


Abstract
In recent years, several radioligands targeting prostate-specific membrane antigen (PSMA) have been clinically introduced as a new class of theranostic radiopharmaceuticals for the treatment of prostate cancer (PC). In the second decade of the 21st century, a new era in nuclear medicine was initiated by the clinical introduction of small-molecule PSMA inhibitor radioligands, 40 y after the clinical introduction of 18F-FDG. Because of the high incidence and mortality of PC, the new PSMA radioligands have already had a remarkable impact on the clinical management of PC. For the continuing clinical development and long-term success of theranostic agents, designing modern prospective clinical trials in theranostic nuclear medicine is essential. First-in-human studies with PSMA radioligands derived from small-molecule PSMA inhibitors showed highly sensitive imaging of PSMA-positive PC by means of PET and SPECT as well as a dramatic response of metastatic castration-resistant PC after PSMA radioligand therapy. This tremendous success logically led to the initiation of prospective clinical trials with several PSMA radioligands. Meanwhile, MIP-1404, PSMA-11, 2-(3-{1-carboxy-5-[(6-fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pentanedioic acid (DCFPyL), PSMA-617, PSMA-1007, and others have entered or will enter prospective clinical trials soon in several countries. The significance becomes apparent by, for example, the considerable increase in the number of publications about PSMA-targeted PET imaging from 2013 to 2016 (e.g., a search of the Web of Science for “PSMA” AND “PET” found only 19 publications in 2013 but 218 in 2016). Closer examination of the initial success of PC treatment with PSMA inhibitor radiotracers leads to several questions from the basic research perspective as well as from the perspective of clinical demands: What lessons have been learned regarding the design of PSMA radioligands that have already been developed? Has an acceptable compromise between optimal PSMA radioligand design and a broad range of clinical demands been reached? Can the lessons learned from multiple successes within the PSMA experience be transferred to further theranostic approaches?
- small-molecule PSMA inhibitors
- theranostic PSMA radioligands
- PSMA radiotracers
- prostate cancer
- radioligand therapy
PROLOG
A new era in nuclear medicine was initiated during the 21st century by the clinical introduction of a new class of small-molecule prostate-specific membrane antigen (PSMA) inhibitor radiotracers, just 40 y after the clinical introduction of 18F-FDG. PSMA radioligands are of utmost clinical interest and show promise for the diagnosis and therapy of prostate cancer (PC). However, what lessons have been learned regarding the design of PSMA radioligands that have already been developed? Has an acceptable compromise between optimal PSMA radioligand design and compliance with clinical demands been found? Can the PSMA radioligand approach be transferred to the development of other targeted theranostic approaches? These key questions are addressed in 4 lessons from the basic research and clinical demand perspectives: lesson 1, retrospective on Glu-ureido–based PSMA radioligands of clinical relevance; lesson 2, need for structure-aided design of Glu-ureido–based PSMA radioligands; lesson 3, elucidation of structure–property relationships with in vitro, in vivo, and ex vivo assays; and lesson 4, consideration of how to transfer the PSMA radioligand approach to other targeted theranostic approaches (e.g., tumor heterogeneity).
LESSON 1: RETROSPECTIVE ON GLU-UREIDO–BASED PSMA RADIOLIGANDS OF CLINICAL RELEVANCE
Recent literature contains comprehensive reviews that summarize the substance class of small-molecule and low-molecular-weight peptidomimetic Glu-ureido–based PSMA inhibitors. A few recently published reviews provide a comprehensive overview of the current status of small-molecule PSMA inhibitor radiotracers (1–4).
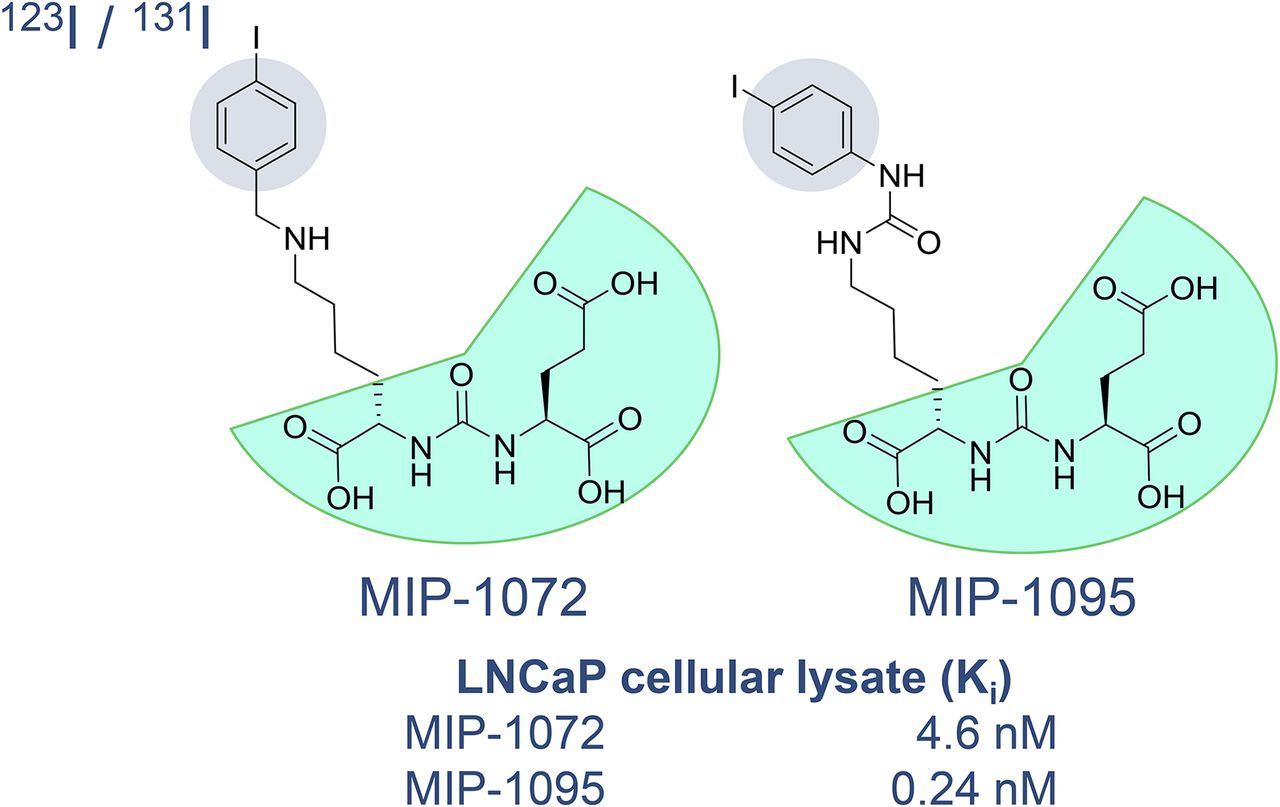
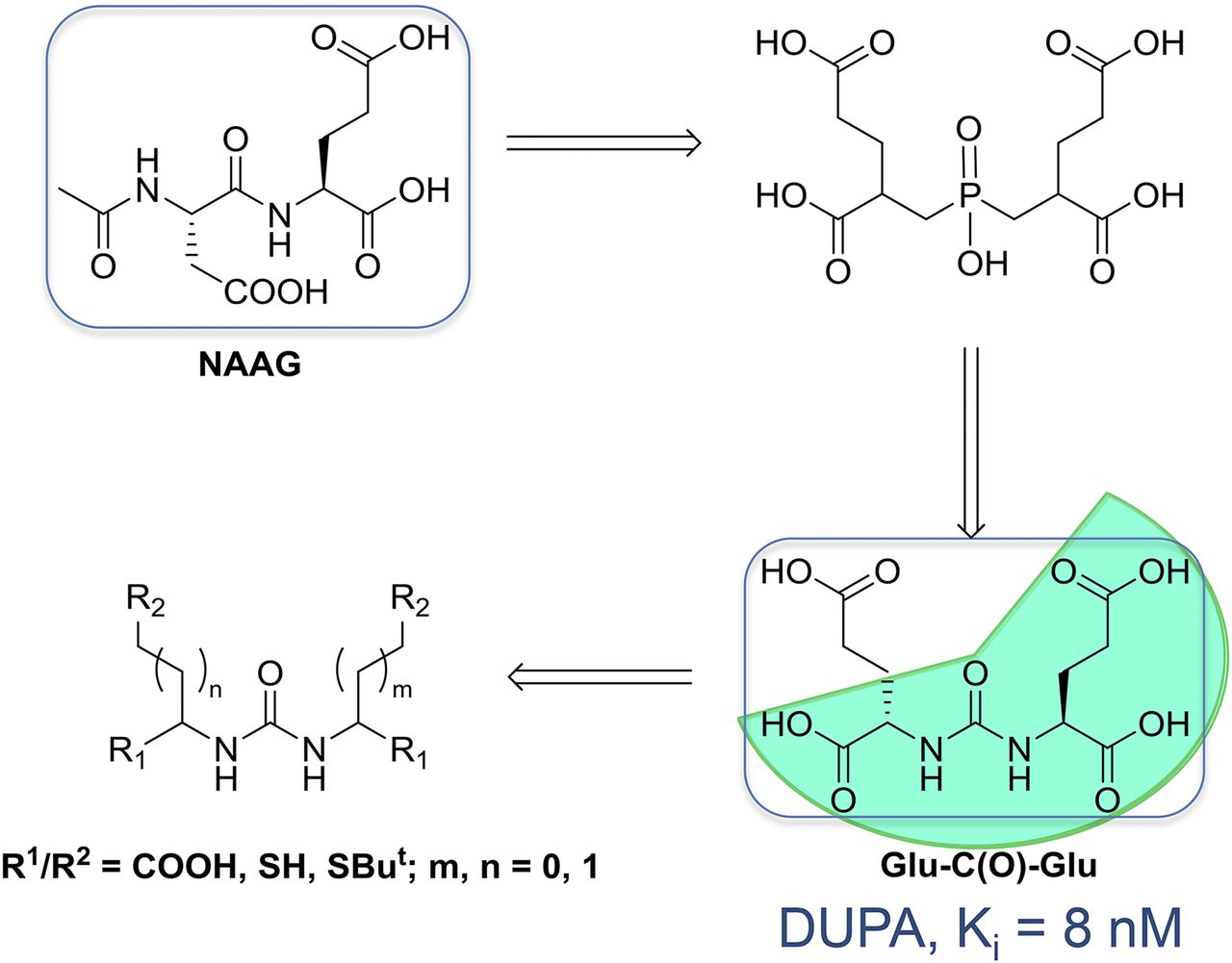
Several PSMA radioligands have already entered clinical settings and are being used for the diagnosis and radioligand therapy (RLT) of PC. The first small-molecule inhibitors of PSMA for the imaging of PC were introduced into clinical settings in 2008 by Molecular Insight Pharmaceuticals, Inc. (MIP, now a subsidiary of Progenics Pharmaceuticals Inc.) (ClinicalTrials.gov identifier: NCT00712829). These ligands, 123I-MIP-1072 and 123I-MIP-1095, were based on the Glu-urea-Lys motif and contained aromatic substituents for stable introduction of the single-photon-emitting radionuclide 123I (half-life, 13.2 h) (5,6). However, worldwide, 68Ga-PSMA-11 currently is the most prominent 68Ga-labeled PSMA radioligand for the PET imaging of PSMA-positive PC. 68Ga-PSMA-11 was clinically introduced several years ago (7,8). PSMA-11 belongs to the class of low-molecular-weight Glu-ureido–based PSMA inhibitors. In general, the class of urea-based PSMA inhibitors was first described by Kozikowski et al. (9,10), building on the work of Jackson et al. (11,12), and introduced as a peptidomimetic counterpart of the most abundant peptidyl neurotransmitter, N-acetyl-l-aspartyl-l-glutamate (Fig. 1). One of the first preclinical imaging studies with small-molecule PSMA inhibitor radioligands in PSMA-positive tumor xenografts was reported by Foss et al. in 2005 (Fig. 2) (13).

Glu-ureido–based PSMA inhibitor exemplifying rational design of urea-based glutamate carboxypeptidase II inhibitors. DUPA = 2-[3-(1,3-dicarboxypropyl)ureido]pentanedioic acid; NAAG = N-acetyl-l-aspartyl-l-glutamate. (Reprinted with permission of (9).)
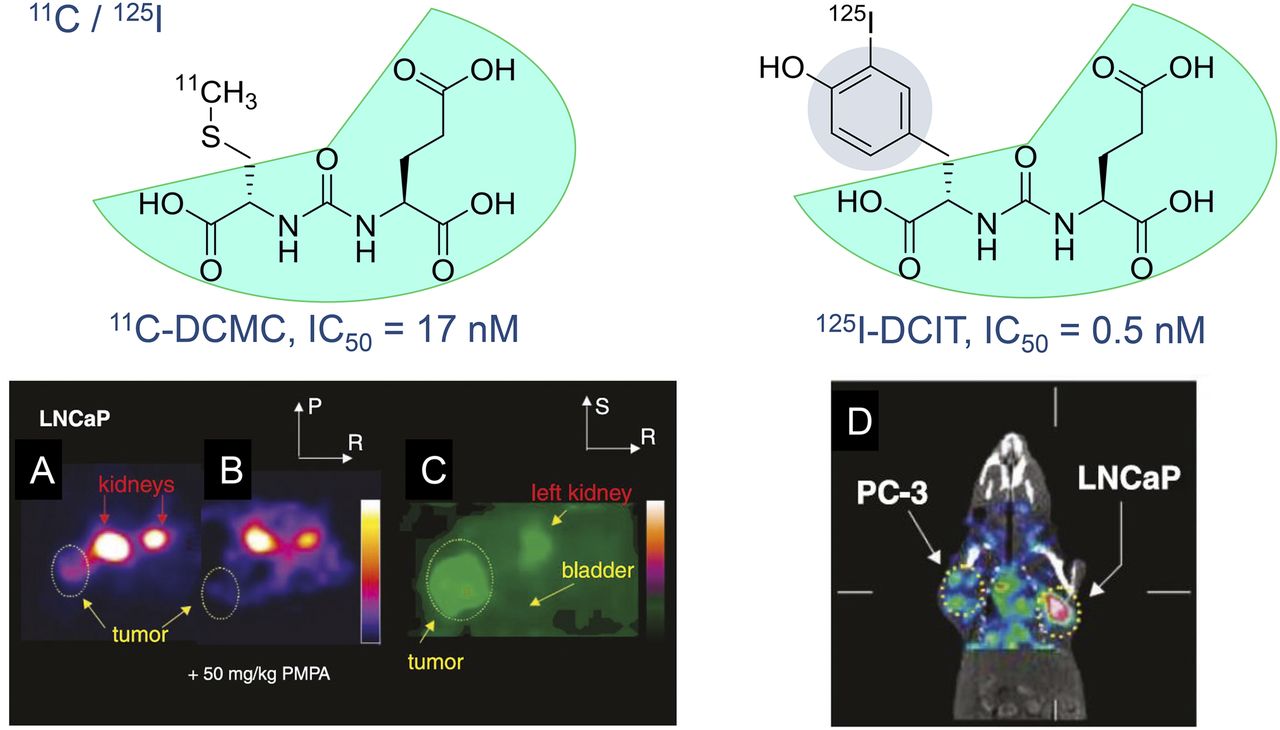
In vivo examinations of radiolabeled small-molecule PSMA inhibitors in experimental models of PC. (A and B) Axial PET images of N-[N-[(S)-1,3-dicarboxypropyl]carbamoyl]-S-11C-methyl-l-cysteine (11C-DCMC) in mouse kidney and LNCaP tumors without (A) and with (B) coinjection of 2-(phosphonomethyl)pentane-1,5-dioic acid (PMPA) at 50 mg/kg to block PSMA. (C) Magnified, coronal reconstructed image of animal in A outside plane of kidneys immediately after urination indicates lack of specific binding to bladder. Tumor and portion of left kidney were clearly visualized. Images in A and B were scaled to same maximum. IC50 = 50% inhibitory concentration; P = posterior, R = right, S = superior. (D) SPECT with CT overlay shows uptake of N-[N-[(S)-1,3-dicarboxypropyl]carbamoyl]-S-3-125I-iodo-l-tyrosine (125I-DCIT) in LNCaP tumor, whereas PC-3 tumor retains only minimal radiotracer. (Reprinted with permission of (13).)
In principle, small-molecule PSMA inhibitors bind to PSMA, also called glutamate carboxypeptidase II or N-acetylated α-linked acidic dipeptidase/N-acetyl-l-aspartyl-l-glutamate peptidase I, which is a binuclear membrane-bound zinc protease (MEROPS [peptidase database; http://merops.sanger.ac.uk/] identifier: M28.010) that is highly upregulated in all stages of PC with nearly no expression in healthy tissues. Moreover, there is a significant correlation between the level of expression of PSMA and the progression of the disease (14–16). PSMA is thus an optimal target for both the imaging and the endoradiotherapy of PC.
The preclinical evaluation of 68Ga-PSMA-11 (also called 68Ga-DKFZ-PSMA-11 or 68Ga-PSMA-HBED-CC) (17) encouraged the aforementioned clinical introduction of this promising PC imaging agent in Heidelberg, Germany. The complexing agent N,N´-bis[2-hydroxy-5-(carboxyethyl)benzyl]-ethylenediamine-N,N´-diacetic acid (HBED-CC) was conjugated to the pharmacophore Glu-urea-Lys via the aminohexanoic acid linker. The resulting conjugate, PSMA–HBED-CC (PSMA-11), could not bind to clinically relevant therapeutic radiometals, such as 177Lu or 225Ac. Therefore, PSMA-11 could be used only for diagnostic purposes. However, it soon became clear that PSMA inhibitors could also be used for PSMA RLT of PC. The first clinical theranostic approach with radioiodinated versions of PSMA inhibitor MIP-1095 for the treatment of patients with late-stage disease (that is, metastatic castration-resistant PC) was reported in 2014 (18). The tracer MIP-1095 was first described by Hillier et al. in 2009 (Fig. 3) (5), and the first-in-human evaluation of the 123I-labeled version was finally published in 2013 (19).
As a consequence of those findings, the group in Heidelberg, Germany, started an initiative to transform the diagnostic tracer PSMA-11 into the theranostic variant PSMA-617, which can also be radiolabeled with the therapeutically relevant trivalent radiometals 177Lu or 90Y for β-therapy and 225Ac for α-therapy. Of 18 DOTA conjugates with optimized linker moieties between the Glu-urea-Lys–binding motif and the DOTA chelator, PSMA-617 was identified as the best candidate for translation to clinical settings (20,21). In parallel, another theranostic PSMA radioligand, PSMA for imaging and therapy (PSMA I&T), was also described (22,23). 177Lu-PSMA-617 and the 225Ac-labeled version are currently the main candidates for endoradiotherapy (i.e., PSMA RLT) of PC (24–27).
Because of large numbers of patients and the hitherto limited capacity for the production of the radiometal-based PET tracer 68Ga-PSMA-11, there is also a need for 18F-labeled PSMA ligands. With PSMA-617 as the lead structure, the PET tracer 18F-PSMA-1007 was recently developed; this agent is an ideal and promising 18F-labeled PSMA inhibitor for prostate PET imaging, especially for the primary diagnosis of PC (28). The clinical potential of this novel PSMA-targeted PET radioligand is currently being explored (29,30). The 18F-labeled PET radioligands N-[N-[(S)-1,3-dicarboxypropyl]carbamoyl]-4-fluorobenzyl-L-cysteine (18F-DCFBC) (31–33) and DCFPyL (18F-DCFPyL) (34,35) were recently introduced into clinical settings as well. In contrast, 18F-PSMA-1007 has the advantage that tracer-associated activity accumulation in the urinary bladder over time is almost not observable; this advantage makes this 18F-labeled PSMA radioligand an ideal candidate for the primary diagnosis of PC and the staging of local recurrent disease (29,30).
Figure 4 shows all of the Glu-ureido–based PSMA radioligands that we consider to be clinically relevant for both SPECT and PET diagnostics and PSMA RLT.

Glu-ureido–based PSMA radioligands of clinical relevance. Cpd. = compound; Ref. = reference; RN = radionuclide.
The promising SPECT PSMA radioligands MIP-1404 (36,37) and PSMA for imaging and surgery (PSMA I&S) (38) are also in clinical development, with MIP-1404 being the first PSMA imaging agent to finalize phase 3 clinical trials. Uniquely, the iodine-containing MIP-1095 can be used for SPECT (the 123I-labeled version), for PET (the 124I-labeled version), and for RLT (the 131I-labeled version and possibly the 211At version for α-therapy). 68Ga-PSMA-11, 18F-DCFBC, 18F-DCFPyL, and 18F-PSMA-1007 are exclusive PET PSMA radioligands. PSMA-617 and PSMA I&T are theranostic PSMA radioligands because they can be radiolabeled with both diagnostic (e.g., 68Ga and 44Sc) and therapeutic (e.g., 177Lu) radiometals (23,39,40). Because PSMA-1007 is derived from PSMA-617, the tracers (18F-PSMA-1007 and 177Lu-PSMA-617) can be used as a theranostic tandem of PSMA radioligands. Other tandem combinations are also possible because the diagnostic component need not be an exact replica of the therapeutic component.
What Have We Learned from Lesson 1?
Taking into account the data about the Glu-ureido–based PSMA radioligands that have already been clinically introduced (Fig. 4), we conclude that a good compromise between optimal PSMA radioligand design and compliance with clinical demands has been found. This conclusion is supported by the fact that prospective clinical trials are ongoing. For example, there are trials with 68Ga-PSMA-11 (e.g., European Union Trials Register EudraCT 2016-001815-19 [German Cancer Consortium trial]; ClinicalTrials.gov NCT02919111 [PSMA PreRP]), 18F-DCFPyL (e.g., ClinicalTrials.gov NCT02981368 [OSPREY]), 131I-MIP-1095 (e.g., ClinicalTrials.gov NCT03030885), 99mTc-MIP-1404 (e.g., European Union Trials Register EudraCT 2012-001864-30; ClinicalTrials.gov NCT02615067 [proSPECT-AS]), and 177Lu-PSMA-617 (e.g., ClinicalTrials.gov NCT03042468). It is expected that 99mTc-MIP-1404 (Trofolastat) will be launched on the market as the first low-molecular-weight SPECT PSMA radioligand because a phase 3 clinical trial with this tracer is already under way. The structure-aided characterization (lesson 2) and the elucidation of the structure–property relationships of the Glu-ureido–based PSMA inhibitors (lesson 3) may help in deciding whether the design of PSMA radioligands that have already been developed is optimal for PC imaging and endoradiotherapy (see the PSMA-binding motif of the PSMA radioligands, shaded in cyan in Figs. 1–3).
LESSON 2: NEED FOR STRUCTURE-AIDED DESIGN OF GLU-UREIDO–BASED PSMA RADIOLIGANDS
Structural studies of PSMA–ligand complexes provided mechanistic insight into interactions governing ligand recognition by the enzyme. They helped in rationalizing a vast amount of available structure–activity relationship data and were later used for the structure-aided design of the next generation of PSMA ligands. The first PSMA crystal structures were reported more than a decade ago (41,42), and now more than 60 x-ray crystallographic structures of PSMA–ligand complexes are publicly available at the Protein Data Bank (PDB) (www.rcsb.org).
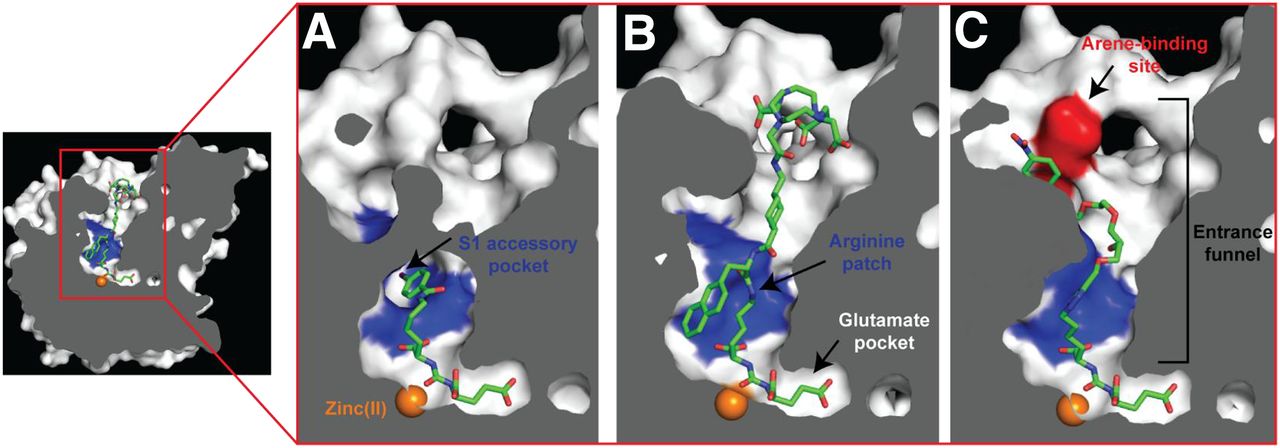
The PSMA internal inhibitor–binding cavity can be roughly divided into 3 continuous parts: the S1′ glutamate recognition pocket, the dinuclear zinc(II) active site, and an irregularly shaped entrance funnel connecting the active site to the external surface of PSMA (Fig. 5) (43). This structural arrangement is reflected in turn in the inhibitor design, for which all currently used Glu-ureido–based PSMA ligands comprise a terminal glutamate moiety connected to a linker/effector part via the zinc-binding ureido functionality. Such ligands therefore can be viewed as a composite of 3 semiindependent modules that can be tailored to suit experimental or clinical needs.

Internal cavity of PSMA. Cross-section of PSMA showing internal inhibitor-binding cavity comprising S1′ glutamate recognition pocket, dinuclear zinc(II) active site, and irregularly shaped entrance funnel. Although S1′ glutamate recognition pocket is restricted in size and shape, spacious entrance funnel can accommodate functional groups of different sizes and physicochemical characteristics. Within entrance funnel, arginine patch, S1 accessory hydrophobic pocket, and arene-binding site—prominent structural features used for inhibitor design—are highlighted. Zinc ions are shown as orange spheres, and PSMA ligands are shown as stick representations: DCIBzL (A) (PDB code 3D7H), PSMA-617 (B) (unpublished data), and ARM-P4 (C) (PDB code 2XEG).
Originally, efforts regarding extensive structure–activity relationships were aimed at identifying permissible substitutions of the terminal glutamate that would improve the physicochemical and biologic characteristics of target ligands. However, all such substitutions reported to date have failed to provide viable leads and instead have resulted in compounds with substantially lower PSMA affinities (9,44–46). Likewise, a search for a new “ultimate” zinc-binding group has not been successful; consequently, ureido-based PSMA inhibitor scaffolds are currently the most prevalent theranostic PSMA-targeting vectors used, followed only by transition-state mimetics, such as phosphoramidates (47).
In recent years, researchers turned their attention to modifications at the linker/functional spacer/effector portion of Glu-ureido–based ligands, and this approach resulted in several clinically validated compounds, such as PSMA-617 (20,21). Such modifications are well tolerated by PSMA because of the fact that, unlike the constricted S1′ glutamate recognition pocket, the entrance funnel is quite spacious and can accommodate many diverse chemical groups (43,48–50). Typically, a flexible aminohexanoyl moiety is used as the proximal segment of the linker. The P1 carboxylate group, which is the common denominator of many, but not all, Glu-ureido–based ligands, is critical for high-affinity PSMA binding and therefore can be viewed as an integral part of the Glu-ureido pharmacophore (45). The flexibility of the proximal linker enables “optimal” positioning of coupled functional spacers or effector moieties within the amphipathic entrance funnel. Additionally, it facilitates the engagement of these moieties with structurally defined pockets in the entrance funnel (e.g., the S1 accessory hydrophobic pocket or the arene-binding site), thus contributing to the increased affinity of such bivalent ligands for PSMA (Fig. 6) and allowing for the structure-assisted design of the next generation of ligands (48,51–55).
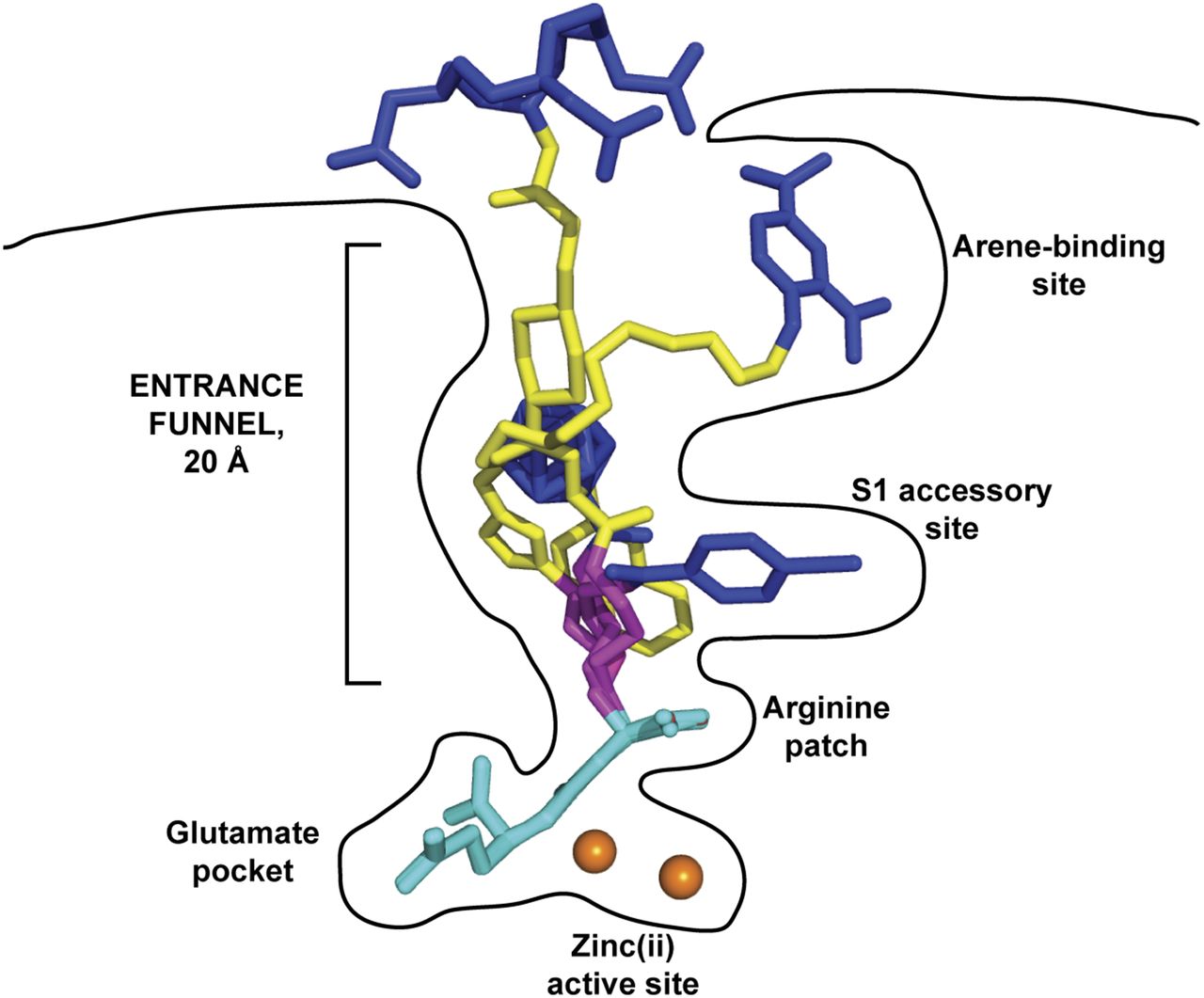
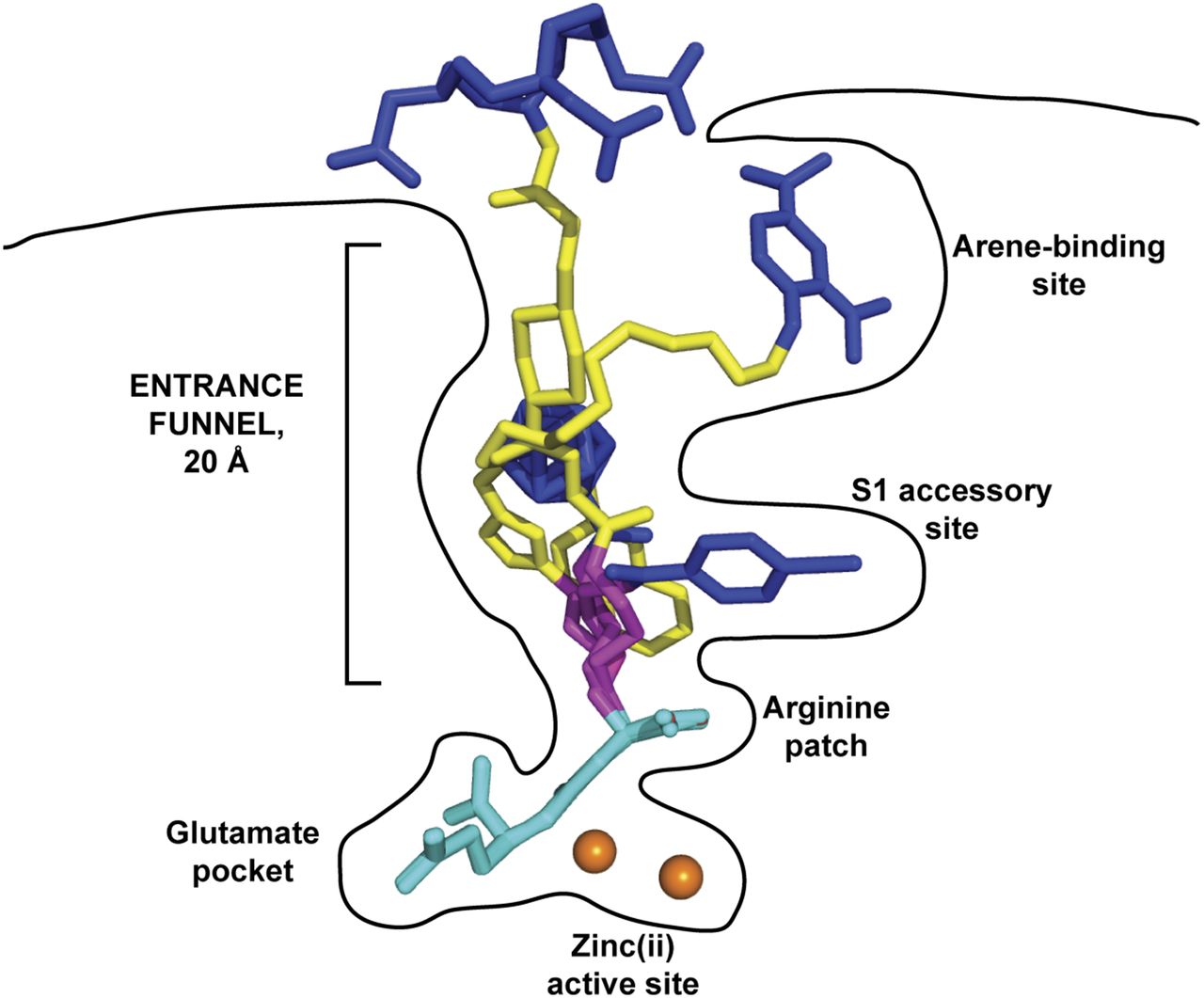
Glu-ureido–based ligands within binding cavity of PSMA. Complexes between PSMA and 4 Glu-ureido–based ligands are superimposed on corresponding Cα atoms of protein. Although there is complete structural overlap of pharmacophore modules (cyan), positioning of flexible proximal linker (magenta), functional spacer (yellow), and effector moiety (blue) is divergent within (and outside) amphipathic entrance funnel. Zinc ions are shown as orange spheres. PSMA–inhibitor complexes used were DCIBzL (PDB code 3D7H) (48), ARM-P4 (PDB code 2XEG) (52), carborane (PDB code 4OME) (55), and PSMA-617 (unpublished data).
What Have We Learned from Lesson 2?
It is obvious that the proximal linker/functional spacer will be a hot spot for modifications aimed at the development of new Glu-ureido–based PSMA ligands. It is hoped that the designed ligands, aided by x-ray crystallography, will tap into structural features of the entrance funnel that have not been thoroughly explored, resulting in their improved inhibitory or biologic characteristics. Additionally, such modifications may help in creating ligands that could be truly PSMA specific by selectively targeting PSMA (i.e., glutamate carboxypeptidase II)—a property that would be advantageous for therapeutic applications. Finally, on the basis of the structural characterization of PSMA–ligand complexes so far, in some cases, the functional spacer or effector moiety contributes substantially to the PSMA affinity of a given ligand and therefore can be viewed as an extension of the original Glu-urea-Lys pharmacophore motif—in other words, an affinity enhancer.
LESSON 3: ELUCIDATING STRUCTURE–PROPERTY RELATIONSHIPS WITH IN VITRO, IN VIVO, AND EX VIVO ASSAYS
The applicability of highly potent PSMA inhibitors can be predicted through systematic preclinical in vitro, in vivo, and ex vivo evaluations.
In vitro experiments with newly designed PSMA inhibitors are usually performed using the PSMA-positive lymph node carcinoma of the prostate (LNCaP) cell line, derived from an androgen-sensitive human lymph node metastatic lesion of prostatic adenocarcinoma (56) or, more recently, using the transfected human PSMA–expressing PC-3 PIP cell line (PC-3 transfected to stably express the human PSMA protein) and the human PSMA–deficient PC-3 flu cell line (39). The main focus is examination of the PSMA-binding affinity and the cellular internalization behavior. Moreover, in vivo imaging of tumor-bearing mice or rats reveals variable tumor visualization as well as clearance of radiolabeled compounds from the kidneys and the whole body. Finally, ex vivo biodistribution demonstrating radioactivity accumulation in tumors, all relevant organs (e.g., salivary glands, spleen, liver, kidneys, and intestines), the blood, and muscles provides the clearest evidence for selection of the most promising compounds.
It is not surprising that significant structure-dependent differences between individual PSMA inhibitors are generally observed. In the development of PSMA-617, PSMA inhibition potency, cellular internalization, and imaging quality could be strongly influenced even by slight differences in the linker moiety between the PSMA-targeting Glu-urea-Lys–binding motif and the DOTA chelator (20). In that study, the in vitro results, especially internalization of the ligand by LNCaP cells, correlated highly with the in vivo tumor imaging properties. Thus, cellular internalization in vitro seems to be necessary for in vivo success, especially for endoradiotherapy (i.e., PSMA RLT). Moreover, it was shown that introducing a hydrophobic group(s) might interfere with PSMA-binding ability because lipophilicity influenced the clearance of radioactivity by the kidneys and from the background (20).
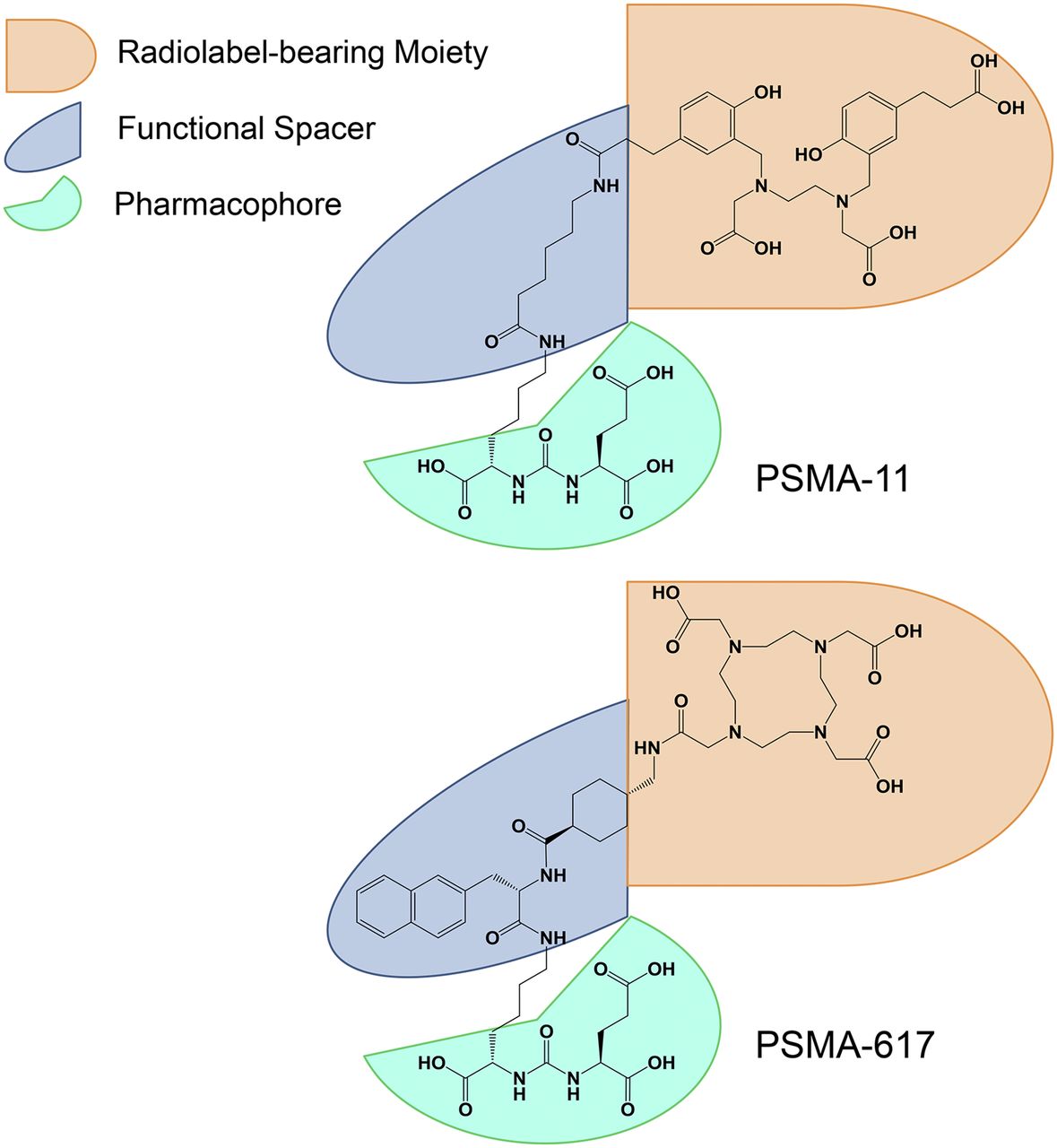
Another example involves the widely used diagnostic radiotracer 68Ga-PSMA-11 (8), which served (among others) as the benchmark for the development of a DOTA-conjugated theranostic counterpart (Fig. 7). Eder et al. reported that direct replacement of the HBED-CC chelator in PSMA-11 by the DOTA chelator resulted in significant diminution of the tumor-targeting properties of the new compound (17). Thus, the original aminohexanoic acid linker in PSMA-11 was redesigned to mimic the proven biologic interactions of HBED-CC with the PSMA-binding pocket(s). An extensive preclinical evaluation provided an effective strategy for producing highly potent DOTA-conjugated PSMA inhibitors attaining sufficient PSMA-dependent cellular internalization, akin to that of PSMA-11, and improving the pharmacokinetic properties. The optimal properties were finally attributed to PSMA-617 (20,21).

Dependence of transformation of diagnostic tracer PSMA-11 into theranostic variant PSMA-617 on structural arrangement of pharmacophore, functional spacer, and radiolabel-bearing/effector moiety.
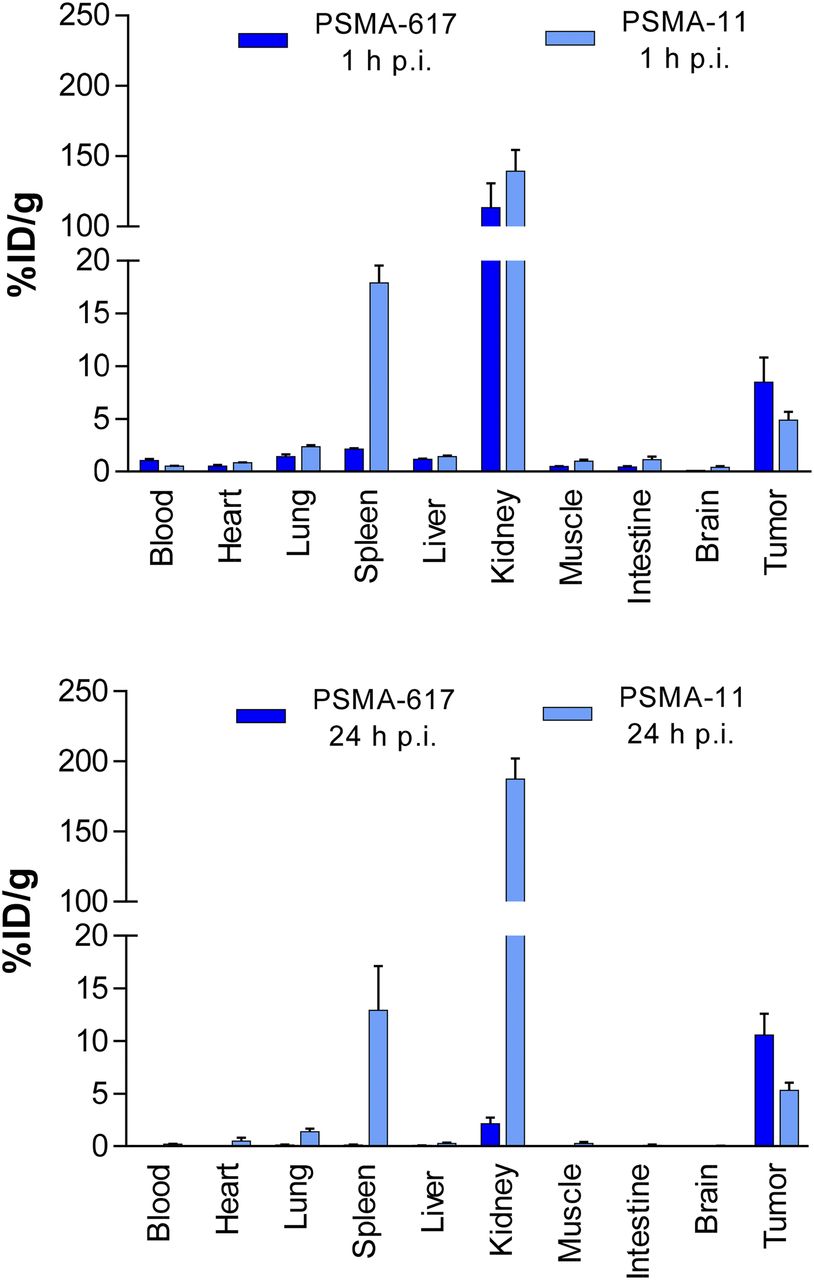
A side-by-side in vitro comparison of PSMA-617 and PSMA-11 is shown in Table 1. The PSMA inhibition potency and internalization into LNCaP cells were greater for PSMA-617. The diverse behavior of these 2 inhibitors was also demonstrated by dynamic small-animal PET imaging (Fig. 8) and ex vivo organ distribution (Fig. 9). Major differences were observed in the spleen (17.88 ± 2.87 [mean ± SD] percentage injected dose/g [%ID/g] for PSMA-11 and 2.13 ± 0.16 %ID/g for PSMA-617) at 1 h after injection and in the kidneys (187.4 ± 25.3 %ID/g for PSMA-11 and 2.13 ± 1.36 %ID/g for PSMA-617) and the tumor (3.20 ± 2.89 %ID/g for PSMA-11 and 10.58 ± 4.50 %ID/g for PSMA-617) at 24 h after injection. Compared with the results at 1 h after injection, the kidney uptake of PSMA-11 was not significantly reduced at 24 h after injection, whereas PSMA-617 was nearly completely cleared. Such rapid renal clearance of radioactivity is necessary to reduce potential radiation toxicity (radiotoxicity) to the kidneys and to avoid possible chronic long-term side effects of PSMA-specific endoradiotherapy (21).
PSMA Inhibition Potency (Ki) and Cellular Internalization of PSMA-11 and PSMA-617, as Determined with LNCaP Cells (21)

PET/CT and SPECT/CT imaging of PC-3 PIP/flu tumor-bearing mice. Tumor-targeting efficacy and pharmacokinetic properties were evaluated with 44Sc-PSMA-617 (A), 177Lu-PSMA-617 (B), 68Ga-PSMA-617 (C), and 68Ga-PSMA-11 (D) 2 h after injection. Bl or bl = bladder; ki = kidney. (Reprinted with permission of (39).)

Organ distribution of 68Ga-PSMA-617 and 68Ga-PSMA-11 1 h after injection (p.i.) and of 177Lu-PSMA-617 and 67Ga-PSMA-11 24 h after injection. (Reprinted with permission of (21).)
Taken together, data from systematic in vitro, in vivo, and ex vivo preclinical evaluations are important for elucidating the structure–property relationships of prospective PSMA inhibitors and for clarifying the effects of structural modifications on the tumor-targeting and pharmacokinetic properties. This strategy might lead to more accurate rational and structure-aided design of new urea-based PSMA-targeted and other radioligands.
What Have We Learned from Lesson 3?
A careful preclinical evaluation of structure–property relationships connects the molecular structure of PSMA ligands with in vitro (e.g., inhibition constants [50% inhibitory concentration and Ki] and dissociation constant [Kd]), ex vivo (e.g., %ID/g), and in vivo (e.g., SUV) data. Further in vitro data, such as lipophilicity, and the optimal specific/molar activity of the radioligand used for imaging and therapy, should also be considered in relationship to PSMA inhibition potency. For transformation of the exclusive diagnostic PET tracer PSMA-11 into a theranostic version (in this case, PSMA-617), elucidation of the structure–property relationships was the main prerequisite for identifying the optimal candidate (PSMA-617). PSMA-617 can now be labeled with both therapeutic radionuclides (e.g., 90Y, 177Lu, and 225Ac), having physical half-lives of several days, and short-lived diagnostic radionuclides (e.g., 68Ga and 44Sc).
The data suggest that rational design initiatives for a new series of PSMA radioligands should include careful examination of their structure–property relationships to reveal which variant is optimal for diagnostic or therapeutic applications or which variant is ideal for both imaging and therapy. Because even slight structural modifications of PSMA ligands can result in a complete loss or alteration of the necessary in vitro, ex vivo, and in vivo properties, a detailed preclinical characterization of a PSMA ligand is needed before clinical translation can be considered. Clinical demands (e.g., for primary diagnosis, diagnosis of relapse, or PSMA RLT of metastatic castration-resistant PC) require the best PSMA ligands for diagnostic or therapeutic PSMA-targeted PC treatment.
LESSON 4: CONSIDERATION OF HOW TO TRANSFER PSMA RADIOLIGAND APPROACH TO OTHER TARGETED THERANOSTIC APPROACHES
For many years, several other classes of theranostic radioligand systems have been in preclinical and clinical development for imaging or peptide receptor radionuclide therapy (in addition to the prominent radioiodine therapy approach). These include, among others, radiolabeled peptide analogs, such as the somatostatin receptor (SSTR) (57,58), cholecystokinin 2 receptor (gastrin receptor) (59,60), gastrin-releasing peptide receptor (GRPr) (61,62), glucagon-like peptide 1 receptor (63), and C-X-C motif chemokine receptor 4 (CXCR4) (64–66) ligands. So far, the SSTR, GRPr, and CXCR4 radioligands have been the most promising in clinical settings.
In the state-of-the-art theranostic SSTR radioligand approach, clinically established radioligands are used for the treatment of refractory neuroendocrine tumors that predominantly upregulate subtype SSTR2, belonging to the G-protein–coupled receptor (GPCR) family (57). Since the 1980s, 111In-diethylenetriaminepentaacetic acid-octreotide (OctreoScan; Mallinckrodt) has been used in clinical settings for neuroendocrine tumor imaging (58). With the introduction of commercially available 68Ge/68Ga generators, several PET tracers followed: 68Ga-DOTATOC (U.S. Food and Drug Administration Orphan Drug Designation in 2014), 68Ga-DOTATATE (U.S. Food and Drug Administration kit approval for NETSPOT [Advanced Accelerator Applications] in 2016), and 68Ga-DOTANOC (67–69). The same DOTA conjugates have been used for peptide receptor radionuclide therapy (i.e., for SSTR2 radioligand therapy) when labeled with β-particle emitters (e.g., 90Y and 177Lu) or an α-particle emitter (e.g., 213Bi) (70,71). For 177Lu-DOTATATE (Lutathera; Advanced Accelerator Applications), expanded access has been granted for the “treatment of patients with inoperable, somatostatin receptor positive, midgut carcinoid tumors, progressive under somatostatin analog therapy” (ClinicalTrials.gov NCT02705313).
Theranostic SSTR antagonists, such as 68Ga-DOTA-JR11 and 177Lu-DOTA-JR11, have been propagated and suggested to have an advantage over SSTR agonists because SSTR antagonists are not readily internalized into tumor cells and therefore can label (putatively) more receptor sites in vivo (72,73). This is a unique and puzzling finding of utmost interest for this class of theranostic GPCR radioligands, and it can only be speculated that the described advantage of SSTR antagonists over SSTR agonists can be explained by factors such as the recognition of a larger number of binding sites by antagonists, slow dissociation of SSTR-bound antagonists, and delayed and slow internalization of antagonists (74).
PSMA does not belong to the GPCR family; it belongs to the membrane-type zinc peptidase family. For PSMA inhibitor radiotracers, there is evidence that the internalization of PSMA ligands into tumor cells must be improved to gain optimal uptake in tumor lesions over time—relevant especially for PSMA RLT (lesson 3) (20,40). Moreover, during preclinical evaluations of theranostic radioligands, the physiologic levels of expression of the target must be carefully considered; these levels should be orders of magnitude lower in the normal state than in the diseased state.
Importantly, only low levels of endogenous PSMA expression have been found in many organs (in addition to the normal prostate), including the proximal tubules of the kidneys, the lacrimal and salivary glands, the spleen, the intestinal brush border membranes, the liver, the testes, the ovaries, and the brain. These findings are among the main prerequisites for concluding that only low receptor levels are present in the normal state and that the chosen biologic target is highly upregulated in the diseased state—leading to the decision to start designing a new class of theranostic radioligands.
PSMA indeed is overexpressed in prostate cancer cells relative to normal prostate cells (>6-fold), the kidneys (>2,000-fold), and the intestines (>900-fold) (75). With 600,000–800,000 PSMA molecules per cancer cell (as determined on LNCaP cells), PSMA represents an important PC marker (76). Higher stages and higher grading of PC are associated with significant upregulation of PSMA; this association may imply a role of PSMA in the transformation and/or invasiveness of PC (15,16,77).
The fact that the physiologic function(s) of PSMA in the aforementioned tissues (other than the nervous and digestive systems) is mostly unknown should not hinder the initiation of efforts to design a new class of theranostic radioligands (15,16,78–83).
The design of an ideal agent for the imaging of a particular type of cancer must begin with the selection of a molecular target that is specific to the cancer in question and that is consistently expressed at high levels in tumor cells throughout the natural progression of the disease, preferably with no alteration in expression during therapy. The signal from tumor cells indicates the viable tumor mass and can be used to monitor disease burden. This information is particularly critical for assessing the presence of a tumor and changes in tumor mass and spread, for determining whether the disease is low-grade localized disease, and for determining the response to treatment in a patient with late-stage metastatic disease.
Ligand selection should be based on rapid uptake and persistent localization at the target site with minimal retention in nontarget tissue. Small molecules have a critical advantage over much larger constructs given their faster rate of clearance from the blood and increased tumor permeability, which allow them to evade physiologic barriers encountered by larger molecules, such as antibodies.
As detailed previously, PSMA was selected as the molecular target of choice because its expression is dramatically upregulated in poorly differentiated, metastatic, and hormone-refractory adenocarcinomas (15) as well as after androgen deprivation therapy (84) and in lymph node metastases (85).
The collective results to date therefore suggest that PSMA is an ideal target for the molecular imaging of PC. This notion has been repeatedly validated because several groups have successfully demonstrated that radiolabeled small-molecule inhibitors of PSMA have the potential to localize PC through common molecular imaging modalities, such as SPECT and PET. These novel PSMA-targeting tracers should affect clinical management by guiding appropriate patient-specific treatment strategies. It is likely that a sensitive and specific means of imaging PC and PC metastases would have a significant impact on the clinical management of PC by providing greater certainty about the presence and extent of disease during the course of a patient’s life.
What Have We Learned from Lesson 4?
Researchers have worked for decades on new classes of theranostic radioligands for the treatment of different tumor entities. Merging knowledge of the rational and structure-aided design of new small-molecule theranostic radioligands with understanding of the structure–property relationships of the ligands in vitro, ex vivo, and in vivo is necessary to find the optimal radiotracer for a particular biologic target to design the best treatment options for different types of cancer. Depending on the type of biologic target (the receptor) to be addressed (e.g., GPCR for SSTR and zinc protease for PSMA), any future class of theranostic radioligands must be designed to detect and treat a particular cancer, thereby addressing various clinical needs in oncology.
It is known that in some cases of PC, PSMA is not upregulated at all or is only heterogeneously upregulated in tumor and metastatic tissues (86,87). As an additional option, clinicians could consider other classes of theranostic radioligands, such as the GRPr radiotracers, to offer the best PC patient–centered care (61).
Diversity of biologic features in tumors indeed presents a problem not only for diagnosis but also for treatment, as shown by molecular analysis of multiple spatially separated samples obtained from primary renal carcinomas and associated metastatic sites. It was found that 63%–69% of all somatic mutations were not detectable across every tumor region. Furthermore, gene expression signatures for a good prognosis and a poor prognosis were detected in different regions of the same tumor (88). These findings mean that cancer cell clones with various phenotypes may exist in spatially separated regions within the same tumor and in different tumors within a single patient (89). The same is true for PC, in that genomic heterogeneity is observed within individual prostate glands and between patients (90). At the phenotype level, this finding has been demonstrated by immunohistochemistry for PSMA by Mannweiler et al. (86). In their analysis, PSMA-positive cells were often surrounded by areas that were negative for PSMA. In primary tumors as well as distant metastases, total or partial PSMA negativity and cytoplasmic positivity were seen, without a correlation with the Gleason score, histologic subtype, or localization of metastases. Therefore, novel strategies targeting 2 or more tumor-associated molecules may be needed to increase the efficacy of diagnosis and treatment of a cancer type.
A combined analysis at the RNA and protein levels for the expression of αvβ3-integrin, neurotensin receptor 1, PSMA, and prostate stem cell antigen (PSCA) was done in PC tumor cells and xenografts (91). Although there were discrepancies between increases at the RNA level and those at the protein level for αvβ3-integrin, neurotensin receptor 1, and PSCA, these proteins represent possible targets for the imaging and therapy of PC. Similar results were obtained in an immunohistochemistry study with antibodies against GRPr, PSCA, and PSMA (92). In lymph node metastases, GRPr positivity was seen in 85.7% of the cases; the corresponding results for PSCA and PSMA were 95.2% and 100%, respectively. In bone metastases, GRPr staining was positive in 52.9% of the cases; the corresponding results for PSCA and PSMA were 94.1% and 100%, respectively.
These data qualify PSCA and GRPr as alternative or better complementary targets for the diagnosis and therapy of PC.
EPILOG
In this article, we have attempted to address several issues related to the new class of small-molecule Glu-ureido–based PSMA inhibitor radiotracers from the basic research and clinical demand perspectives. We briefly discussed the diversity of biologic features in tumors (tumor heterogeneity), suggesting potential complementary biologic targets that could result in further theranostic radioligand approaches, at least for the treatment of PC (lesson 4).
First-in-human (phase 0 and 1) studies and prospective clinical trials in general are already under way to confirm the preclinical proof-of-concept validations for the human situation (lesson 1). First proof-of-concept studies with PSMA radioligands are highlighted in detail in Figure 2 of the article by Eiber et al. in this supplement issue of The Journal of Nuclear Medicine; side-by-side, only slight, but distinct, differences in the biodistribution behaviors of 68Ga-PSMA-11, 68Ga-PSMA I&T, 18F-DCFBC, 18F-DCFPyL, and 18F-PSMA-1007 (Fig. 4) were seen in PC patients. This side-by-side comparison should be the next step in clinical settings. Phase 0 comparison studies were performed in the first human trials with PSMA inhibitors, namely, MIP-1072 and MIP-1095 (19), and in the initial clinical evaluation of MIP-1404 and MIP-1405 (36). Interindividual (or even intraindividual) patient examinations helped determine the optimal PSMA radioligand for each intended PC option (imaging or therapy). Tracer accumulation in normal organs, such as the salivary and lacrimal glands, the kidneys, the spleen, and the intestines (which is not directly comparable to the preclinical in vivo and ex vivo results [lesson 3]), must be assessed in patients as well, especially from the safety (radiation dosimetry) perspective. The outcome of such assessments (18,26,29,40,93–97) will show whether future generations of PSMA radioligands should still be considered for first-in-human studies or whether efforts targeting new complementary molecules should be undertaken to further increase the efficacy of the diagnosis and treatment of PC.
Transferring knowledge gained from the development of the new class of theranostic PSMA radioligands to further theranostic approaches in nuclear medicine is possible. This concept is underlined by the classes of theranostic radioligands (e.g., targeting SSTR, GRPr, and CXCR4) that have already been clinically established for the treatment of other cancer types. In general, the movement of radiotracer development (i.e., radiopharmaceutical drug development) in cancer research towards theranostic radioligand approaches has already occurred in nuclear medicine and is attracting other disciplines in the area of (radio)oncology. This approach can generate international consortium project groups focused on unique prospective theranostic clinical trials with the aims of providing cancer patients with the optimal treatment option and obtaining regulatory acceptance by the respective health care systems.
DISCLOSURE
This publication was supported in part by the CAS (RVO: 86652036), project BIOCEV (CZ.1.05/1.1.00/02.0109) from the ERDF, and the GACR to Cyril Bařinka (301/12/1513). Klaus Kopka, Martina Benešová, and Uwe Haberkorn hold property rights to PSMA-617 and PSMA-1007. John Babich is an inventor of MIP-1095, MIP-1072, MIP-1404, and MIP-1405. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Sandra Casula, from the German Cancer Research Center, who helped in the editing and careful review of the manuscript. We also thank Springer Nature, the American Chemical Society, and the American Association for Cancer Research for permission to reprint Figures 1, 2, and 8.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- 1.↵
- 2.
- 3.
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.
- 50.↵
- 51.↵
- 52.↵
- 53.
- 54.
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.
- 66.↵
- 67.↵
- 68.
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.
- 80.
- 81.
- 82.
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.
- 95.
- 96.
- 97.↵
- Received for publication February 16, 2017.
- Accepted for publication March 27, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- PROLOG
- LESSON 1: RETROSPECTIVE ON GLU-UREIDO–BASED PSMA RADIOLIGANDS OF CLINICAL RELEVANCE
- LESSON 2: NEED FOR STRUCTURE-AIDED DESIGN OF GLU-UREIDO–BASED PSMA RADIOLIGANDS
- LESSON 3: ELUCIDATING STRUCTURE–PROPERTY RELATIONSHIPS WITH IN VITRO, IN VIVO, AND EX VIVO ASSAYS
- LESSON 4: CONSIDERATION OF HOW TO TRANSFER PSMA RADIOLIGAND APPROACH TO OTHER TARGETED THERANOSTIC APPROACHES
- EPILOG
- DISCLOSURE
- Acknowledgments
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- Targeting prostate cancer by new bispecific monocyte engager directed to prostate-specific membrane antigen
- Radionuclide Therapy in Prostate Cancer: From Standalone to Combination PSMA Theranostics
- Intraindividual Comparison of 18F-PSMA-1007 and 18F-DCFPyL PET/CT in the Prospective Evaluation of Patients with Newly Diagnosed Prostate Carcinoma: A Pilot Study
- A Perspective on the Evolving Story of PSMA Biology, PSMA-Based Imaging, and Endoradiotherapeutic Strategies
- Theranostic Concepts: More Than Just a Fashion Trend--Introduction and Overview