased by over 50% in 57% of patients (11). PSMA-targeted α-particle emitters may be even more promising, as evidenced by treatment with an 225Ac-labeled agent producing a significant tumor response in patients who were unresponsive to prior β-emitter therapy (12,13). However, the side effect of xerostomia from uptake of the agents in the salivary glands, and the potential of long-term renal toxicity, remain possible limitations.
A possible issue with α-emitters such as 225Ac is that multiple α-emitting daughters are generated from α-emitting parents, and in each case the energy imparted by the nuclear recoil effect is orders of magnitude greater than chemical bonds. That energy makes the release of the radioactive daughters from the targeting vector extremely likely, which can then lead to unintended irradiation of nontarget tissues (14,15). Our approach has been to use the radiohalogen 211At, which emits a single α-particle per decay. That strategy may permit greater control on the targeting of the therapeutic radiation, thereby reducing the chance of off-target effects. It should be noted that 58% of the α-particles emitted during 211At decay do involve a chemical transformation of astatine to polonium before α-emission. In that case, the parent decays by electron capture, not α-emission; the α-emitting 0.52-s half-life 211Po daughter is therefore not nuclear-recoil–afflicted. Even with worst-possible-case assumptions—that 211Po escapes immediately from the cell surface and can freely diffuse—nearly 100% of 211Po atoms should decay within 2 cell diameters from the original cell surface (16). The second 211At decay branch (42%) is by direct α-particle emission to 207Bi, which has a 32.9-y half-life. That long-lived radioactive daughter is not of concern because about 100,000 decays of 211At are needed to produce a single decay of 207Bi. Accordingly, a 370-MBq (10 mCi) hypothetical patient dose of an 211At-labeled PSMA agent would yield approximately 3.7 kBq (∼0.1 μCi) of 207Bi, a level that is only 0.1% of the annual limit of intake recommended for 207Bi by the Nuclear Regulatory Commission (3.7 MBq [100 μCi]) (17). Despite these issues, we believe 211At remains the best option for α-therapy.
Our previous studies used the following compounds, shown in Figure 1: DCABzL, HS-549, GV-620, GV-904, and YC-550. The initial compound studied, 211At-DCABzL, showed high and prolonged uptake in PSMA-expressing (PSMA+) tumor xenografts and renal cortex, with moderate uptake in thyroid and stomach, likely from dehalogenation (18). Despite that suboptimal biodistribution, we were able to demonstrate a treatment-related increase in survival in both flank tumor xenograft and micrometastatic models with a single dose of 0.74 MBq (20 μCi) and 0.11–0.37 MBq (3–10 μCi), respectively (18). From long-term toxicity studies, we determined that the dose-limiting toxicity was late radiation nephropathy (18). Using the 211At-labeled analogs YC-550, HS-549, GV-904, and GV-620 (Fig. 1), we observed faster renal clearance in mice than was seen with 211At-DCABzL. However, in vivo dehalogenation or off-target organ uptake remained an issue (19).

PSMA-targeted agents.
Here, we describe a new 211At-labeled PSMA-targeted compound with high stability in vivo and rapid clearance from off-target tissues (including kidneys, salivary and lacrimal glands) in mice. We also demonstrate a dose-dependent therapeutic effect in flank xenograft and metastatic tumor models of prostate cancer.
MATERIALS AND METHODS
Reagents, Cell Lines, and Animal Models
Chemistry
The syntheses of compounds 4 (Fig. 1) and its tin precursor, 15, as well as of unlabeled compounds 3, 3-Lu, and their tin precursor, 9, are outlined in Figures 2 and 3 and are described in detail in the supplemental materials (available at http://jnm.snmjournals.org [20–22]). The PSMA-binding affinity of compounds 3 and 4 was determined using a fluorescence-based assay we have previously reported (18).

Synthesis of compound 3, 3-Lu, radiolabeling of precursor 9, 125I-3, 125I-3-Lu, and 211At-3-Lu. Reagents and conditions: 4-(tributylstannyl)benzaldehyde, methanol, sodium cyanoborohydride (a); 5-(Fmoc-amino)valeric acid, O-(N-succinimidyl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate, N,N-diisopropylethylamine, dimethylformamide (b); 20% piperidine in dimethylformamide (c); DOTA-N-hydroxysuccinimide tri-tert-butylester, N,N-diisopropylethylamine, dimethylsulfoxide (d); I2, CH2Cl2 (e); 1/1 trifluoroacetic acid/CH2Cl2 (f); 0.2 M NH4OAc, dimethylsulfoxide, 5 mM 175Lu(NO3)3·H2O in 0.1N HCl, 70°C, 20 min (g, step i), ethylenediaminetetraacetic acid (g, step ii); Na125I or 211At, methanol, glacial acetic acid, room temperature, 20 min (h); and trifluoroacetic acid, 60–70°C, 30–45 min (j).

Synthesis of radiolabeling precursor 15, 4, 125I-4, and 125I-4-Lu. Reagents and conditions: triethylamine, dimethylsulfoxide, room temperature, 2 h (a); I2, CH2Cl2, room temperature, 2 h (b); 1/1 trifluoroacetic acid/CH2Cl2, room temperature, 2 h (c); Na125I, N-chlorosuccinimide, glacial acetic acid, methanol, room temperature, 20 min (d); concentrated formic acid, 60°C, 1 h (e); 0.1 M sodium acetate, pH 4.5, 5 mM 175Lu(NO3)3 in 0.1 M HCl, 60°C, 20 min (f, step i); and 5 mM ethylenediaminetetraacetic acid (f, step ii).
Radiochemistry
Sodium 125I-iodide in 10 μM NaOH (pH 8–11) was purchased from Perkin Elmer. 211At was produced on the Duke University CS-30 cyclotron via the 209Bi(α,2n)211At reaction on natural bismuth targets (23,24).
We investigated 2 methods (A and B) for the preparation of 125I-3-Lu and 211At-3-Lu (Fig. 2). Method A included purification of 125I/211At-3 before complexation with 175Lu(III), whereas in method B lutetium complexation was performed in situ without purification of the intermediate. In both methods, the final compound was purified by high-performance liquid chromatography. Radiosynthesis of 211At-3-Lu by method B began with a solution of 211At in 0.02% N-chlorosuccinimide in methanol (600 μL; 638 MBq [17.3 mCi]) that was added to 310 μg (204 nmol) of compound 9 in a borosilicate screw cap vial followed by 12 μL of glacial acetic acid. The vial was capped, shaken, and allowed to stand at room temperature for 10 min. The reaction mixture was concentrated to dryness using a stream of nitrogen at 60°C. A 95:5 mixture of trifluoroacetic acid:water (200 μL) was added, and the vial was heated at 60°C for 30 min. Volatiles were evaporated using a stream of nitrogen at 60°C. Sodium acetate buffer (0.1 M), pH 4.5 (500 μL), and a solution of Lu(NO3)3 in 0.1 M HCl (65 μL; 325 nmol) were added to the residue, and the solution was mixed with a micropipette. That solution was heated at 60°C for 20 min, 100 μL of 5 mM ethylenediaminetetraacetic acid was added, and the reaction mixture was diluted with 600 μL of water. The final product was purified by high-performance liquid chromatography. For this, a Phenomenex Luna C18 column (250 × 4.6 mm, 10 μm) was eluted at a flow rate of 1 mL/min with a gradient consisting of 0.1% trifluoroacetic acid in both water (solvent A) and acetonitrile (solvent B). The proportion of B was linearly increased from 15% to 40% over 30 min. Under those conditions, 211At-3-Lu [54 MBq (1.47 mCi)] eluted at 22.5 min. Pooled high-performance liquid chromatography fractions containing 211At-3-Lu were diluted to 20 mL with water and were loaded onto a Waters Oasis HLB Light Sep-Pak. The cartridge was washed with 5 mL of water and dried under a stream of nitrogen, and the product was eluted with 0.5 mL of ethanol. The eluate was concentrated using a stream of nitrogen, and the activity was reconstituted in saline. Detailed radiosyntheses of 211At-3-Lu by method A, and syntheses of 125I-3, 125I-3-Lu, 125I-4, and 125I-4-Lu, are presented in the supplemental materials. Radiolabeling yields for 125I/211At-3 and 125I/211At-3-Lu are summarized in Tables 1 and 2.
Radiolabeling Yields for 125I/211At-3
Radiolabeling Yields for 125I/211At-3-Lu
Cell Lines and Culture Conditions
PSMA+ PC3 PIP and PSMA-negative (PSMA−) PC3 flu cells were maintained as previously described (25–27). For the experimental metastatic model, parental PC3-ML-Luc cells were obtained from Dr. Mauricio Reginato (Drexel University). Those cells are characterized in Supplemental Figure 1. Cell lines were maintained Mycoplasma-free through biweekly testing with the MycoAlert Mycoplasma detection kit (Lonza).
Animals
Animal studies conformed to protocols approved by the Johns Hopkins Animal Care and Use Committee. Johns Hopkins University has an approved Public Health Service Policy, and the approved protocols follow this and Animal Welfare Act regulations. NSG (NOD/SCID/IL2Rγnull) mice were obtained from the Animal Resources Core of the Johns Hopkins Sydney Kimmel Comprehensive Cancer Center.
In Vitro Studies
PSMA+ PC3 PIP cells were plated at 5 × 105 cells per well and incubated overnight. Cells were then incubated with 211At-3 in medium (∼3.7 kBq/100 μL) at 37°C for 0.5, 1.0, 2.0, and 4.0 h. Cell culture supernatant was removed, and the cells were processed as before (18). Cell-associated radioactivity was calculated as percentage of input dose. The internalized fraction of radioactivity was determined by solubilizing cells with 1% SDS cell lysis buffer after removing the unbound fractions and cell surface–bound fractions (by washing with glycine-HCl buffer). To determine binding specificity, PSMA+ PC3 PIP cells were coincubated with 211At-3 and the known PSMA inhibitor (R, S)-2-(phosphonomethyl)pentanedioic acid (100 μM) (28).
In Vivo Studies
Biodistribution
Six- to 8-wk-old male NSG mice were implanted subcutaneously with PSMA+ PC3 PIP (1.5 × 106) and PSMA− PC3 flu cells (1 × 106) in 100 μL of Hanks balanced salt solution (Cellgro; Corning) at the forward right and left flanks, respectively. Mice were used in ex vivo biodistribution assays when the xenografts reached 5–7 mm in diameter. Biodistribution experiments were performed in the above mice bearing both PSMA+ PC3 PIP and PSMA− PC3 flu flank xenografts after an intravenous bolus of 37 kBq (1 μCi) of 125I-4, 125I-4-Lu, 125I-3, 125I-3-Lu, or 211At-3-Lu. Tissues harvested at 1, 4, and 24 h after injection (n = 5 per time point) included blood, heart, lung, liver, spleen, pancreas, stomach, small intestine, large intestine, fat, muscle, salivary gland, lacrimal gland, kidney, bladder, PSMA+ PC3 PIP tumor, and PSMA− PC3 flu tumor. Each tissue was weighed, and the associated radioactivity was measured with an automated γ-counter (2480 Wizard; Perkin Elmer). The percentage injected dose (%ID) was calculated using a known dilution of %ID. All measurements were corrected for decay. Data are expressed as %ID/g of tissue or per organ (%ID) for organs that were too small for accurate dissection. All data are expressed as mean ± SD.
Antitumor Efficacy in Subcutaneous Xenograft Model
PSMA+ PC3 PIP and PSMA− PC3 flu cells were implanted subcutaneously in male NSG mice as described above. When tumor diameter reached 5–7 mm, a single intravenous injection was performed with saline or with 0.24 MBq (6.6 μCi), 0.74 MBq (20 μCi) , 1.48 MBq (40 μCi), or 3.7 MBq (100 μCi) of 211At-3-Lu (n = 5 per group). Tumor progression was monitored by measuring subcutaneous tumor volume [(width2 × length)/2 mm3] using a caliper. A tumor volume increase of more than 4-fold was scored as death of the animal, at which point it was euthanized.
Antitumor Efficacy in Metastatic Model
Four- to 6-wk-old NSG mice were injected intravenously with 1 × 106 PC3-ML-Luc cells suspended in 200 μL of Hanks balanced salt solution to form micrometastatic deposits. One week after injection of cells, the mice were injected intravenously with 0 MBq (0 μCi; saline), 0.185 MBq (5 μCi), 0.37 MBq (10 μCi), 0.74 MBq (20 μCi), 1.48 MBq (40 μCi), or 3.7 MBq (100 μCi) of 211At-3-Lu (n = 5 per group). Metastatic tumor progression was monitored by in vivo bioluminescence imaging and survival of injected animals. Weekly bioluminescence imaging was performed using the IVIS Spectrum in vivo imager (Perkin-Elmer). Mice were sacrificed and scored as death if they lost more than 20% of body weight or had signs of discomfort, such as hunched posture, anorexia, or dehydration. For both animal models, the probability of survival was characterized by Kaplan–Meier curves using Prism software (GraphPad Software).
Long-Term Toxicity
Healthy 11-wk-old male CD1 mice (Charles River) weighing 35–40 g received intravenous injections of 0 MBq (0 μCi; saline), 0.24 MBq (6.6 μCi), 0.74 MBq (20 μCi), or 1.48 MBq (40 μCi) of 211At-3-Lu (n = 5 per group). Mice were monitored for 13 mo with a health inspection daily and a weight measurement twice per week. Monthly urinalysis was performed for specific gravity and urine protein content using Chemstrip test strips (Roche Diagnostics). After 13 mo, the mice were euthanized in a CO2 chamber, and blood, kidney, salivary glands, and lacrimal glands were collected. Complete blood counts, including white blood cells, red blood cells, hemoglobin, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, and platelets, were measured using the scil Vet ABC hematology analyzer (scil Animal Care Co.). Blood chemistry testing for blood urea nitrogen, glucose, alkaline phosphatase, total protein, alanine aminotransferase, and creatinine was performed using a Spotchem EZ chemistry analyzer (Arkray USA). Histopathologic evaluation was performed by a certified veterinary pathologist for kidneys, salivary glands, and lacrimal glands with hematoxylin and eosin–stained slides of each tissue.
Statistics
Survival analyses for the metastatic and subcutaneous models were performed using Prism software (version 9; GraphPad Software). P values were calculated by the log-rank (Mantel–Cox) test and were considered significant if less than 0.05.
RESULTS
Chemistry
Because of the promising results obtained with 177Lu-1 and 177Lu-2, our strategy was to replace the nonradioactive bromine or iodine atom with 125I and evaluate their biodistribution in tumor-bearing animals with or without chelated nonradioactive lutetium (125I-3 and 125I-3-Lu [inhibition constant, 0.09–34 nM (29)]; Fig. 2). For comparison, we also synthesized and evaluated 125I-4 and 125I-4-Lu (Fig. 3), where the radioiodine was incorporated into the linking group, and each contained a 4-bromobenzyl moiety as in compound 1. The 211At-labeled analog of the most promising iodinated compound was synthesized and evaluated. The binding affinity of unlabeled compounds 3 and 4 was as follows: half-maximal inhibitory concentration, 1.51 nM (95% CI, 0.62–3.64 nM); inhibition constant, 0.30 nM (95% CI, 0.13–0.73 nM), and half-maximal inhibitory concentration, 7.82 nM (95% CI, 6.16–9.94 nM); inhibition constant, 0.30 nM (95% CI, 1.23–1.99 nM), respectively.
Radiochemistry
Syntheses of 125I-4 and 125I-4-Lu were each performed once. The yield of 125I-4 from 125I-iodide was 14%, and that for the conversion of 125I-4 to 125I-4-Lu was 82%. Radiolabeling conditions and yields for 125I/211At-3 and 125I/211At-3-Lu are given in Tables 1 and 2, respectively. In general, fresh batches of 211At provided higher overall yields of either compound. Purification of the nonmetallated intermediate (125I/211At-3) did not enhance yield and actually provided lower overall yields of the final product (125I/211At-3-Lu).
Cell Uptake and Internalization
In vitro studies of 211At-3-Lu demonstrated total uptake within PSMA+ PC3 PIP cells of 13.4% ± 0.5% of the input dose after 4 h of incubation and an increasing internalized fraction over time, namely, 15.8% ± 0.7%, 19.0% ± 0.7%, 24.5% ± 1.0%, and 27.7% ± 2.2% at 0.5, 1.0, 2.0, and 4.0 h, respectively. Coincubation with (R, S)-2-(phosphonomethyl)pentanedioic acid showed an average of 0.6% uptake at all time points, confirming PSMA-specific uptake (Supplemental Fig. 2) (18).
Biodistribution
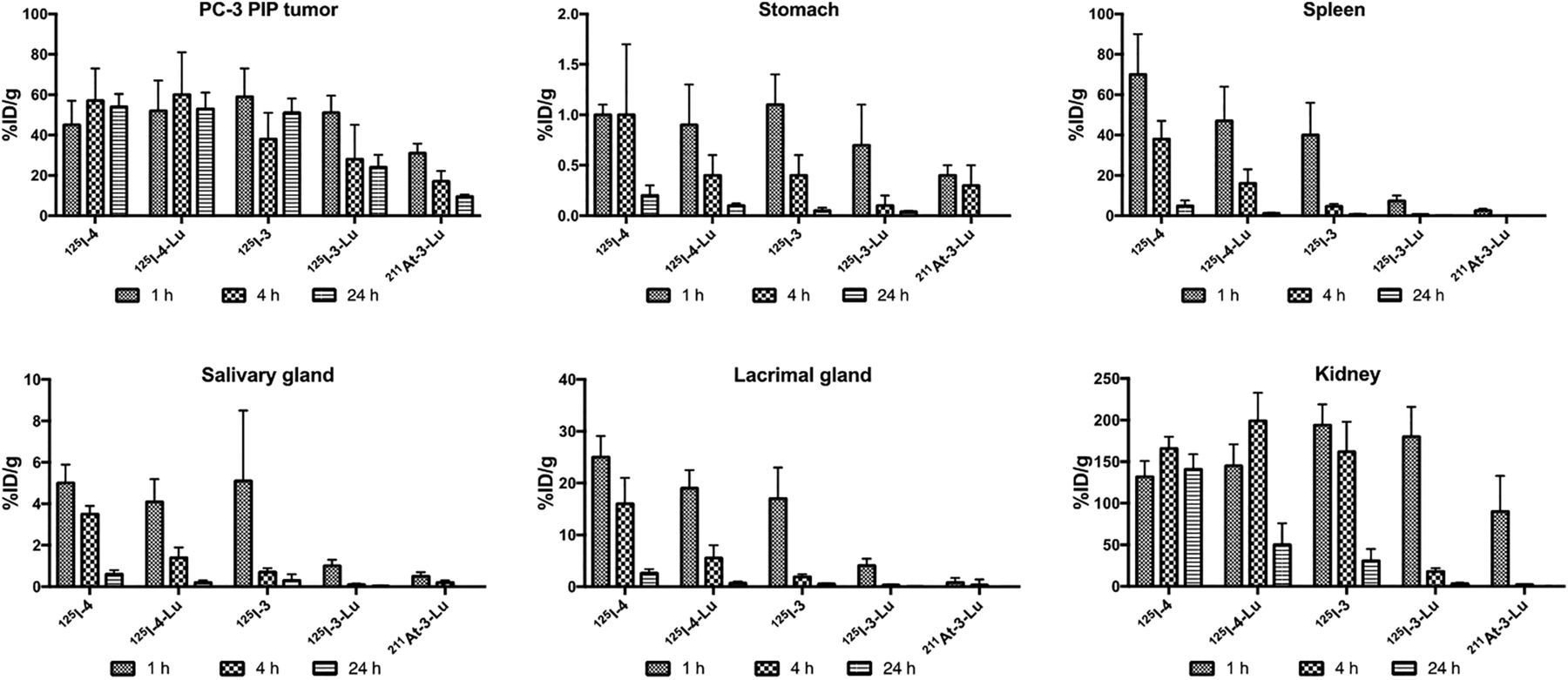
Detailed biodistribution data, represented as %ID/g for all radiolabeled compounds, are given in Supplemental Tables 1–6. There was little deastatination of 211At-3-Lu in vivo as evidenced by low uptake of radioactivity in stomach (0.39 ± 0.12 %ID/g), salivary glands (0.47 ± 0.19 %ID/g), and spleen (2.51 ± 0.94 %ID/g) at 1 h after administration, which decreased further by 4 h for stomach and salivary glands (<0.05 %ID/g in spleen) (Supplemental Table 3). Uptake in PSMA+ PC3 PIP tumor and selected nontarget organs is shown in Figure 4. All compounds had high tumor uptake at 1 h (30–60 %ID/g). Although tumor activity remained at that high level out to 24 h for 125I-4, 125I-4-Lu, and 125I-3, 50% and 67% of 125I-3-Lu and 211At-3-Lu activity, respectively, cleared from the tumor by 24 h. Importantly, 211At-3-Lu was nearly undetectable in normal organs at 24 h. Renal uptake of 211At-3-Lu at 1 h (89.5 ± 42.7 %ID/g) was 30%–50% lower than that seen for other compounds. By 4 h, renal activity levels for 125I-3-Lu and 211At-3-Lu decreased to 18.2 ± 3.9 and 2.1 ± 0.6 %ID/g, respectively, whereas activity in kidneys for the other compounds remained high (162–199 %ID/g). By 24 h, activity in kidneys from 125I-3-Lu and 211At-3-Lu decreased to 3.30 ± 1.15 %ID/g and 0.02 ± 0.20 %ID/g, respectively. On the other hand, renal activity was 141 ± 18, 50.4 ± 25.9, and 30.6 ± 14.0 %ID/g for 125I-4, 125I-4-Lu, and 125I-3, respectively. Despite the lower %ID/g values of 211At-3-Lu in tumor, its considerably faster renal clearance resulted in tumor-to-kidney ratios of 8 and 130 at 4 and 24 h, respectively (Fig. 5). Those values are roughly 5- and 16-fold higher than achieved with 125I-3-Lu and 20- to 200-fold higher than achieved with the other compounds. The uptake in spleen at 1 h was much lower for 125I-3-Lu and 211At-3-Lu (7.41 ± 2.57 and 2.51 ± 0.94 %ID/g, respectively) than for the other compounds (40–70 %ID/g). Radioactivity from spleen cleared rapidly for all agents, resulting in tumor-to-spleen ratios at 4 h of 1.3, 4.4, 6.8, 43, and 97 for 125I-4, 125I-4-Lu, 125I-3, 125I-3-Lu, and 211At-3-Lu, respectively. The uptake of 125I-3-Lu and 211At-3-Lu in salivary and lacrimal glands was lower than observed for any other compound at any time point, resulting in tumor– to–salivary gland ratios of 58 and 75 and tumor–to–lacrimal gland ratios of 13 and 44 at 1 h after injection, respectively (Supplemental Tables 5 and 6, respectively). At 4 h, those values were 365 and 129 for salivary gland and 92 and 164 for lacrimal gland.


α-Therapy with 211At-3-Lu
We first evaluated the efficacy of 211At-3-Lu by scoring growth inhibition of subcutaneous xenograft tumors of both PSMA+ PC3 PIP and PSMA− PC3 flu cells implanted in the same animal. A single intravenous injection of 4 different doses (0 MBq [0 μCi; saline], 0.24 MBq [6.5 μCi], 0.74 MBq [20 μCi], 1.48 MBq [40 μCi], or 3.7 MBq [100 μCi]) did not affect tumor growth of PSMA− PC3 flu tumors, as the median survival of the tumor-bearing mice was 9, 14, 11, 11, and 13 d, respectively. On the other hand, median survival compared with untreated controls for animals harboring PSMA+ PC3 PIP tumors at doses of 0 MBq (0 μCi; saline), 0.24 MBq (6.5 μCi), 0.74 MBq (20 μCi), 1.48 MBq (40 μCi), or 3.7 MBq (100 μCi) were 11 (not statistically significant), 27 (P = 0.0015), 39 (P = 0.0005), 29 (P = 0.0005), and not reached (P = 0.0005), respectively, indicating that 211At-3-Lu was capable of PSMA-specific tumor growth control and enhancement of survival (Fig. 6). We also tested the efficacy of a single dose of intravenously administered 211At-3-Lu for treating metastatic deposits of PSMA-expressing tumors. Higher doses (1.48 and 3.7 MBq) provided survival benefits compared with the untreated group (Fig. 6B). Median survival for animals treated with 0 MBq (0 μCi; saline), 0.186 MBq (5 μCi), 0.373 MBq (10 μCi), 0.74 MBq (20 μCi), 1.48 MBq (40 μCi), or 3.7 MBq (100 μCi) were 48, 49 (not statistically significant), 48 (P = 0.5769, not statistically significant), 52 (P = 0.0699, not statistically significant), 57, (P = 0.0286), and 58.5 d (P = 0.2718, due to an early mouse death), respectively.

Kaplan–Meier curves showing survival in flank (A) and PC3-ML-Luc experimental metastatic (B) models at dose provided. PC3-PIP = PSMA+ PC3 PIP tumors; PC3-flu = PSMA− PC3 flu cell–derived tumors.
Long-Term Radiotoxicity
Eleven-week-old male CD1 mice were injected with 0.24 MBq (6.6 μCi), 0.74 MBq (20 μCi), or 1.48 MBq (40 μCi) of 211At-3-Lu as a single intravenous injection. We also included an untreated group for the entire duration of the study as an age-matched control. All groups of mice consistently gained weight for the 13-mo period of monitoring (Supplemental Fig. 3). Blood chemistry data for creatinine, blood urea nitrogen, glucose, alkaline phosphatase, alanine aminotransferase, and total protein for all treated groups were similar to those for age-matched untreated controls (Supplemental Fig. 4). A complete blood count also indicated that treated groups remained within normal limits (Supplemental Fig. 4). Monthly evaluation of urine protein level (Supplemental Table 7) and specific gravity (Supplemental Table 8) showed no sign of renal impairment compared with untreated controls for the duration of the study. Histopathologic examination of kidneys, salivary glands, and lacrimal glands revealed no treatment-specific pathologic abnormalities at any dose studied (Fig. 7). We observed mild inflammation, a few dilated tubules with protein deposits, and mild multifocal fibrosis in kidneys from all groups (including controls), which were age-related phenomena. Mild age-related inflammation was also observed in salivary and lacrimal glands from all groups.

Representative microscopic images of kidney, salivary gland, and lacrimal gland (size bar, 100 μm). Black arrows indicate inflammation; white arrow indicates dilated tubule with protein deposit.
DISCUSSION
Banerjee et al. reported a series of 4-halobenzyl derivatives of Lys-Glu-urea inhibitors of PSMA containing a linking group that connects the PSMA-targeting urea pharmacophore with a metal chelator (29). Two of the most promising compounds were 1 and 2 (Fig. 1). When 177Lu-1 and 177Lu-2 were administered intravenously to tumor-bearing mice, they exhibited high uptake in PSMA+ PC3 PIP tumor xenografts (29), low uptake in the salivary glands, rapid renal clearance, and dose-dependent tumor growth delay. We previously demonstrated in a head-to-head preclinical study that our scaffold bearing the β-particle emitter 177Lu was inferior to that delivering 225Ac, an α-emitter, providing a rationale for our focus on PSMA-targeted compounds bearing an α-emitting warhead (30).
Reports of PSMA-targeted therapy with the α-emitter 225Ac-PSMA-617 have been encouraging, even in late-stage disease. Some trials reported PSA declines of at least 90% in roughly half of patients (31) and overall survival of more than 15 mo (32). Such results may exceed those of new chemo- or hormonal therapies. However, those results have come at the costs of decreased quality of life, including nontransient, treatment-halting xerostomia and substantial hematologic toxicity, according to 1 retrospective trial (33). A greater mitigation of off-target effects, which has proved challenging to date, is needed for PSMA-targeted radiopharmaceutical therapy to develop a niche in the management of prostate cancer. Two ways to do so are by optimizing the pharmacokinetics and choosing the correct α-particle emitter. We have attempted both by focusing on the type II Lys-Glu-urea scaffold we have previously reported (29), a close structural analog of which is currently under investigation in a phase 1–2 clinical trial (NCT0349083800), and on using 211At, which produces only 1 α-particle per decay and has a tractable physical half-life of 7.2 h (34–37).
For convenience of handling, we initially studied the 125I-labeled surrogates of the intended 211At compounds to gauge pharmacokinetics and in vivo stability. Compounds of the 4 series (Fig. 3) enabled us to explore the effect of halogen location in the molecule on pharmacokinetics, as well as the influence of a metal within the chelator, which we previously showed enhanced affinity for PSMA (29). Although 125I-4 and 125I-4-Lu behaved similarly in PSMA+ PC3 PIP tumor, 125I-4-Lu in kidney substantially decreased by 24 h, indicating a positive effect of the presence of lutetium in the chelator on renal clearance. Because even higher tumor-to-kidney ratios were observed for 125I-3-Lu than for 125I-4-Lu, we continued with the former for further in vivo testing. Relative to 125I-3-Lu, 211At-3-Lu demonstrated lower tumor uptake but also lower off-target uptake. There was also moderate uptake within stomach, consistent with some deastatination (38); however, levels remained below 0.5 %ID/g and tumor-to-stomach ratios were approximately 100 (Figs. 4 and 5). By contrast, 211At-DCABzL never had tumor-to-stomach ratios that rose above a few percentage points out to 18 h (18), and a recently published 211At-labeled minibody targeting prostate cancer had a ratio that only exceeded 1 on treatment with perchlorate at 5 h (1.2) and 9 h (1.4) after injection (39).
The lack of uptake within stomach and salivary glands of 211At-3-Lu could in part be due to the stability of the low-molecular-weight, Lys-Glu-urea–based targeting scaffold (40). There was also little radioactivity in blood (Supplemental Table 3) and no significant change in white blood cell counts (Supplemental Fig. 4). Accordingly, neither treatment with perchlorate (39) nor blocking agents were required to mitigate off-target effects. In vivo, 211At-3-Lu treatment caused a PSMA- and dose-dependent increase in survival compared with control animals in both the flank and metastatic models (Fig. 6). Comparison of this result with other reported compounds is challenging because of the different model systems used. If we focus on our own earlier therapy studies, we find that 211At-3-Lu provided survival effects at much lower doses than 211At-DCABzL (18), which had a maximum tolerated dose (MTD) of 37 kBq (1 μCi). However, that study was performed on nude rather than SCID mice, which we used here. We did not reach the MTD for 211At-3-Lu, as the highest dose administered in the long-term (13 mo) toxicity study with normal mice was only 1.48 MBq (40 μCi). Nonetheless, even at a dose up to 40 times higher than the MTD for 211At-DCABzL in the same mouse strain, our toxicity data showed only mild changes at all doses and in all organs studied (Fig. 7). Perhaps a more relevant comparison is to 225Ac-L1 (30), which demonstrated an MTD of 9.3 kBq (0.25 μCi) (fractionated × 4) because the scaffold is the same as that of 211At-3-Lu. Compound 212Pb-L2 (41), which has a similar scaffold, demonstrated an MTD of 1.5 MBq (∼40 μCi). For that compound, there were concerns of long-term renal toxicity, which may be due to the 212Bi daughter released and localized to the kidney, which is not a problem with 211At-based agents.
Our goal in this work was to find a suitable α-particle–emitting agent to treat PSMA+ prostate and other cancers that had minimal off-target toxicity, namely, an agent that would be more effective than the corresponding β-particle emitter but not as toxic as those radiolabeled with 225Ac. We have achieved that with 211At-3-Lu, attesting to the potential benefits of this potentially tamer α-emitter. As with 212Pb, 211At emits only 1 α-particle per decay. As such, these 2 radionuclides do not produce daughter α-emissions outside the intended target site and will not be expected to have the toxicity—for example, in liver—attendant on such emissions, such as those seen with 225Ac. However, there are differences between 212Pb and 211At that make us favor the latter. The therapeutic potential of 212Pb will be diminished if the daughter nuclei do not remain at the target site so that their energy can also be captured (7). For example, Ackerman et al. have calculated that migration of daughters with 212Pb could reduce its relative biological effectiveness to that of conventional external-beam radiation and β-particle emitters (42). Furthermore, 211At can be introduced to targeting ligands using chemistry very similar to that for other halogens to minimize perturbation of the targeting scaffold if a fastidious cellular target is sought, or a chelator can be deliberately introduced to enhance pharmacokinetics, as in this case (36,37). Lack of off-target toxicity, including to salivary and lacrimal glands, would obviate cumbersome coadministration of blocking agents. However, although the natural bismuth target material is inexpensive and widely available, 211At requires a 28-MeV α-particle cyclotron beam for efficient production—a requirement that has curtailed its use (43). There are concerted efforts under way to increase the supply of 211At worldwide at academic institutions and research institutes, with commercial sources emerging in the not-too-distant future (https://ionetix.com/why-alpha-therapy).
A limitation of this study was the use of cells that may not reflect the natural abundance and heterogeneity of PSMA in human cancer. That issue has been discussed in detail elsewhere (44). However, PSMA+ PC3 PIP and PSMA− PC3 flu cells have the advantage of being isogenic, except for PSMA expression, enabling us to answer questions about pharmacokinetics with a minimum of variables present. The superior performance of 211At-3-Lu in the flank model rather than the metastatic model may reflect, in part, the supraphysiologic and 10-fold higher PSMA expression in the PSMA+ PC3 PIP cells relative to the PSMA+ PC3-ML-Luc cells used to generate the metastatic deposits (18), which express PSMA at about the same level of LNCaP cells (27).
CONCLUSION
In this small series, 211At-3-Lu proved to have an excellent combination of properties: a pharmacokinetic profile matching the physical half-life of 211At, the ability to improve survival in tumor-bearing animals, and lack of off-target toxicity as demonstrated by hematopoietic stability, unchanged tissue chemistries, weight gain rather than loss throughout treatment, and favorable histopathology. This compound or close analogs are promising for translation if and when an α-particle emitter is to be considered in the therapeutic journey of the patient.
DISCLOSURE
Financial support was received from CA184228, EB024495, CA134675, and the Commonwealth Foundation. Under a license agreement between D&D Pharmatech and Johns Hopkins and Duke Universities, the Universities’ Ronnie Mease, Martin Pomper, Sangeeta Banerjee, Ganesan Vaidyanathan and Michael Zalutsky are entitled to royalty distributions related to the technology described in the study discussed in this publication. Martin Pomper, Sangeeta Banerjee, and Michael Zalutsky hold equity in D&D Pharmatech; Martin Pomper and Sangeeta Banerjee are cofounders. They and Michael Zalutsky are also paid consultants to the company. This arrangement has been reviewed and approved by Johns Hopkins University and Duke University in accordance with their conflict-of-interest policies. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can a PSMA-targeted, α-emitting small molecule be designed with few off-target toxic effects while retaining therapeutic efficacy?
PERTINENT FINDINGS: Lead compound 211At-3-Lu was able to control tumor growth and enhance survival in animals treated at doses of 1.48 MBq or greater and did so without toxicity.
IMPLICATIONS FOR PATIENT CARE: Compounds such as 211At-3-Lu provide a further rationale for the use of 211At in targeted α-emitting radiopharmaceuticals. 211At may be an effective and nontoxic alternative to other α-emitters in use for management of prostate and other PSMA-expressing cancers. Its ease of incorporation in a variety of cancer affinity agents, including small molecules, as well as its convenient physical half-life could provide a safe, practical, and new method to treat a variety of intractable malignancies.
Footnotes
Published online June 04, 2021.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication February 6, 2021.
- Revision received May 5, 2021.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}