


Abstract
In this review we examine the mechanisms (causes) underlying the increased glucose consumption observed in tumors within a teleological context (consequences). In other words, we will ask not only “How do cancers have high glycolysis?” but also, “Why?” We believe that the insights gained from answering the latter question support the conclusion that elevated glucose consumption is a necessary component of carcinogenesis. Specifically we propose that glycolysis is elevated because it produces acid, which provides an evolutionary advantage to cancer cells vis-à-vis normal parenchyma into which they invade.
In 1976, Sidney Weinhouse, a pioneer in the investigation of cancer metabolism (Fig. 1), stated that, “Since our perspectives have broadened over the years, the burning issues of glycolysis and respiration in cancer now flicker only dimly” (1). 18F-FDG PET imaging has rekindled interest in the metabolic control of cancer by demonstrating a remarkable preponderance of metastatic cancers to trap far more glucose than normal tissue. This observation has been the starting point for Darwinian-based mathematic models to answer the question: “Why do metastatic cancers have such high rates of glucose consumption”? Driving this research is the assumption that all aspects of tumor biology including progression, invasion, and metastases are the result of a continuous optimization process governed by Darwinian dynamics (Fig. 2). This leads to a fundamental assumption that any common phenotype (including aerobic glycolysis) must always confer a selective advantage on the cells that possess such a phenotype. Within an evolutionary/bioenergetic context, aerobic glycolysis would appear to be a conundrum because glycolytic metabolism is very inefficient (producing only 2 mol of adenosine triphosphate [ATP] per mole of glucose vs. 36 mol of ATP per mole of glucose for the citric acid cycle) and generates an acidic, potentially toxic environment. Thus, there does not appear to be a significant adaptive advantage of using glycolysis when oxygen is present (the Warburg effect). Yet, its prevalence in the overwhelming majority of metastatic tumors is compelling evidence that aerobic glycolysis plays a very significant role in promoting tumor development.

Sidney Weinhouse. (Courtesy of the Office of NIH History, National Institutes of Health.)

Charles Darwin, circa 1870, by Julia Margaret Cameron (1815–1879).
To address this, the evolutionary dynamics of carcinogenesis have been modeled using several mathematic methods, including information theory, evolutionary game theory, reaction–diffusion models, and modified cellular automata (2–12). Insights from these models combined with modern imaging, what we have termed “imag(in)ing,” have demonstrated that hypoxia and acidosis develop inevitably in the microenvironment of premalignant epithelial tumors, such as ductal carcinoma in situ (DCIS). This results from aberrant proliferation that carries cells away from their underlying blood supply, which remains on the opposite side of an intact basement membrane (BM). These conditions serve as microenvironmental selection forces that govern cellular adaptation during the later stages of somatic evolution. The models demonstrate that, after this evolutionary pathway, aerobic glycolysis does in fact confer a significant growth advantage because it produces a toxic, acidic environment which is more toxic to adjacent tissue compared with the cancer cells themselves. In the process, corollaries have been uncovered that explain other observed phenomena in cancers, namely the extreme variability in gene expression patterns.
This review will discuss causes and consequences of increased aerobic glycolysis by connecting mechanistic information to teleology. It will begin with an introduction to canonical views of carcinogenesis and its molecular controls. This will be followed by a brief review of the evidence that 18F-FDG uptake is elevated in metastatic cancers and a discussion of the molecular causes and consequences of this elevated glycolysis in cancer. Evolutionary Game Theory models and their predictions will then be described and the subsequent imaging of cancer histopathology, tumor hypoxia, and tumor pH. These data have led to additional conceptual models, derived from mathematic models, which attempt to describe carcinogenesis as a series of adaptations to environmental selections.
CARCINOGENESIS AND MICROENVIRONMENT
Cancer Progression
Before we consider the pathophysiology of cancer, the developmental origin of cancer cells and their normal physiologic niches must be discussed. In adults, there are essentially 2 types of tissues: epithelia and stroma. These are behaviorally distinct tissues in that epithelia are highly polarized and will grow in monolayers along a single axis, whereas stromal cells are phenotypically plastic, highly motile, and can grow in response to morphogenetic gradients in any direction. Neoplasia of epithelia gives rise to carcinomas, such as breast, lung, pancreatic, prostate, or melanoma, which have a high prevalence in adults. Stromal cancers include sarcomas, gliomas, and liquid cancers, which are more prevalent in the young (Table 1). In mature organs, epithelia and stroma are physically separated by a BM made of laminin, collagen IV, proteoglycan, and various growth factors and proteases (13). On one side of the BM, epithelial cells grow in a mono- or bilayer of polarized cells, forming a duct that is contiguous with the outside world. On the other side of the BM lies a stroma of connective tissue containing fibroblasts, muscle, blood, and lymphatic vessels, which are constantly remodeling (Fig. 3). Epithelial and stromal components are established during embryogenesis from the 3 primordial germ cell layers of ectoderm, endoderm, and mesoderm that give rise to skin and nervous system, epithelia, and mesenchymal tissues, respectively (Fig. 4). Epithelia are generally derived from endoderm and ectoderm (dermis), and stromal components are generally derived from mesoderm. However, there is significant plasticity, and cell fates are more dependent on spatial and conditional factors than their cell of origin (14). This plasticity has significance to cancer, as epithelial cells undergo a transition to a mesenchymal phenotype (an epithelial-to-mesenchymal transition) concomitant with their adoption of an invasive phenotype (15).
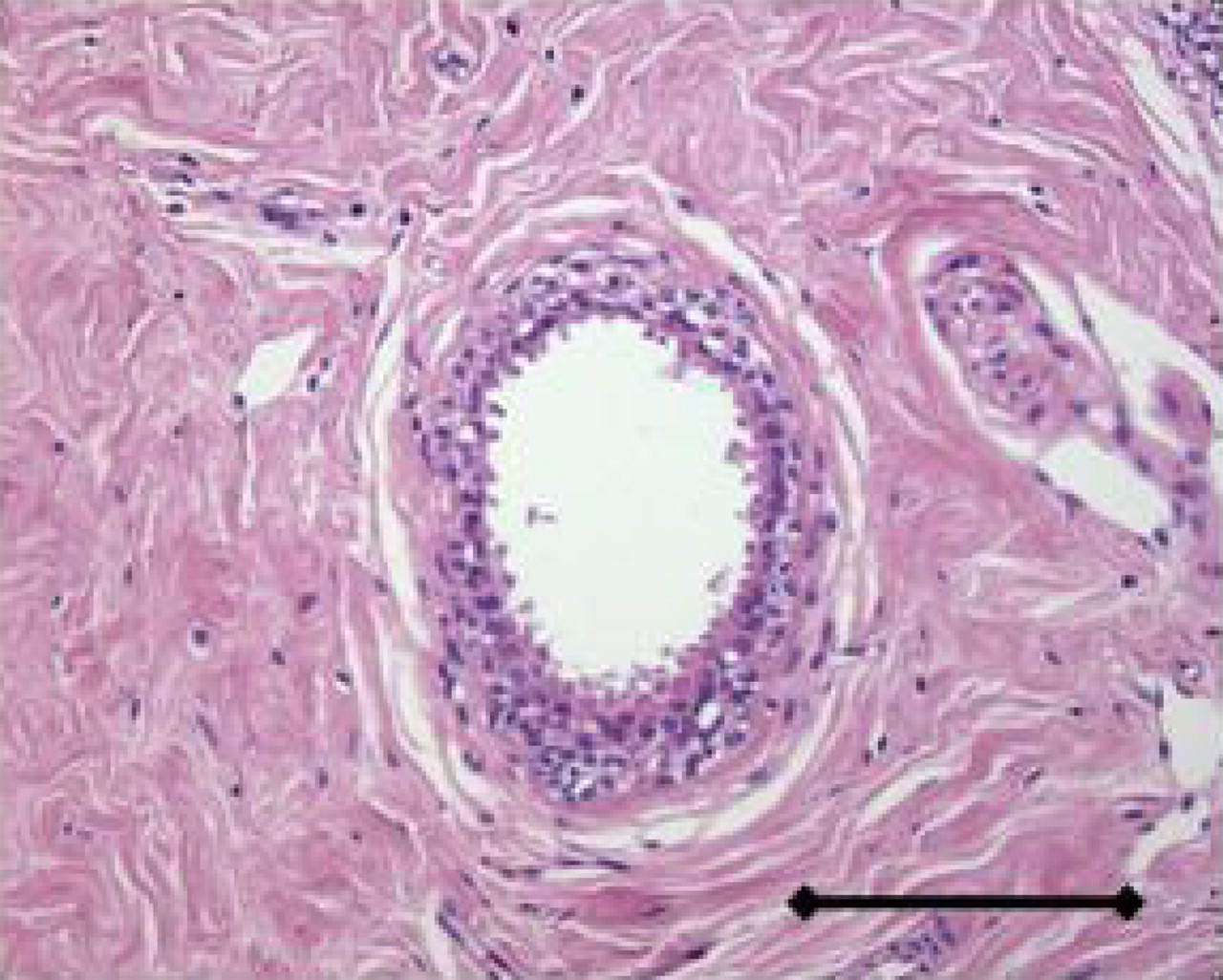
Normal human breast duct. Bar = 0.2 mm.
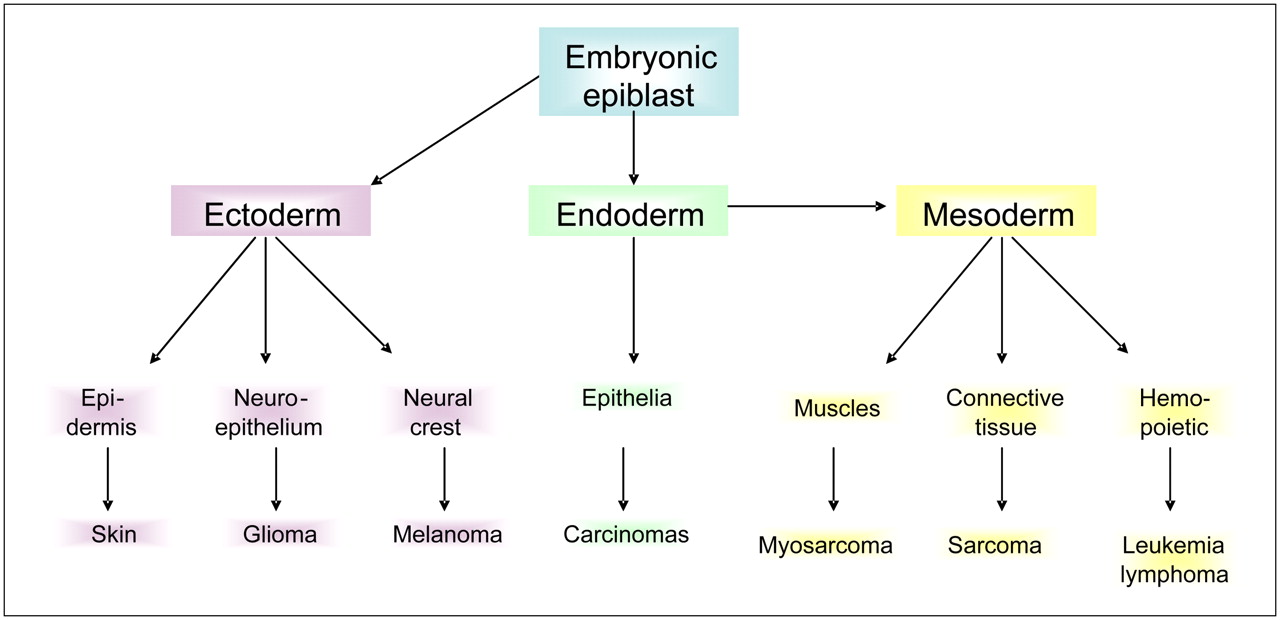
Developmental origins of tissues and cancers.
Incidence of Cancers and Mortality in U.S. Adults and Children (All Races, per 100,000)
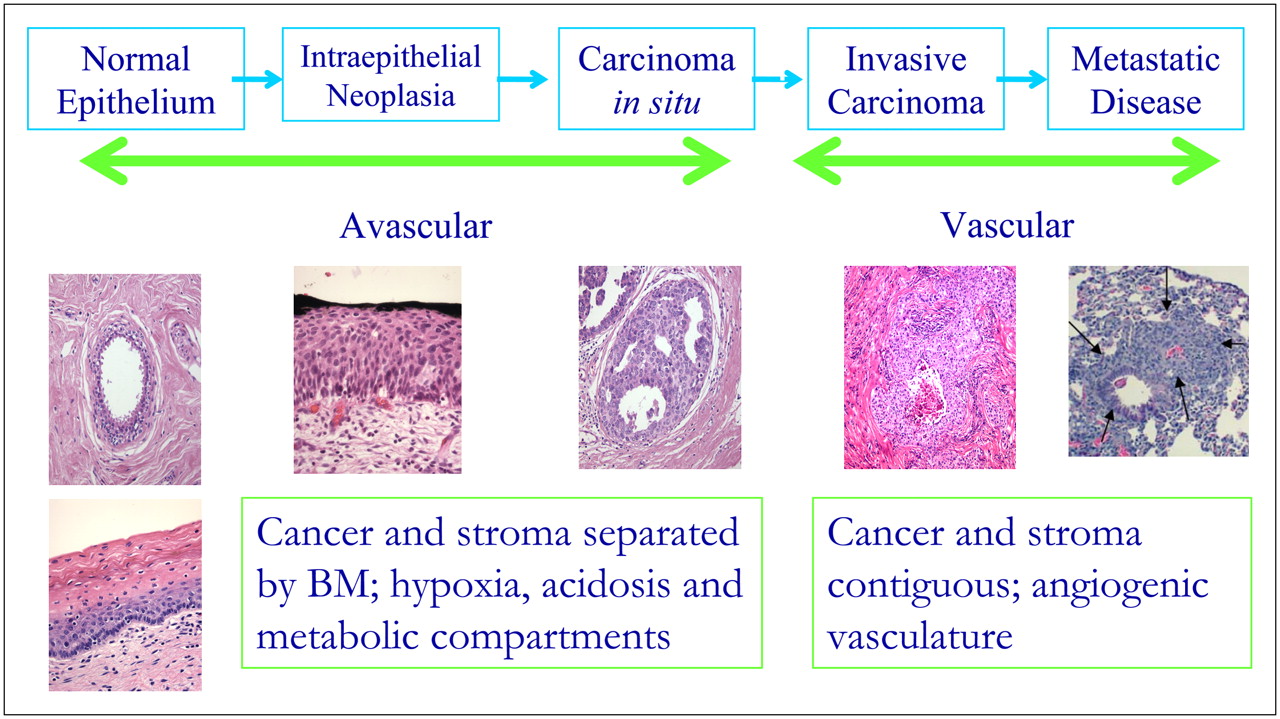
Development of carcinomas proceeds through distinct stages, as shown for colorectal cancer in Figure 5. The initial genetic changes result in hyperplasia that progresses into dysplasia within the lumen of the ducts. The dysplastic condition, known as carcinoma in situ (CiS), or adenoma in the case of colorectal cancer (CRC), progresses through early, middle, and late phases yet still remains separated from the stroma by the BM. Once the BM is breached, the cells make contact with the stroma to become a locally invasive cancer. Further evolution is then required to allow the avascular microscopic disease to “partner” with the stromal cells to promote angiogenesis and extracellular matrix remodeling. The invasive tumor cells, either directly or indirectly via lymphatics, enter the bloodstream (intravasation). A small fraction of these are able to extravasate and colonize distant sites, forming metastatic cancer.
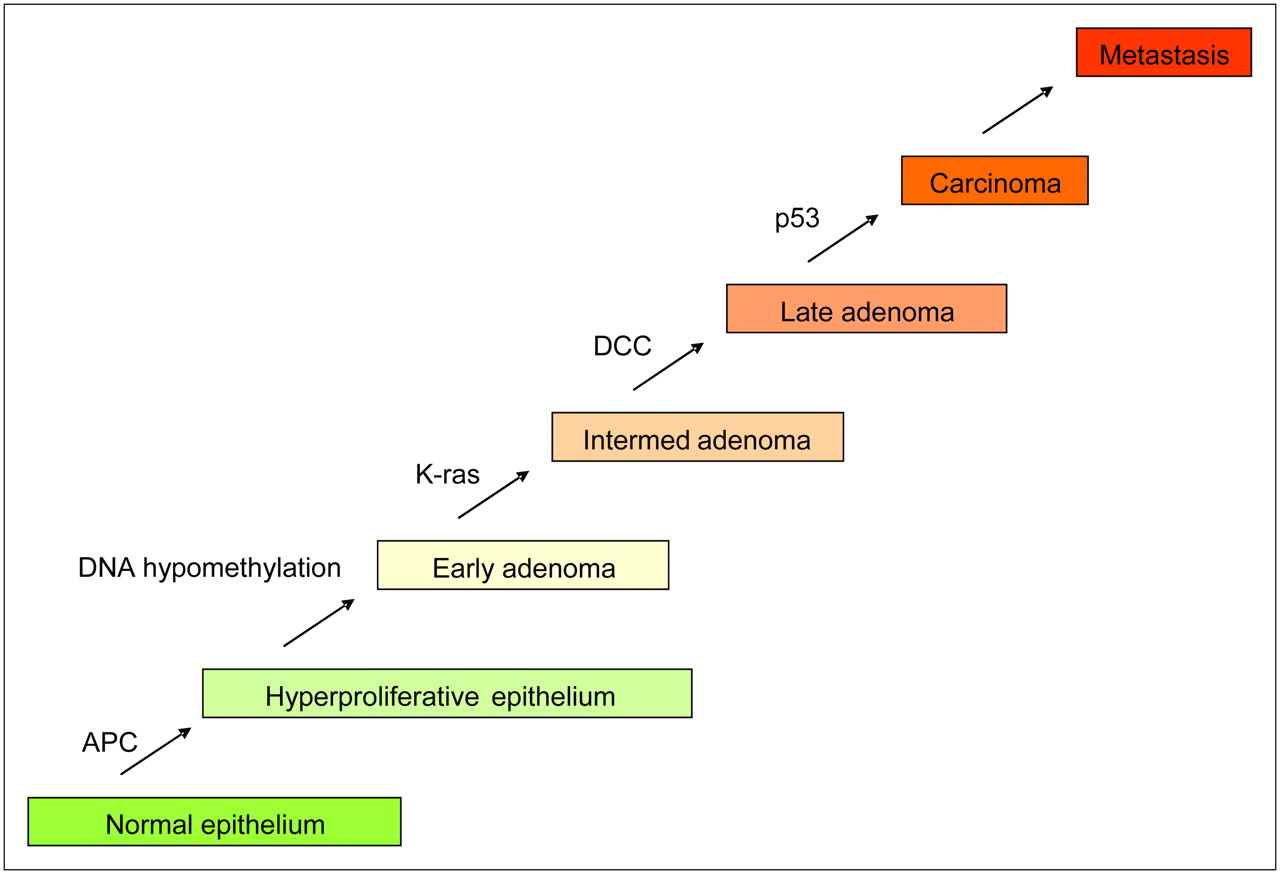
Genetic changes during colorectal carcinogenesis. APC = adenomatosis polyposis coli; DCC = deleted in colorectal cancer; Intermed = intermediate. (Modified from (23).)
Although significant differences among cancers exist, there are some phenotypic elements common to all cancers, which are known as the “hallmarks of cancer”: evading apoptosis, self-sufficiency in growth signals, insensitivity to antigrowth signals, tissue invasion and metastasis, limitless potential to replicate, and sustained angiogenesis (16). We propose that all commonly observed traits must confer specific adaptive advantages to cancer cells as they progress. In other words, the hallmarks of cancer can be viewed as the phenotypic adaptation necessary to overcome all of the proliferation barriers that evolving tumor populations encounter during their progression through carcinogenesis.
Early Carcinogenesis
The initial steps in carcinogenesis result in the transformation of normal epithelium to epithelium that is hyperplastic, which results in cellular proliferation away from the BM, forming multilayer growth. We argue that, whatever the mechanism, these steps have to involve a deregulation of cell–cell and cell–matrix growth inhibitory interactions that maintain the epithelia in a monolayer (17). Despite the importance of this step, the molecular mechanisms of initiation and promotion have remained elusive. It is commonly assumed that this step requires genotoxic stress, as cancers can be initiated by chemicals that form DNA adducts (18). Hence, radiation damage or inflammation-derived reactive oxygen species, combined with deficiencies in DNA repair, could also be initiating events. The initiating events likely involve mutational activation of proto-oncogenes to oncogenes. Proto-oncogenes are normal cellular constituents that are involved in regulation of cell cycle and proliferation that, when activated, can give rise to unregulated growth, a hallmark of cancers. These can be activated through chemical carcinogens, and some have homologs in RNA viruses. Fusion of these 2 approaches with characterization of the src oncogene by Varmus and Bishop was awarded the Nobel Prize in 1986 (19). Table 2 lists common classes of proto-oncogenes and the incidence of their representative oncogenes.
Representative Oncogenes and Tumor Suppressor Genes
Chronic activation of pro-proliferative pathways would normally be opposed by the action of proliferation suppressor gene products that include cell cycle checkpoint genes (20,21). Hence, carcinogenesis is also associated with loss of function of tumor suppressor genes, some of which are listed in Table 2. Typically, humans that are heterozygous for tumor suppressor genes have increased susceptibilities to cancers after loss of heterozygosity. This heritable loss of function can occur through genetic mutations, through chromosomal loss, or through epigenetic mechanisms, such as DNA methylation (22).
Genetic Models of Carcinogenesis
The developmental progression of many cancers can be evaluated and predicted according to their complement of induced oncogenes and repressed tumor suppressor genes. Fearon and Vogelstein were the first to document this for colorectal cancer, CRC (23). In this work, they identified distinct stages of CRC from normal epithelium to metastatic disease (Fig. 5). In samples from each of these stages, they identified chromosomal and genetic alterations associated with each stage. As cancers progressed, they accumulated genetic alterations such that, for example, the incidence of mutated ras increased from ∼7% in early adenoma to ∼55% in intermediate, ∼60% in late, and ∼45% in invasive carcinomas. Thus, it can be proposed that this mutation caused cells to be more aggressive but that it was not necessary once cells metastasized. These data support a clonal, evolutionary model of carcinogenesis, such as that diagrammed in Fig. 6. Environmental or endogenous factors induce genotoxic stress, which is normally repaired. However, with relative frequency, genetic alterations can be fixed in a cell (green) that will render it more proliferative. These expand and accumulate successive mutations (blue, red) that each contribute to development of a more aggressive cancer phenotype. This model represents current thinking, which is decidedly genocentric.
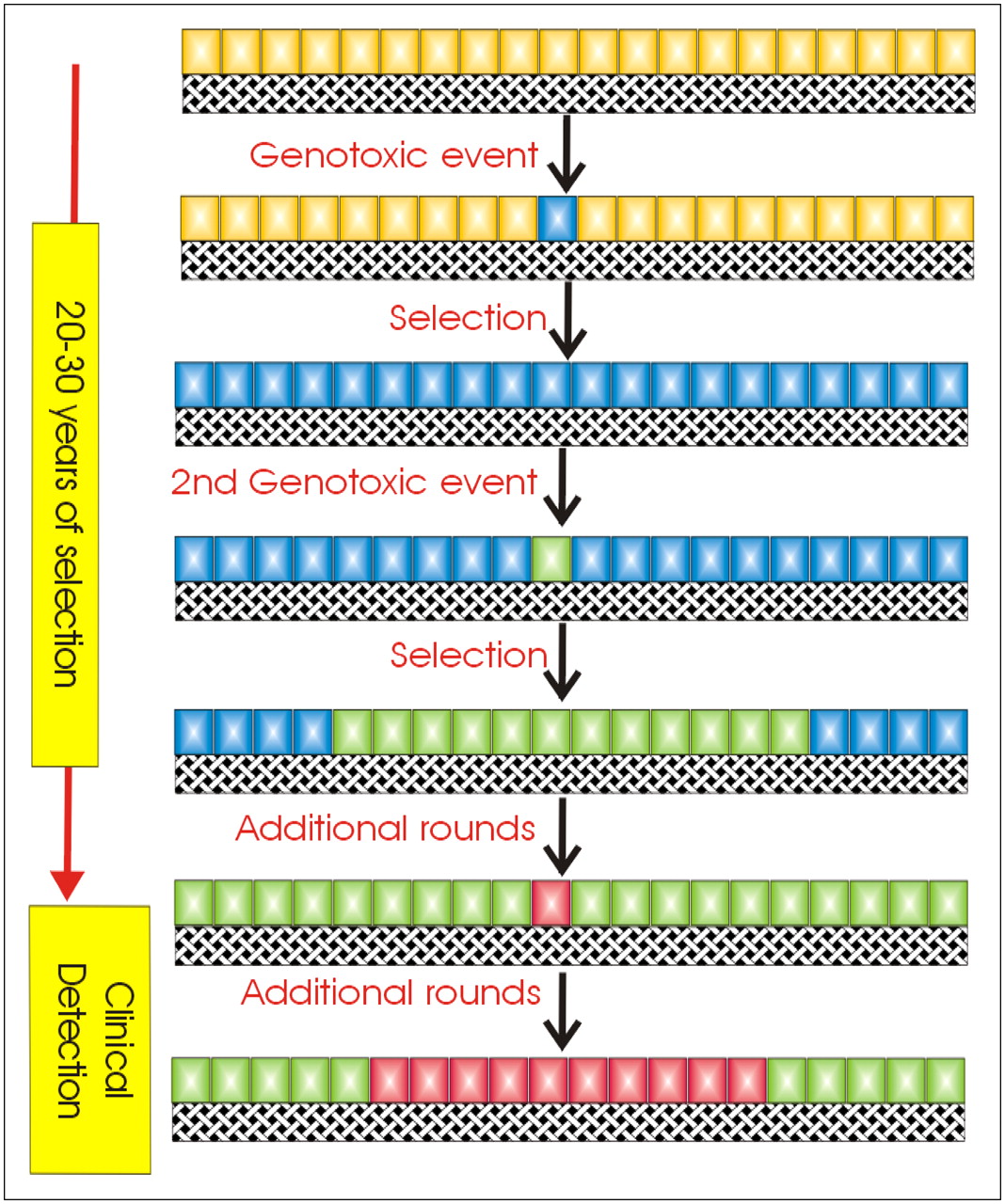
Model for clonal growth of cancers.
Although this model alludes to the clonal expansion of tumor cells as an evolutionary process, it virtually ignores the altering environmental landscape that leads to selection. The interaction between gene products and their environment in carcinogenesis has been the subject of recent models from our group and others.
Microenvironment During Cancer Progression
As preinvasive cancers proliferate and evolve, their microenvironment is altered, as shown in Figure 7. Normal epithelia are single cell layers growing on a BM surface, surrounded by stroma. These cells are adjacent to blood vessels on the other side of the BM and so are well perfused with serum growth factors and nutrients. Hence, the proliferation of normal epithelial cells into the lumen must be constrained through cell–cell and cell–BM interactions. If epithelial cells die, they are sloughed off into the lumen. The lack of cell–cell contact will induce an adjacent cell to divide to fill the space. Notably, proliferating cells require attachment to the BM to proliferate and survive. If this attachment is abrogated, the cell will die through a process termed “anoikis,” which is a specialized form of cell death (24,25). Thus, the earliest phenotypic changes observed in carcinogenesis must be adaptations that suppress anoikis. These include acquisition of the ability to proliferate when not in contact with the BM as well as relaxation of cell–cell contact inhibition so that cells can continually proliferate under conditions that would ordinarily suppress growth. Such alterations can be triggered by a multitude of genetic changes, which introduces the principle of “functional equivalence.” This principle states that multiple genetic alterations can produce similar phenotypic sequelae. Thus, at this early stage, many genetic alterations can initiate cancer growth, but they must have in common a phenotype of resistance to anoikis and reduced cell–cell contact inhibition.

Morphologic changes associated with cancer progression.
With increased hyperplasia (dysplasia), the growing epithelium becomes many layers thick. As we have pointed out in prior work, this results in steeper gradients for delivery of substrates (13) as well as stromal-derived growth factors. A notable consequence of this increasing gradient is that the periluminal cells become increasingly oxygen deprived as hyperplasia transitions to dysplasia in CiS lesions. In breast cancers, staining for hypoxia-induced gene products, such as glucose transporter isoform 1 (Glut-1) or carbonic anhydrase (CA) IX (CA-IX), shows that periluminal hypoxia becomes evident in late-stage DCIS (comedo) lesions (26). Thus, during transition from early to late dysplastic lesions, regional variations in the local microenvironment occur with, among other things, substantial periluminal hypoxia. This heterogeneity in the microenvironment alters the selection forces in the “adaptive landscape” and, thus, the adaptive properties of the evolving cancer phenotypes.
Hypoxia and acidosis will select for cells that are apoptosis- resistant (27–32). Notably, the genetic alterations that occur early during carcinogenesis (i.e., to relax contact inhibition) may or may not increase the fitness for this second round of selection. If they do, then they are said to exhibit “multibarrier effects.” For example, K-ras not only will reduce contact inhibition, it will also make cells more resistant to apoptosis and thus would be an effective multibarrier oncogene. Alterations to integrins can reduce anoikis but will not necessarily render cells apoptosis-resistant.
A consequence of hypoxic selection may be increased glucose consumption through stabilization of hypoxia-inducible factor (HIF) 1α (HIF-1α) (30). Hence, it is possible that the glycolytic phenotype is established during this phase of cancer progression. Indeed, increased 18F-FDG uptake in mouse tumors occurs before transition to an invasive phenotype (33). Glucose metabolism invariably leads to increased acid production, and the effect of this on acidification of the interstitial pH can be exacerbated by reduced perfusion in the periluminal volume of in situ lesions (34,35). Acid pH has been shown to increase invasive behavior (36–42). Thus, one mechanism of cancer progression may involve elevated glycolysis leading to acidosis that leads to invasion. However, although it is often observed, according to the equivalence principle, elevated glucose consumption need not be a prerequisite for cancers to transition from a CiS to an invasive carcinoma. Alternative mechanisms can be envisioned whereby cells can acquire an invasive phenotype without glycolysis and acidosis.
Transitioning to an invasive cancer involves breaking through the BM, giving epithelial cells direct access to the vasculature and the stroma for the first time in their natural history (vide supra). It is at this time, through unknown mechanisms, that these cancer cells undergo an epithelial-to-mesenchymal transition. These mesenchymelike cells are more highly motile than their epithelial precursors and can be characterized by expression of stroma-specific markers (43–45).
With direct access to the stroma, cancer cells can disperse through the heme or the lymphatic vasculature. Migration of these cells through lymphatic vessels results in colonization of the draining lymph nodes in a characteristic pattern. Thus, in breast cancer, the draining axillary lymph nodes are generally the first to become involved and are thus known as sentinel nodes. Either directly or indirectly via lymphatics, invasive cancer cells eventually find themselves in the bloodstream. It has been estimated that a 1-cm3 tumor will shed as many as 106 cancer cells per day into the blood (46). Through specific endothelial cell interactions, cancer cells will lodge in ectopic sites that are specific for different cancers. The occurrence of metastases in specific organs led to Paget's “seed–soil” hypothesis, wherein the host organ provides factors necessary for the growth of the metastatic lesion (47). Despite intensive study for more than a century, these factors remain unknown.
In order to form successful metastases, the cancer cells must survive, colonize, and grow in the host tissue. Survival is a rare event, estimated at less than 1 cell per 1,000, although this may be dependent on the relative population of cells with “stemlike” properties (48). Colonization results in micrometastases, which can be abundant and are less than 1.0 mm in diameter. The switch from micro- to macrometastases is incompletely understood but likely involves neoangiogenesis (49).
The acid-mediated invasion hypothesis proposes that successfully invasive tumor lesions export glucose-derived H+, which leads to degradation of the extracellular matrix, thereby allowing more efficient invasion of the cancer into a host organ. This has been shown both theoretically (11) and empirically using window chambers (50). Figure 8 shows periodic acid–Schiff staining of a biopsy from an invasive metastatic colon cancer in the liver, showing a “halo” of less dense extracellular matrix surrounding the lesion. Hence, we propose that successful metastatic cancers are those that can produce H+. As these H+ are glucose-derived, then it follows that successful metastatic cancers consume high amounts of glucose, to produce acid. This appears to be the case, as evidenced from 18F-FDG PET.

Periodic acid–Schiff staining of hepatic colorectal metastasis.
18F-FDG PET OF AEROBIC GLYCOLYSIS IN METASTASES
Perhaps no other imaging test has had as much impact on the fundamental understanding of human cancer as has 18F-FDG PET. Although 18F-FDG avidity can be highly variable in primary cancers, it has an extremely high sensitivity and specificity for metastases. This was comprehensively reviewed in 2001 (51) and discussed within the context of drug development in 2005 (52). Table 3 shows a recapitulation of data from Gambhir et al. (51). Although the ranges are broad, the average sensitivity and specificity are high. Generally speaking, once a cancer progresses to distant metastases beyond lymph nodes, the sensitivity and specificity are greater than 90% (except for prostate cancers).
Sensitivity and Specificity of 18F-FDG PET for Detection and Staging of Metastases
These data are mirrored in Table 4, which includes studies that are more recent than the 2001 review. Note that the sensitivity of cervical and prostate lymph node metastases are not high, but most other metastatic carcinomas have sensitivities and specificities of greater than 90%. As discussed in previous publications, reductions in sensitivity are primarily caused by small lesions with motion, and reductions in specificity can be due to local inflammation (53).
Detection of Metastases by 18F-FDG PET
The common observation of enhanced 18F-FDG uptake indicates that it is a fundamental property of metastases (vide infra). This could suggest a commonality of mechanisms, yet this does not appear to be the case, as described later in this article.
MOLECULAR MECHANISMS REGULATING GLUCOSE UPTAKE
Glucose metabolism is essential to generate both catabolic and anabolic precursors. Thus, it is not surprising that there are a multitude of pathways that activate glucose metabolism. It has long been assumed that the primary function of glucose is to provide energy to the cell in the form of ATP as an alternative to mitochondrial respiration. Hence, a major regulator of glucose consumption is the availability of oxygen, which was first described by Pasteur (Fig. 9) in 1856 (54,55). Thus, glucose consumption is higher in hypoxic tissues (55,56). Although 18F-FDG uptake can be elevated in response to hypoxia, seminal studies comparing 18F-FDG uptake with retention of hypoxic markers (18F-misonidazole, 18F-pimonizaole) have shown little correlation between hypoxia and 18F-FDG uptake in humans (57,58), indicating that cancers continue to consume glucose at high rates, even in the presence of adequate oxygen, an effect first described by Warburg (Fig. 10) in 1930 (59).

Louis Pasteur.

Otto Warburg.
Hence, “aerobic glycolysis” can be described as a cancer hallmark (60). Elevated rates of 18F-FDG uptake are strongly negatively correlated to patient outcome in many cancers (51,61), as are elevated lactate levels (62,63). Hence, elevated glucose consumption may play an essential role in cancer progression. Genes of glycolysis are ubiquitously overexpressed in cancers (64) (65). Thus, it is of fundamental importance to understand the molecular mechanisms regulating the metabolic switch to aerobic glycolysis. The biochemical uptake mechanisms of 18F-FDG were reviewed by Pauwels et al. in 2000 (66). As discussed later in this article, there does not appear to be a single mechanism for control of aerobic glycolysis in all cancers. A conceptual framework for interpreting this observation is discussed at the end of this section.
GLUT and HK
The 2 major proteins that are unequivocally associated with increased 18F-FDG uptake in cancers are the glucose transporter, primarily GLUT-1, and hexokinase, primarily HK-2 (67–69). Thus, insights can be obtained by examining the molecular controls over the expression and activity of these proteins. Given the physiologic importance of glucose uptake, it is not surprising that GLUT-1 expression is regulated by several hormones, both extracellular, such as insulin, prolactin, follicle stimulating hormone, noradrenaline, and vasopressin, and intracellular, such as testosterone, estrogen, progesterone, glucocorticoids, retinoic acid, and thyroxin (70,71). Some of these effects are likely mediated via activity of the SP1 transcription factor (72,73) or cyclic adenosine monophosphate (cAMP) levels (74), both of which can be dysregulated in cancers.
In addition to control by glucose levels, HK-2 expression can be affected by hormones such as insulin, and second messengers such as cAMP (75,76). The HK-2 isoform is notable because it is reversibly bound to mitochondria and is a key regulator of glycolysis and survival in multiple cancers, where it is a poor prognostic indicator (77–79). When bound to mitochondria, HK-2 preferentially uses mitochondrion-generated ATP to phosphorylate glucose very efficiently (80). Drugs, such as clotrimazole, can induce a dose-dependent detachment of hexokinase from mitochondria and inhibit glycolysis (81). Notably, the binding site for HK-2 on the mitochondrial membrane is the voltage-dependent anion channel (VDAC), a mitochondrial protein (porin) involved in exchange of metabolites for oxidative phosphorylation, as well as the release of cytochrome c during intrinsic apoptosis cascade (82). This is one of many instances wherein elevated glycolysis is correlated to apoptosis resistance, although delineation of a function relationship between these 2 processes has been elusive.
Mitochondrial Defects
Given the importance of glucose in energy metabolism, it is perhaps not surprising that mitochondrial defects would result in increased “aerobic” glycolysis (83). Thus, even under normoxic conditions, the cell machinery sensing the presence of oxygen would be ineffective. Indeed, Warburg's original hypothesis stated that increased aerobic glycolysis was due to a deficiency in respiration (84). Despite elegant and sophisticated methods (at the time), he was not able to prove his hypothesis during his lifetime. However, with more modern molecular techniques, mitochondrial defects have been correlated with increased aerobic glycolysis. For example, increased 18F-FDG uptake in vivo and glucose consumption in vitro correlate strongly to a loss of mitochondrial markers, such as the F1-ATPase, in lung cancers (85). Mitochondrial impairment is often seen in clear cell renal carcinomas (86). In colorectal cancers, transcriptome and proteome analyses have observed that expression of glycolytic and tricarboxylic acid cycle proteins were inversely proportional (87–89). Also, in breast cancer, the metabolic proteome undergoes a pronounced shift toward an enhanced glycolytic phenotype concurrent with a profound reduction in the F1-ATPase levels with a classification sensitivity of approximately 97% to overall and disease-free survival (90). Notably, the expression of F1-ATPase alone allowed identification of a subgroup of breast cancer patients with significantly worse prognosis. In more empiric studies, directed depletion of mitochondrial DNA resulted in reduced electron transport activity and increased glycolysis (91), or accumulated mutations in the NADH dehydrogenase subunit 2, ND2, led to increased HIF-associated aerobic glycolysis (92).
The molecular signals between mitochondria and glycolysis are likely identical to the “retrograde signaling” involved in the Pasteur effect. A central mechanism appears to involve the activity of pyruvate dehydrogenase (PDH), which is responsible for importing pyruvate into the mitochondria as a substrate for the tricarboxylic acid cycle (also known as the Krebs cycle or the citric acid cycle). The mammalian pyruvate kinase M2 isoenzyme is regulated by ATP, adenosine diphosphate, AMP, ribose-5-phosphate, fructose-1,6-bisphosphate, and the regulatory enzyme pyruvate dehydrogenase kinase 1 (PDK1). Recently, it has been shown that phosphotyrosine residues can bind to and inactivate the M2 isoform and that this diverts carbons from energy production into anabolic processes (92a). A companion article showed that knockdown of the M2 (embryonic) form of PK and replacement with the M1 (adult) isoform resulted in inhibition of aerobic glycolysis in 3 different tumor cell lines (92b). Inhibition of PDH kinase with, for example, dichloroacetate, leads to increased aerobic metabolism, decreased glycolysis, and reversal of apoptosis resistance (93). The hypoxia-inducible factor, HIF-1 (discussed later in this article), directly transactivates the gene encoding PDK1, which will lead to inhibition of PDH activity (94). Notably, reductions in PDH activity through the aforementioned mechanisms may result in increased pyruvate and lactate levels, and these will stabilize HIF-1 in an oxygen-independent manner (95). The AMP-activated kinase (AMPK) may also play an important role in transducing mitochondrial signals to the cytoplasm. For example, upregulation of glycolysis in response to mitochondrial inhibition is dependent on AMPK activity (96). Physiologically, the AMPK family of proteins comprises heterotrimeric enzymes that enhance uptake and oxidative metabolism of fatty acids and glucose, and coordinately inhibit anabolic pathways (97). They are activated by LKB1, a kinase that is inactivated in a rare hereditary form of cancer, Peutz-Jeghers syndrome (98). Mitochondrial biogenesis itself is stimulated by oncogenic signals (e.g., c-myc) and this is inhibited by chronic HIF activation (99).
It is also notable that the mitochondria are also central to the intrinsic apoptotic pathway (100). Thus, it is tempting to ascribe both increased aerobic glycolysis and apoptosis resistance (both hallmarks of cancer) to a single, central mechanism. However, empirically proving this hypothesis has proven elusive (101).
Hypoxia-Inducible Factor
HIF-1α and HIF-2α are independently paired with HIF-1β to create basic helix–loop–helix heterodimeric transcription factors that are differentially expressed in a variety of cancers (102). HIF-1α is constitutively synthesized and is degraded in an oxygen-dependent manner (103). Besides the aforementioned hypoxia and substrate-level stabilization by pyruvate and lactate, HIF proteins can be stabilized by multiple genetic factors, such as epidermal growth factor and insulin receptor activation, nitrous oxide, Akt (kinase B) activity, and ras/MEK activity (104–113). Because of this, HIF can be characterized as being reversibly increased by physiologic stress (e.g., hypoxia) or by hormonal growth factor stimulation or as being constitutively active under normoxic conditions through heritable alterations, such as activated oncogenes. Activation of HIF regulates a pleiotropic response that can involve the altered expression of up to 454 genes (114). Among the most commonly observed pathways affected are those that would be predicted to be part of a response to hypoxia, for example, increased glycolysis, increased angiogenesis, and increased iron metabolism. A major consequence of HIF activation is the stimulation of glycolysis through increased transcription of glycolytic genes (115). It is clear that HIF proteins are expressed in cancer and that their prevalence coincides with severity of disease (113,116,117). Its centrality to the regulation of glucose metabolism has led to the general belief that it alone can be responsible for the Warburg or Pasteur effects (118,119).
MYC
myc was first characterized as the transforming gene of the avian myelocytomatosis virus (v-myc). The mammalian homolog c-myc gene was first discovered in Burkitt's lymphoma, associated with a translocation in chromosome 8. The protein MYC is an oncogenic transcription factor that contains a basic helix–loop–helix leucine zipper domain. It is half of a heterodimeric transcription factor complex, usually associated with Max, another basic helix–loop–helix transcription factor. The c-myc proto-oncogene is upregulated, primarily by amplification, in from 6% (breast) to 60% (hepatocellular carcinoma; colon) of cancers and is strongly correlated to outcome (120,121). Among the many MYC/Max transcriptional targets are the genes for glycolysis (122), including lactate dehydrogenase (123,124). MYC expression leads to increased lactate production rates even under aerobic conditions, and this has been implicated to play a role in sensitivity to apoptosis (125).
In addition to directly transactivating glycolytic genes, MYC also affects glycolysis through an interaction with HIF-1. A seminal study investigating this relationship showed that HIF-1α indirectly activates p21cip1, a key cyclin-dependent kinase inhibitor that controls a G1 checkpoint, by physically binding to the MYC N-terminal region and subsequently derepressing p21cip1 function (126). The heterodimeric HIF-1 transcription factor has also been shown to activate the MXI1 gene, which encodes a repressor of MYC transcriptional activity, as well as promoting the proteosomal degradation of MYC independently of MXI1 (127). Thus, under normal conditions, HIF and MYC appear to be antagonistic, as one transcription factor signals deprivation and the other signals for growth and proliferation, respectively. However, if MYC is overexpressed, as in many cancers, HIF appears to cooperate with it in inducing transcription of HK-2 and PDH kinase, which cooperatively result in increased aerobic glycolysis, as well as transactivation of VEGF (128).
PI3K/Akt/mTOR
Phosphatidylinositol-3-kinase (PI3k) is a key regulator of fundamental cellular functions including transcription, translation, proliferation, growth, and survival (129,130). PI3k phosphorylates the 3′-OH group of phosphatidylinositol-4,5-bisphosphate (PIP2) to produce phosphatidylinositol-3,4,5-triphosphate (PIP3) (131). PIP3 acts as a binding site for plekstrin homology domain-containing proteins to translocate to the inner leaflet of the plasma membrane (132). The PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor is a PIP3 phosphatase that acts to reconvert PIP3 back to PIP2. It is inactive through deletion or mutation in many cancers, resulting in constitutively high levels of PIP3.
Akt (protein kinase B) is a 57-kDa Ser/Thr kinase that exists in 3 isoforms, with Akt-1 being most associated with epithelium-derived cancers (133). Akt contains a plekstrin homology domain and preferentially binds to PIP3, which translocates it to the plasma membrane where it is phosphorylated on Thr308 and Ser473 by membrane kinases, such as the phosphoinositide-dependent kinase, PdK-1 (134,135). In breast cancers, the Akt pathway is activated in late DCIS and in the majority of invasive breast cancers, and this is moderately correlated to PTEN loss (136,137).
Activated P-Akt phosphorylates several targets involved in aerobic glycolysis and antiapoptosis. Phosphorylated Akt stimulates glucose consumption in transformed cells without affecting the rate of oxidative phosphorylation (138). When the BAD protein is phosphorylated by Akt, it is prevented from inhibiting the antiapoptotic BCL-2 and BCL-XL proteins (139). The activation of Akt by transforming mutations, such as the amplification of HER-2/neu in breast cancer and the formation of the BCR/ABL fusion gene in chronic myelogenous leukemia, seems to be essential for the transforming activity of these oncogenes (140). High expression of Akt activity makes cells more susceptible to death after glucose withdrawal, causing this to be termed “glucose addiction” (141).
p53/TIGAR/PFKB3
p53 is a tumor suppressor transcription factor that is mutated in up to 80% of cancers (142). Its physiologic function is to produce cell cycle arrest or apoptosis in response to DNA damage (143,144). It is dysfunctional in mutated forms and hence fails to induce checkpoint arrest in response to genotoxicity, thus increasing the accumulation of potentially carcinogenic mutations (145). It has only recently been recognized that wild-type p53 plays a key role in regulating glucose consumption (146). For example, p53-induced apoptosis involves the systematic activation of multiple pathways, including those involved in glucose metabolism (147). Wild-type p53 can repress GLUT1 transcription, and mutations within the DNA-binding domain of p53, which are usually associated with malignancy, impair this repressive effect, thereby resulting in increased glucose metabolism and cell energy supply (148). Like MYC, p53 also directly affects mitochondrial biogenesis. For example, wild-type p53 affects the synthesis of cytochrome c oxidase 2, which has been ascribed to being responsible for aerobic glycolysis in p53-deficient tumors (149).
The effects of p53 on glycolysis may be mediated by a recently characterized protein, the TP53-induced glycolysis and apoptosis regulator (TIGAR), which inhibits glycolysis by modulating fructose-2,6-bisphosphate (F26bP) levels (150). F26bP increases the rate of glycolysis by allosterically activating phosphofructokinase. F26bP levels are regulated by a family of bifunctional phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFB1–PFKFB4). Especially relevant is isozyme 3, PFKFB3, which is activated by mitogenic stimuli and steroid hormones, and is constitutively expressed by several human tumor cells, possibly through activation by the oncogene, Ras (151,152). The gene for PFKB3 is also HIF-induced (153) and is found to be markedly elevated in multiple aggressive primary neoplasms, including colon, breast, ovarian, and thyroid carcinomas (154). Once transcribed, the protein can be activated by phosphorylation (155). Inhibition of inducible PFK-2 protein expression decreases 5-phosphoribosyl-1-pyrophosphate, a product of the pentose phosphate pathway (PPP) and an important precursor for nucleic acid biosynthesis (156).
Ras
The Ras superfamily of low-molecular-weight GTP-binding proteins includes H-Ras, K-Ras, and N-Ras, which are all active in human cancer (157). Ras activation in tumors is frequently the result of a point mutation in the Ras gene (158), which leads to constitutive activation of downstream effector enzymes that regulate cell cycle progression, survival, and migration. One of the most studied signaling pathways is through serine/threonine kinase Raf, which phosphorylates and activates a cascade of mitogen-activated protein kinases (MAPKs) and cell cycle regulatory proteins (159,160). Additionally, Ras interacts directly with the catalytic subunit of type I PI3k (161,162), which stimulates the activity of Akt (163).
Among their many functions, Ras proteins have been implicated in the regulation of aerobic glycolysis (164–167). Pharmacologic inhibition of Ras (164,168) with farnesylthiosalicylate or dominant-negative expression (169) result in the inhibition of Ras signaling to both the Raf/MEK/ERK and the PI3k pathways (170), which result in a substantial downregulation of glycolytic enzymes, including Glut-1. Likewise, stable transfection with H-Ras promotes increased glucose uptake and lactate production (171–173). In these cases, H-Ras upregulated 6-phosphofructo-1-kinase (PFK-1) activity through induction of F26bP (171,173). Increased PFK-1 activity in rat kidney cells stably transfected with Ras and human papillomavirus type 16 E7 oncoprotein leads to higher levels of fructose-1,6-bisphosphate, which allosterically activates the tetramerization of pyruvate kinase type M2 (174). This results in increased AMP levels, which suppressed DNA synthesis and cellular proliferation, probably though activation of AMPK (165,175,176). Similar results were observed in mouse fibroblasts bearing K-Ras codon-specific mutations (167).
Teleology
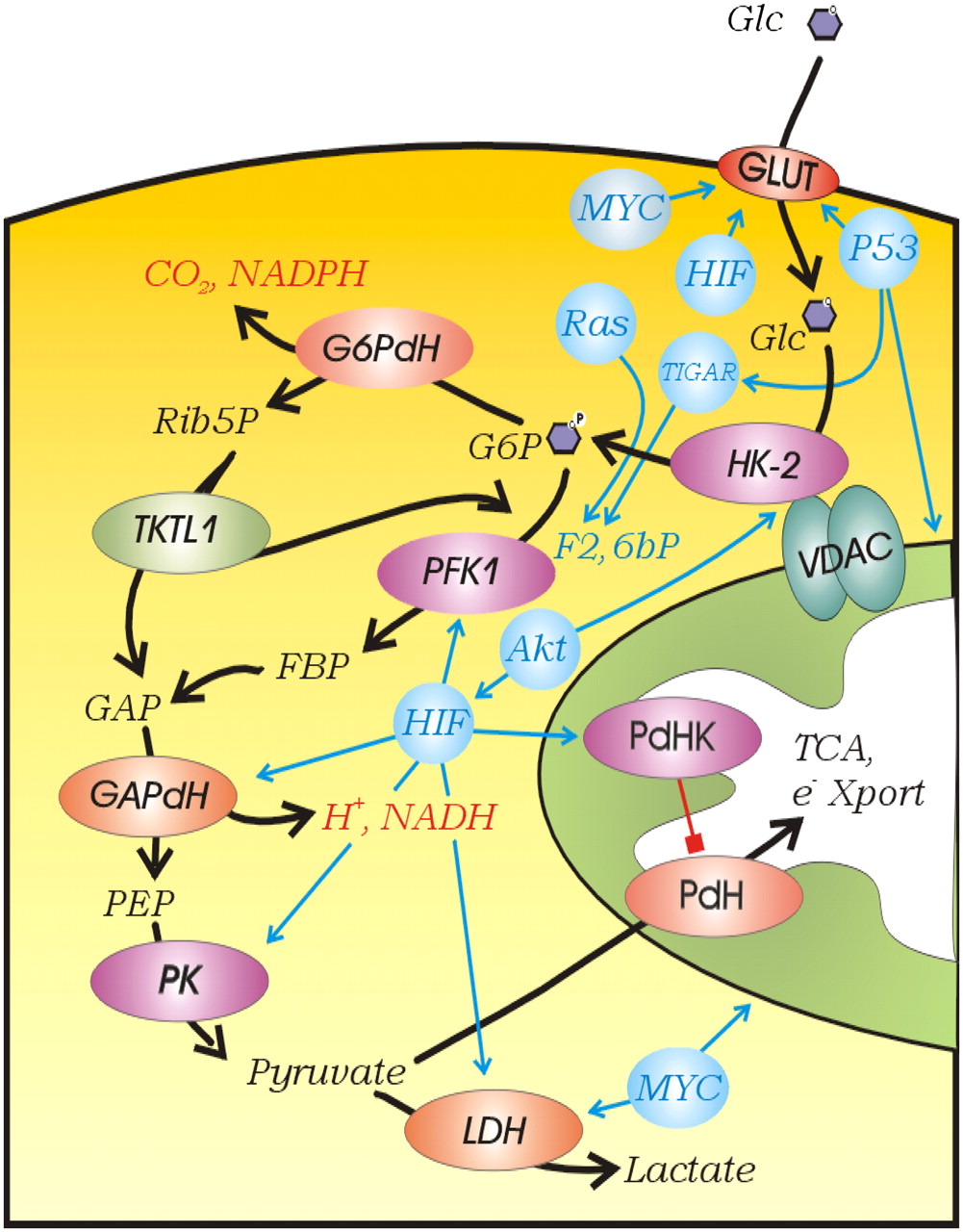
The aforementioned observations are consistent with the unsurprising conclusion that uptake and use of glucose can be controlled by a multitude of factors, all of which have been observed to participate in the increased aerobic glycolysis of cancers (Fig. 11). This stimulates the question: Is increased aerobic glucose consumption merely an epiphenomenon of oncogenic transformation or is it necessary for cancer progression? The sheer prevalence of increased 18F-FDG uptake in metastatic tumors would suggest that it is necessary. Evolutionary models are based on the assumption that any common phenotype must impart a selective advantage. Darwinian evolution is based on selection on phenotypes, not genotypes. A corollary to this is the concept of functional equivalence, in which multiple (epi)genetic mechanisms can give rise to similar phenotypes (4), and this principle can explain the well-documented genetic heterogeneity of cancers (177–180). Although it is tempting to coalesce all of the regulatory mechanisms of glycolysis into a single unified mechanism, such an endeavor may prove futile, if functional equivalence is involved. The heterogeneity of regulatory mechanisms itself conveys the message that the glycolytic phenotype itself is selected, and the mechanism by which it is induced is evolutionarily irrelevant. Like the blind men and the elephant (Fig. 12), all of the aforementioned descriptions of glycolytic control are likely correct yet incomplete, and the only common element is glycolysis itself.

Regulation of glycolysis. Kinase reactions are shown in purple, dehydrogenases are shown in red, and regulatory elements are shown in blue. Abbreviations are as in text. e− Xport = electron transport.

Blind Monks Examining an Elephant, by Itcho Hanabusa (1652–1724). (Ukiyo-e print illustration from digital catalog of U.S. Library of Congress [3g08725u.tif]).
We have previously presented evidence that the glycolytic phenotype is selected in early in situ cancers because they are faced with intermittent hypoxia (181). However, this does not explain why the glycolytic phenotype is maintained after these cancer cells have access to blood vessels and a normoxic environment during the process of metastasis (182–184). If cancers progress via Darwinian somatic evolution, it can be argued that, if it were no longer needed, the glycolytic phenotype would be selected against because it is inefficient and produces toxic by-products. However, it is also possible that these “by-products” are not toxic at all but provide important components for survival and metastasis.
CONSEQUENCES OF INCREASED GLUCOSE UPTAKE
Consumption and metabolism of glucose can produce several by-products, including ATP, reduced nicotinamide adenine dinucleotide (NADH) phosphate (NADPH), and acids. The relevant pathways are shown in Figure 11. Any or all of these may provide important components that could result in positive Darwinian selection and result in increased cancer progression and metastasis: production of ATP, production of acids (H+), production of NADPH (glutathione), and production of lactate.
Bioenergetics
The dominant perception of energy metabolism in cancer has been that ATP production is fueled by oxidative metabolism of glutamine and glucose and by the anaerobic metabolism of glucose through the Embden–Meyerhof pathway (EMP) of glycolysis, and that this ATP is needed to support a high level of oncogene-induced proliferation. However, the available data are equivocal. In a careful review of the available data from careful mass balance studies, it was concluded that, on average, cancers derive 17% of their ATP turnover from EMP glycolysis, whereas normal tissues derive around 20% (185). The total ATP turnover in MCF-7 breast cancer cells is 80% oxidative and 20% glycolytic and the entire contribution from glucose (glycolysis and oxidation) to total ATP turnover was about 23%, or twice that of glutamine (186). Uterine, lung, breast, liver, and skin cancers all derive a significant amount of energy from oxidative phosphorylation (187). A review of the mass balance of the end products of metabolism (lactate and CO2) showed that acid loads were approximately equally split between oxidative and nonoxidative products (188). The mole stoichiometries are 1 ATP per lactate and 5–6 ATP per CO2, suggesting that approximately 15% of the ATP turnover was derived from glycolysis. Thus, although the role of glycolysis in providing ATP is often invoked as a rationale for its increase in cancers, such a claim is not substantiated by available evidence. Hence, if increased ATP production is not the reason for selection of a glycolytic phenotype, other sequelae of glucose metabolism might be.
PPP
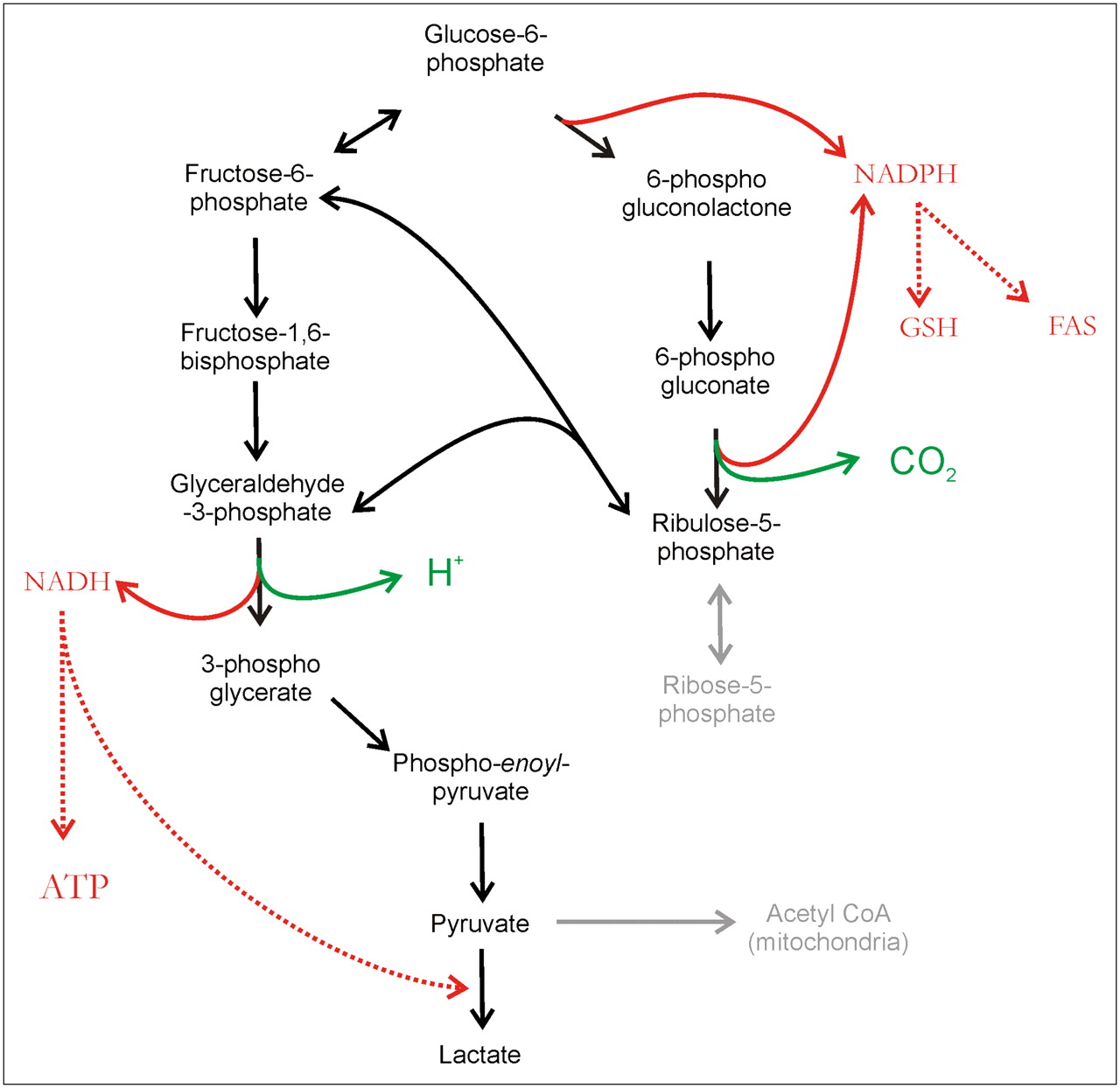
It has been alternatively proposed that some cancers, notably colorectal cancers, upregulate the PPP to convert glucose to pyruvate (189–194). Figure 13 illustrates the relationship between the PPP and EMP, which differ primarily in their redox products. The EMP produces NADH, whereas the PPP produces NADPH as well as ribose-5-phosphate (195). The production of ribose-5P for nucleic acid biosynthesis had been proposed as a rationale for the increase in the PPP by Warburg. NADPH and NADH are equivalent in terms of their ability to maintain an oxidizing cytoplasm in the face of anaerobiosis (196). NADPH can be used for reducing glutathione for antioxidant defenses and there is evidence that PPP activity makes cells more resistant to reactive oxygen species–induced apoptosis (197). NADPH is also a necessary cofactor for fatty acid synthesis and, indeed, increased rates of fatty acid synthesis have been observed in many cancers (198,199). NADH can be used to reduce pyruvate to lactate or to make ATP via mitochondrial oxidative phosphorylation (200). Entry into the upper (oxidative) part of the PPP is regulated by glucose-6-phosphate dehydrogenase (201). The lower, nonoxidative part of the PPP is controlled by transketolase enzyme reactions. A mutated transketolase transcript (TKTL1) has been detected in human malignancies and correlated to poor patient outcome in CRC (202). Thus, the PPP-derived metabolites include antioxidants, nucleic acid precursors, and substrates for fatty acid synthesis, any one of which could be selected to enhance cancer cell survival.

Pathways of glycolysis. Glucose-6-phosphate can be metabolized by Embden–Meyerhoff pathway (left) or PPP (right). In PPP, conversion to ribulose-5-phosphate (oxidative phase) is accompanied by elaboration of CO2 and generation of 2 NADPH molecules, which are used to reduce glutathione (GSH) or fatty acid synthase (FAS). Ribulose can reenter glycolysis via transketolase conversion to fructose-6-phosphate and GAP. CoA = coenzyme A.
Acidosis
Whether glucose is metabolized aerobically or anaerobically, the secreted end products are acids: CO2 and lactate, respectively. CO2 is hydrated to (HCO3−+H+) in a reaction catalyzed by CA. An extracellular CA isozyme, CA-IX, is upregulated by HIF and promotes this hydration to contribute to extracellular acidification (203–205). Although the end-product lactate is an acid, the H+ is actually produced at the level of GAPdH during glycolysis. Hence, production of lactate and H+ is not tightly coupled, an effect that has been observed with in vivo pH and metabolite mapping (206). Lactate is efficiently removed from cells via a monocarboxylate transporter, MCT-4, which can also be HIF-inducible (207). The hydrogen ions are removed by ion transporters, notably the sodium-hydrogen exchanger, NHE-1 (208), although other transporters, such as V-ATPase and anion exchangers, may also be used to export H+ (209,210). Increased acid production and export, combined with poor perfusion, gives rise to an extracellular pH (pHe) of tumors that is undeniably acidic (211–214). Notably, the production of acid is not dependent on the EMP, as cells deficient in glucose-6-phosphate isomerase (which converts glucose-6-phosphate to fructose-6-phosphate) continue to produce acid, primarily glutamine-derived CO2, at a high rate (215–217).
Acidosis has long been viewed as a toxic consequence of increased glycolysis, but recently it has been proposed that it is selectively more toxic to normal parenchymal tissues compared with invading cancers. In many tumor types, acute or chronic treatment with low pH stimulates in vitro invasion and in vivo metastases. This has been shown most dramatically for melanoma cancer wherein lowering culture pH to 6.7 resulted in a significant increase in the incidence of in vivo metastases, after tail vein injection of treated cells (42). This acid treatment leads to increases in interleukin-8 (IL-8), vascular endothelial growth factor (VEGF), and matrix metalloproteinases 2 and 9, and inhibiting these factors reduced both in vitro invasion and in vivo metastasis (218). In breast cancer, acid treatment has been shown to induce release of matrix-degrading proteases, for example, cathepsins B and L (39,219).
Interest in measuring tumor pH has a long history, which was recently reviewed (220). Early measurements used microelectrode measurements in both human and experimental cancers, which showed that the extracellular pH (pHe) was acidic (211). Subsequently, radioactive weak acid tracers, such as 14CO2 and 14C-dimethyloxazolidinone (DMO), showed that the intracellular pH (pHi) of tumors was alkaline (221,222). 31P MRS studies agreed with tracer studies in showing that tumor pHi was neutral to alkaline (223,224). These observations were confirmed by the simultaneous measurement of pHi and pHe with an exogenous 31P-labeled pHe indicator, 3-aminopropylphosphonate (225). Subsequent measurements have shown unequivocally that the pHe of tumor xenografts is acidic, whereas the pHi is neutral to alkaline (226). Although 31P MRS can be spatially localized, the low gyromagnetic ratio limits voxel sizes to approximately 8 × 8 × 8 mm. Higher resolution can be obtained with other nuclei, such as 1H and 19F (227–230), which have been used to measure localized tumor pH using spectroscopic imaging (MRSI) with spatial resolution approaching 1 × 1 × 1 mm (228,231,232). Although these provided unparalleled resolution, drawbacks to these MRSI approaches include long acquisition times even at a high field (∼40 min at 4.7 T). In the early 2000s, pH-dependent T1 relaxometry was introduced, with the promise that it could generate pH maps with high spatiotemporal resolution. Several pH-dependent contrast reagents (CR) have been synthesized (233,234). An issue with these measurements is the need for simultaneous pixel-by-pixel correction for concentration (235). Nonetheless, methods have been developed to image pHe with imaging resolution using MRI relaxometry (236,237). An alternative method uses the rate of acid-catalyzed exchange of exogenous or endogenous amide hydrogens with bulk water to measure pH (238–240). This approach, chemical exchange saturation transfer (CEST), is relatively insensitive, requiring greater than 50 mM exchangeable amides. Although it has been made more sensitive using pH-sensitive paramagnetic lanthanide chelates (ParaCEST) (241–243), this approach still has a lack of sensitivity, requiring greater than a 10 mM contrast agent and, like pH-dependent relaxometry, also requiring knowledge of the CA concentration.
Lactate
It is also possible that lactate itself is a signal for increased survival, invasion, and metastasis. Lactate levels can be measured in human tumors by 1H magnetic resonance spectroscopy (MRS) (244). Such measures have shown that lactate levels are strong indicators of poor prognosis in brain, breast, lung, and liver cancers (245–248). Notably, lactate levels did not correlate with histopathologic stage of oligodendrogliomas, suggesting that this may not be a universal observation (249). As described previously, lactate can directly lead to the stabilization of HIF-1α (250). Lactate can also induce an increase in the expression of hyaluronan and hyaluronidase (CD44) in stromal fibroblasts and tumor cells, and these are important components of increased invasion and metastasis (251).
CONCLUSION
Imag(in)ing is the interplay between mathematically based teleologic models and empiric data. Empiric 18F-FDG PET data have emphasized the sheer preponderance of elevated glucose trapping in cancers, a phenomenon first described half a century ago. Teleology modeling has asked why this must be so and has come to the conclusion that elevated glucose consumption must be a necessary phenotype for the formation of metastatic cancers and therefore must be considered a hallmark of cancer. This is supported not only by the commonality of the observation but also by the multitude of molecular mechanisms that can give rise to this phenotype, a principle known as functional equivalence. In then asking “Why do cancers consume glucose?” 3 metabolic sequelae were considered: bioenergetics, PPP formation of NADPH and ribose, and production of acid. Although bioenergetic production of ATP is often cited as an important consequence of elevated glucose consumption, this is not supported by available empiric evidence. Although PPP is elevated in some cancers, this is not universally observed. Production of acid is an inevitable consequence of increased glucose consumption. Although mechanistic descriptions are not fully developed, increased acid production has been shown to correlate with and cause an increased capacity of cancer cells to invade host tissues. Thus, it is proposed that the phenotype of elevated glucose consumption in cancers is selected through somatic evolution because it leads to increased acid production.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- 1.↵
- 2.↵
- 3.
- 4.↵
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.
- 29.
- 30.↵
- 31.
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.
- 39.↵
- 40.
- 41.
- 42.↵
- 43.↵
- 44.
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 92a.↵
- 92b.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.
- 106.
- 107.
- 108.
- 109.
- 110.
- 111.
- 112.
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.
- 167.↵
- 168.↵
- 169.↵
- 170.↵
- 171.↵
- 172.
- 173.↵
- 174.↵
- 175.↵
- 176.↵
- 177.↵
- 178.
- 179.
- 180.↵
- 181.↵
- 182.↵
- 183.
- 184.↵
- 185.↵
- 186.↵
- 187.↵
- 188.↵
- 189.↵
- 190.
- 191.
- 192.
- 193.
- 194.↵
- 195.↵
- 196.↵
- 197.↵
- 198.↵
- 199.↵
- 200.↵
- 201.↵
- 202.↵
- 203.↵
- 204.
- 205.↵
- 206.↵
- 207.↵
- 208.↵
- 209.↵
- 210.↵
- 211.↵
- 212.
- 213.
- 214.↵
- 215.↵
- 216.
- 217.↵
- 218.↵
- 219.↵
- 220.↵
- 221.↵
- 222.↵
- 223.↵
- 224.↵
- 225.↵
- 226.↵
- 227.↵
- 228.↵
- 229.
- 230.↵
- 231.↵
- 232.↵
- 233.↵
- 234.↵
- 235.↵
- 236.↵
- 237.↵
- 238.↵
- 239.
- 240.↵
- 241.↵
- 242.
- 243.↵
- 244.↵
- 245.↵
- 246.
- 247.
- 248.↵
- 249.↵
- 250.↵
- 251.↵
- 252.↵
- 253.↵
- 254.↵
- 255.↵
- 256.↵
- 257.↵
- 258.↵
- 259.↵
- 260.↵
- 261.↵
- 262.↵
- 263.↵
- 264.↵
- 265.↵
- 266.↵
- 267.↵
- 268.↵
- 269.↵
- 270.↵
- 271.↵
- 272.↵
- Received for publication January 30, 2008.
- Accepted for publication March 19, 2008.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- The harsh microenvironment in early breast cancer selects for a Warburg phenotype
- Water as a reactant in the differential expression of proteins in cancer
- The Harsh Microenvironment in Early Breast Cancer Selects for a Warburg Phenotype
- Targeting tumor phenotypic plasticity and metabolic remodeling in adaptive cross-drug tolerance
- Collagen Production and Niche Engineering: A Novel Strategy for Cancer Cells to Survive Acidosis and Evolve
- The Utility of PET/CT in the Planning of External Radiation Therapy for Prostate Cancer
- RhoC GTPase Is a Potent Regulator of Glutamine Metabolism and N-Acetylaspartate Production in Inflammatory Breast Cancer Cells
- Expanding the Scope of Fluorine Tags for PET Imaging
- High glucose and insulin differentially modulates proliferation in MCF-7 and MDA-MB-231 cells
- Quantitative Proteomic Analysis in Metastatic Renal Cell Carcinoma Reveals a Unique Set of Proteins with Potential Prognostic Significance
- Tumor Aggressiveness and Patient Outcome in Cancer of the Pancreas Assessed by Dynamic 18F-FDG PET/CT
- Targeting Hypoxia, HIF-1, and Tumor Glucose Metabolism to Improve Radiotherapy Efficacy
- Chronic Autophagy Is a Cellular Adaptation to Tumor Acidic pH Microenvironments
- Relationship between 18F-Fluorodeoxyglucose Accumulation and KRAS/BRAF Mutations in Colorectal Cancer
- Preparation and Characterization of L-[5-11C]-Glutamine for Metabolic Imaging of Tumors
- PET Imaging of Glutaminolysis in Tumors by 18F-(2S,4R)4-Fluoroglutamine
- Multimodal Imaging with 18F-FDG PET and Cerenkov Luminescence Imaging After MLN4924 Treatment in a Human Lymphoma Xenograft Model
- Overcoming Trastuzumab Resistance in Breast Cancer by Targeting Dysregulated Glucose Metabolism