


Visual Abstract
Abstract
Progressive supranuclear palsy (PSP) is a neurodegenerative disorder characterized by neuroglial tau pathology. A new staging system for PSP pathology postmortem has been described and validated. We used a data-driven approach to test whether postmortem pathologic staging in PSP can be reproduced in vivo with 18F-flortaucipir PET. Methods: Forty-two patients with probable PSP and 39 controls underwent 18F-flortaucipir PET. Conditional inference tree analyses on regional binding potential values identified absent/present pathology thresholds to define in vivo staging. Following the postmortem staging approach for PSP pathology, we evaluated the combinations of absent/present pathology (or abnormal/normal PET signal) across all regions to assign each participant to in vivo stages. ANOVA was applied to analyze differences among means of disease severity between stages. In vivo staging was compared with postmortem staging in 9 patients who also had postmortem confirmation of the diagnosis and stage. Results: Stage assignment was estimable in 41 patients: 10, 26, and 5 patients were classified in stage I/II, stage III/IV, and stage V/VI, respectively, whereas 1 patient was not classifiable. Explorative substaging identified 2 patients in stage I, 8 in stage II, 9 in stage III, 17 in stage IV, and 5 in stage V. However, the nominal 18F-flortaucipir--derived stage was not associated with clinical severity and was not indicative of pathology staging postmortem. Conclusion: 18F-flortaucipir PET in vivo does not correspond to neuropathologic staging in PSP. This analytic approach, seeking to mirror in vivo neuropathology staging with PET-to-autopsy correlational analyses, might enable in vivo staging with next-generation tau PET tracers; however, further evidence and comparisons with postmortem data are needed.
Progressive supranuclear palsy (PSP) is a severe neurodegenerative disorder resulting in diverse clinical phenotypes with restricted eye movements, an akinetic–rigid syndrome, falls, and cognitive and behavioral deficits (1). The neuropathology of PSP is characterized by intracellular aggregates of 4-repeat tau in neurons and glia (2–5); these aggregates are distributed in a progressive sequence starting in the substantia nigra, globus pallidus and subthalamic nucleus, then pons, striatum and the precentral gyrus in the cerebral cortex, before reaching the cerebellum and frontal cortex (6). Later, the neuroglial pathology might extend to the occipital cortex (7).
A new neuropathologic staging system for PSP tau pathology postmortem was recently introduced and independently validated (7,8). This method confirms an association between pathology stage and clinical severity before death. To stage disease severity antemortem requires a different methodology. For the tauopathy of Alzheimer disease, for example, 18F-flortaucipir PET can reproduce staging in vivo (9–16).
Here, we test whether regional binding of the radioligand 18F-flortaucipir (also known as 18F-AV-1451), quantified using nondisplaceable binding potential, can be used to replicate the staging of PSP pathology in vivo. We validated the staging in 2 ways: correlation with clinical severity at the time of 18F-flortaucipir PET and neuropathologic staging of a subset of participants postmortem.
MATERIALS AND METHODS
Participants
We recruited 42 patients with a clinical diagnosis of probable PSP using Movement Disorder Society PSP 2017 criteria (1) (19 women and 23 men; mean age, 70.3 y [SD, 7.0 y; range, 50–84 y]; 35 with PSP Richardson syndrome and 7 with other phenotypes) and included data from 39 cognitively healthy controls (16 women and 23 men; mean age, 65.8 y [SD, 8.2 y; range, 48–84 y]; mean revised Addenbrooke’s Cognitive Examination score, 96.2 [SD, 2.9; range, 89–100]). Disease severity was measured using the PSP rating scale (PSPRS) (mean, 36.6 [SD, 14.2; range, 10–74]). To date, 9 of the 42 patients donated their brain to the Cambridge Brain Bank, after a mean of 2.45 (SD, 0.98) years from PET. All of these patients had postmortem pathologic confirmation of PSP pathology.
All participants underwent dynamic PET imaging for 90 min after 18F-flortaucipir injection (GE Signa PET/MRI for 22 patients; GE Discovery 690 PET/CT for 13 patients; GE Advance PET for 7 patients; GE Signa PET/MRI for 24 controls; GE Discovery 690 PET/CT for 7 controls; GE Advance PET for 8 controls) (all scanners were from GE Healthcare). The sensitivity advantage of the PET/MRI scanner was used to reduce the target injection activity by 50% compared with that used in the PET and PET/CT scans, leading to a comparable signal-to-noise ratio in the acquired data across the scanners. Full details of the imaging protocols were published elsewhere (17,18). Seven of the 9 patients who donated their brains underwent 18F-flortaucipir imaging with GE Discovery 690 PET/CT; the other 2 were scanned with GE Advance PET.
Relevant approvals were granted by the Cambridge Research Ethics Committee (references: 13/EE/0104, 16/EE/0529, and 18/EE/0059), the East of England–Essex Research Ethics Committee (16/EE/0445), and the Administration of Radioactive Substances Advisory Committee. All participants provided written informed consent in accordance with the Declaration of Helsinki.
Determination of Regional 18F-Flortaucipir Binding
18F-flortaucipir nondisplaceable binding potential was calculated in regions of interest corresponding closely to those used for postmortem staging of PSP by Kovacs et al. (7): globus pallidus, cerebellum (white matter and dentate nucleus), middle frontal gyrus, and occipital lobe (lingual gyrus and cuneus) (Supplemental Fig. 1A) (supplemental materials are available at http://jnm.snmjournals.org). The striatum and subthalamic nucleus were excluded because of 18F-flortaucipir off-target binding or challenges in defining the PET signal. Regional values were quantified using a modified version of the n30r83 Hammersmith atlas (http://brain-development.org/brain-atlases/adult-brain-atlases/adult-brain-maximum-probability-map-hammers-mith-atlas-n30r83-in-mni-space/), which includes parcellation of the brain stem and cerebellum, and a basis function implementation of the simplified reference tissue model (19), with cerebellar cortex gray matter as the reference region. Before kinetic modeling, regional PET data were corrected for partial-volume effects from cerebrospinal fluid by dividing the regional PET value by the mean regional gray matter plus white matter fraction determined from Statistical Parametric Mapping (SPM12, https://www.fil.ion.ucl.ac.uk/spm/) segmentation. Left and right regional nondisplaceable binding potential values were averaged bilaterally. Using regional mean and SD values from controls, we calculated w-scores (z-scores adjusted for the effect of covariates, Supplemental Fig. 1B), accounting for phenotypic and systematic differences, such as age and scanner type (PET/MRI vs. non-PET/MRI); see Malpetti et al. (17) for a discussion on harmonization of PET and PET/CT data.
In Vivo Staging Based on 18F-Flortaucipir Binding
Data-Driven Severity Thresholds
To quantify pathology severity in each region, we used a conditional inference tree analysis to define in a data-driven manner region-specific 18F-flortaucipir binding thresholds of w-scores, entering both patients and controls in the model. This method is similar to that used previously for imaging-based pseudo-Braak staging of Alzheimer disease (9). Specifically, region-specific thresholds were identified using nonparametric binary recursive partitioning with the function “ctree” in R (v. 4.0.0, R Core Team - R Foundation for Statistical Computing) and running this tree analysis on w-scores for each region separately. Using these region-specific thresholds, we assigned binary severity scores to individual regional w-scores (w-score ≤ regional threshold: 0 or absent; w-score > regional threshold: 1 or present).
In Vivo Staging
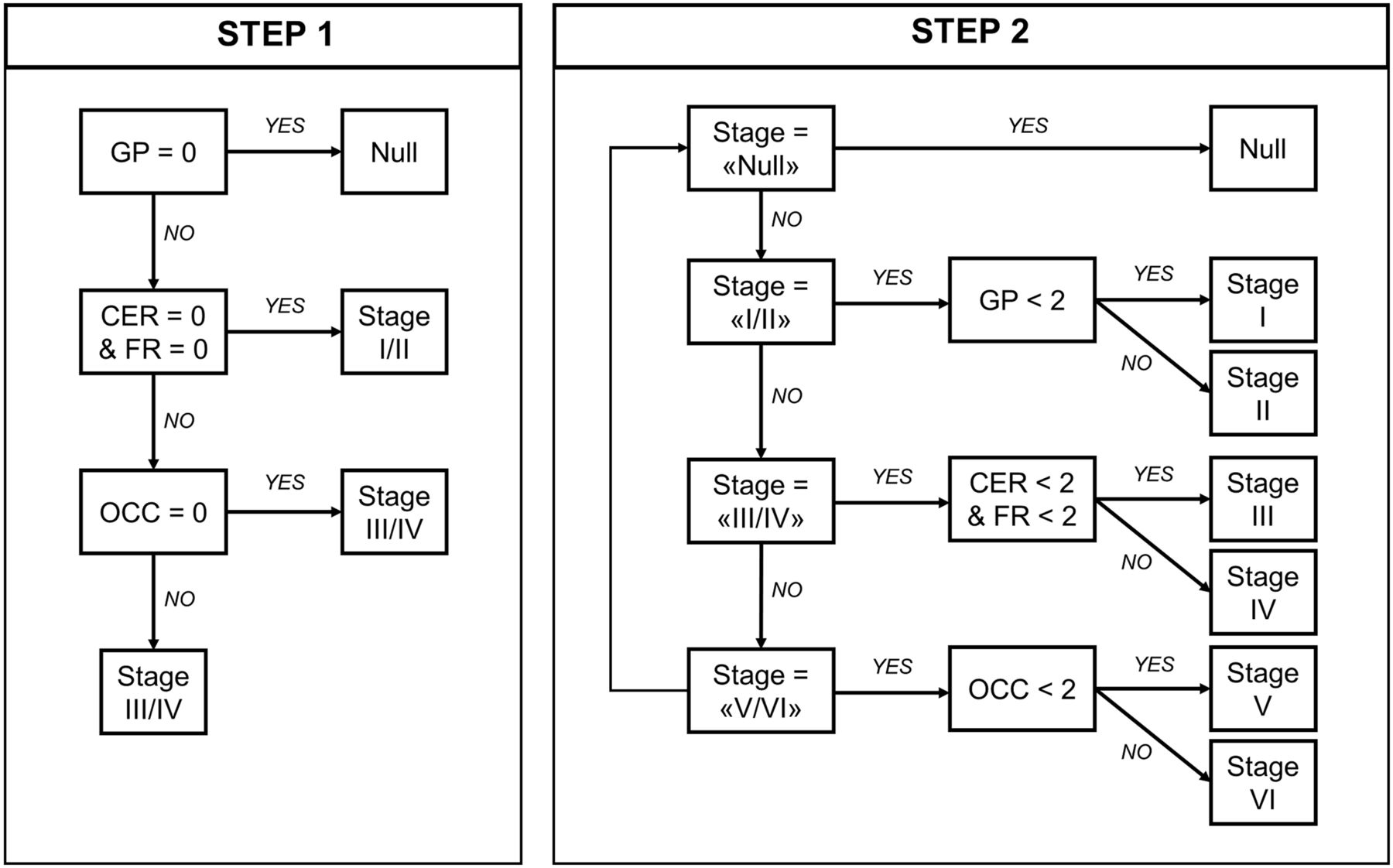
First, using the staging system described by Kovacs et al. (7), which is based on cumulative and progressive pathology severity, we evaluated the combination of absent/present values across all 4 regions to assign each participant to stages I/II, III/IV, or V/VI (step 1 on Fig. 1). Second—in an explorative analysis—within each stage defined in the previous step, a 3-point pathology severity system was applied to each region (w-score ≤ regional threshold: absent, coded as 0; w-score > regional threshold: mild/moderate pathology, coded as 1; w-score > 2 times the threshold: moderate/severe pathology, coded as 2), and 1 of the 6 stages was assigned accordingly (stages I–VI; step 2 on Fig. 1). We repeated these staging analyses with a second analytic approach, using a preselected number of SD values from region-specific nondisplaceable binding potential control means to define pathology severity (Supplemental Fig. 2). ANOVA was applied to analyze differences among means of disease severity (PSPRS) between stages.

In vivo staging rules. Step 1: in vivo stages are defined with cumulative evidence of absence (region = 0) or presence (region = 1) of pathology in each of 5 regions considered, as defined by region-specific thresholds (regional w-score > threshold = 1; regional w-score ≤ threshold = 0). Step 2: in vivo substages are defined within each step 1 stage, considering 3-level pathology severity scale (0 = none; 1 = mild/moderate pathology; 2 = moderate/severe pathology). Regions: cerebellum (CER; white matter and dentate nucleus), middle frontal gyrus (FR), globus pallidus (GP), and occipital lobe (OCC; lingual gyrus and cuneus).

In vivo staging based on data-driven thresholds. (A) Severity scores are reported for each group of regions considered to define in vivo stages (step 1: 0 = absent; 1 = present) and substages (step 2: 0 = none; 1 = mild/moderate pathology; 2 = moderate/severe pathology). (B and C) Box plots of PSPRS scores by stages defined with step 1 (B) and step 2 (C). CER = cerebellum (white matter and dentate nucleus); FR = middle frontal gyrus; GP = globus pallidus; OCC = occipital lobe (lingual gyrus and cuneus); PSP-CBS = PSP-corticobasal syndrome; PSP-F = PSP-frontal; PSP-OM = PSP-oculomotor; PSP-PGF = PSP-progressive gait freezing; PSP-RS = PSP-Richardson syndrome.
Postmortem Diagnosis and Staging Based on Immunohistochemistry
Tissue blocks of the left hemisphere were sampled according to National Institute of Neurological Disorders and Stroke standard guidance for neurodegenerative diseases from the brain stem, subcortical, and cortical areas. These were evaluated for the initial pathologic diagnosis of PSP (hyperphosphorylated tau; AT8, MN1020; Thermo Scientific, possible concomitant pathologies of amyloid β (clone 6F/3D, M0872; Dako), α-synuclein (SA3400; Enzo Life Sciences), and TDP-43 (TIP-PTD-P02; Cosmo Bio Co. Ltd.); and vascular pathology. Using the previously described staging scheme (7,8), we evaluated neuronal and oligodendroglial tau pathology in the globus pallidus, subthalamic nucleus, and cerebellar white matter and dentate nucleus and astrocytic tau pathology in the striatum, middle frontal gyrus, and occipital cortex. The regional cytopathologies were rated on a 4-level system (none, mild, moderate, and severe) using the guidelines proposed by Briggs et al. (8). In vivo staging results with both data-driven and SD approaches were compared with postmortem staging in the 9 patients who donated their brain.
RESULTS
The conditional inference tree analysis identified region-specific pathologic thresholds of 18F-flortaucipir binding for the globus pallidus (w-score, >0.795), cerebellar white matter (w-score, >0.783) and dentate nucleus (w-score, >0.845), and middle frontal gyrus (w-score, >1.416). For the occipital lobe, the analysis did not identify the threshold, so we used 1.645 as the w-score critical value (P = 0.05). A simple set of decision rules (Fig. 1) enabled plausible Kovacs stages to be estimated in 41 patients (Fig. 2A): 10 patients were classified in stage I/II because of increased 18F-flortaucipir binding limited to the globus pallidus, 26 were classified in stage III/IV because of additional increased 18F-flortaucipir binding in the frontal or cerebellar regions, and 5 were classified in stage V/VI because of additional increased 18F-flortaucipir binding in the occipital lobe; 1 patient could not be classified because no increased binding was found in the globus pallidus. The explorative substaging (6 stages) identified 2 patients in stage I (mild/moderate pathology in the globus pallidus), 8 in stage II (moderate/severe pathology in the globus pallidus), 9 in stage III (mild/moderate pathology in the frontal lobe or cerebellum), 17 in stage IV (moderate/severe pathology in the frontal lobe or cerebellum), and 5 in stage V (mild/moderate pathology in the occipital lobe). When the same approach was applied to controls, 31, 5, 1, and 2 participants were classified in no stage, stage I, stage II, and stage III, respectively. Four patients (Fig. 2A, patients 6, 35, 36, and 39) showed an atypical severity pattern that was discordant with the description of Kovacs et al. (7).
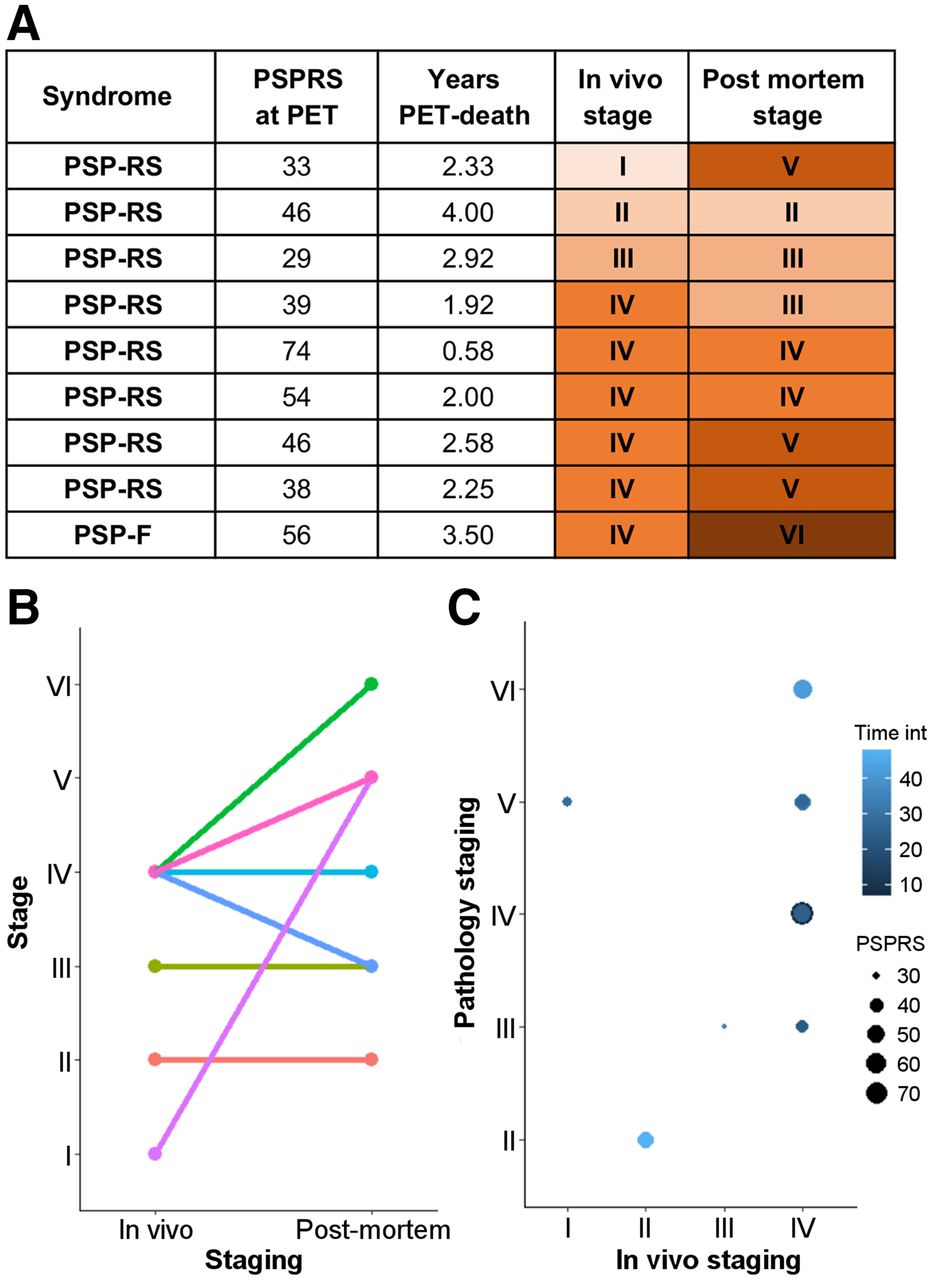
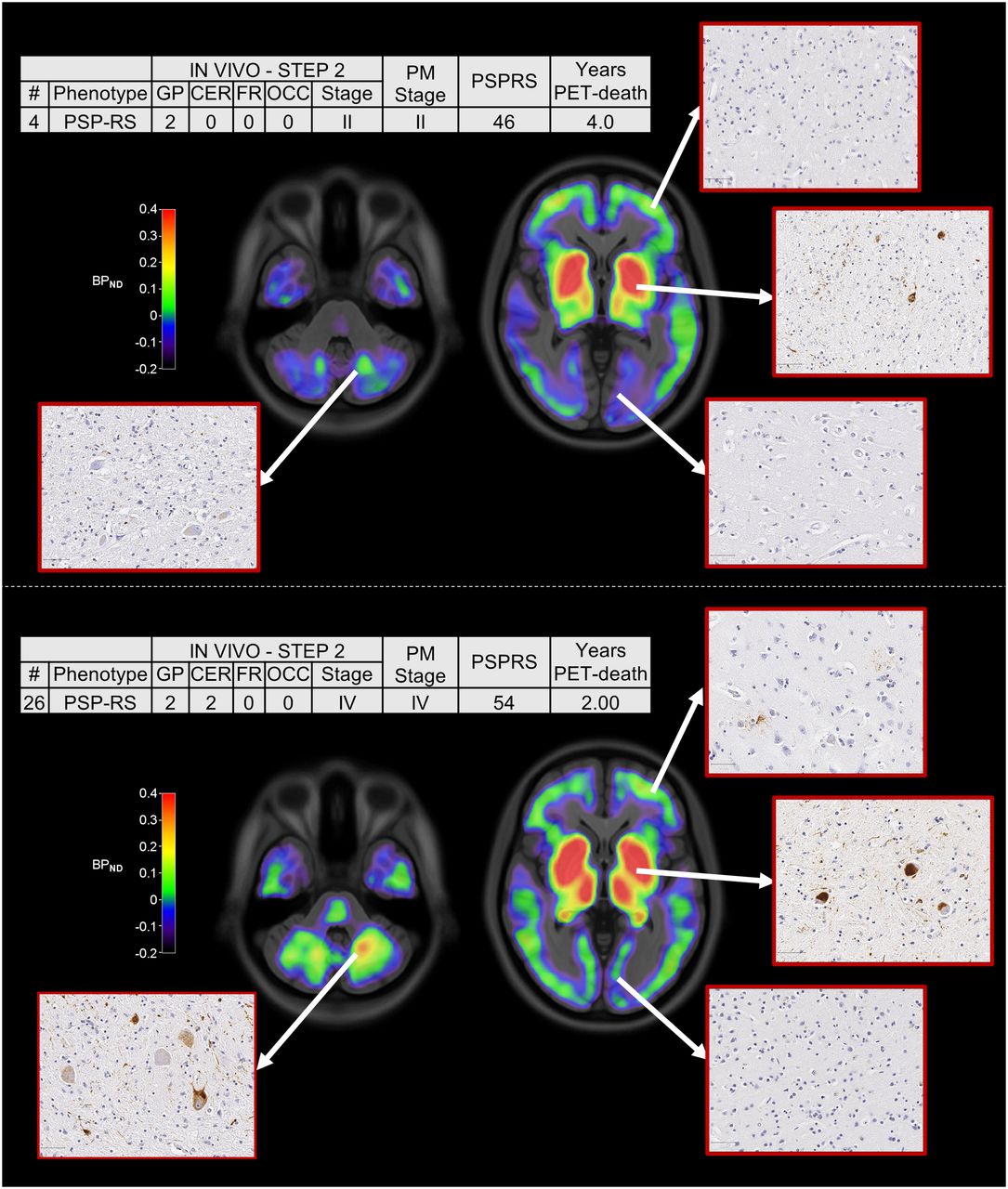
Across all patients, the estimated in vivo stages did not relate to clinical severity (P > 0.05 in an ANOVA) (Figs. 2B and 2C). In 8 of the 9 patients who donated their brains, pathology stage as determined by in vivo 18F-flortaucipir PET was less than or equal to that determined postmortem (Fig. 3). In vivo staging and postmortem staging were not significantly correlated (Spearman r, 0.168; P = 0.67). Correlation analyses were also used to test the residuals of each staging variable (in vivo and postmortem staging) after regressing out clinical severity (PSPRS scores) and the interval from PET to time of death. The correlation was not statistically significant (Spearman r, 0.150; P = 0.70). Figure 4 shows examples of 18F-flortaucipir nondisplaceable binding potential maps and corresponding postmortem staining data for patients who were classified in stage II (patient 4) and stage IV (patient 26) by both in vivo staging and postmortem staging.

Comparison of in vivo and postmortem stages for 9 patients who underwent 18F-flortaucipir PET and pathology autopsy. (A) Clinical and staging details. (B) Single-subject (lines) comparisons of in vivo and postmortem staging. (C) Graphical representation of the effect of interval from PET to time of death (Time int) and clinical severity on association between in vivo staging and postmortem staging. PSP-F = PSP-frontal; PSP-RS = PSP-Richardson syndrome.
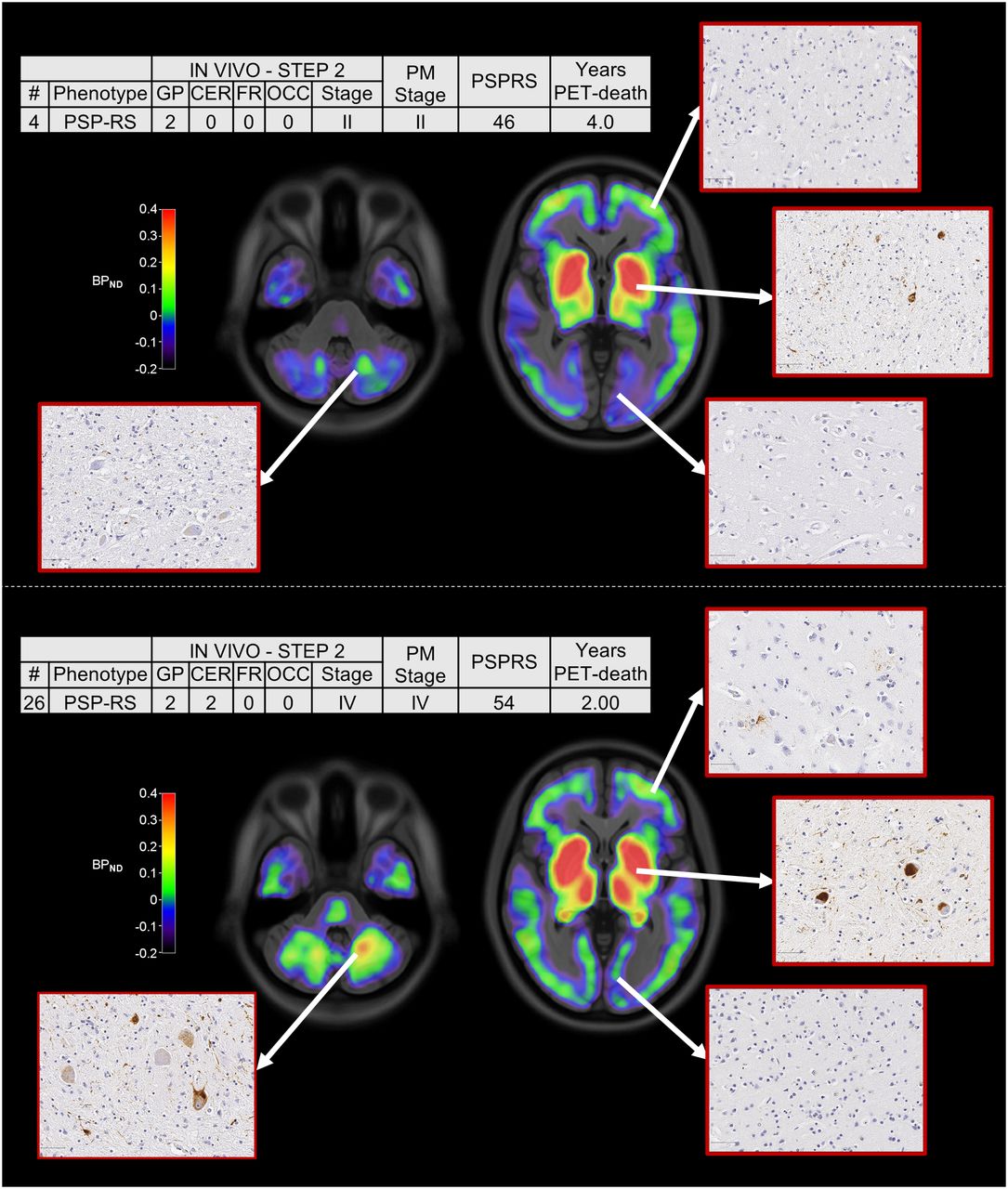
18F-flortaucipir nondisplaceable binding potential (BPND) maps, postmortem staining, and related clinical details for 2 patients classified in stage II (top) and stage IV (bottom) with both in vivo staging and postmortem staging. Spatially normalized BPND maps are shown in radiologic format overlaid on ICBM MNI152 2009a T1 MRI template (https://www.bic.mni.mcgill.ca/ServicesAtlases/ICBM152NLin2009). CER = cerebellum; FR = middle frontal gyrus; GP = globus pallidus; OCC = occipital lobe; PM stage = postmortem stage; PSP-RS = PSP-Richardson syndrome.
DISCUSSION
The principal finding of the present study was that 18F-flortaucipir PET does not provide accurate in vivo staging corresponding to neuropathologic staging for PSP. The nominal stage derived from 18F-flortaucipir PET did not correlate with disease severity or relate to staging postmortem.
As a result of the data-driven in vivo staging system, compared with controls, we observed higher 18F-flortaucipir binding in the globus pallidus in all but 1 patient, with a few patients showing increased 18F-flortaucipir binding in the occipital cortex (Fig. 2A). This regional distribution of 18F-flortaucipir binding was in line with the pathologic description of PSP and with what was previously described for 18F-flortaucipir in PSP (13,17,18,20). Whereas the 18F-flortaucipir binding patterns allowed us to nominally apply PSP pathology staging in vivo, the in vivo staging was not systematically predictive of pathology staging postmortem. As expected because of the time interval between the PET scan and autopsy, in 8 of 9 cases with autopsy, the individual in vivo staging was less than or equal to the postmortem staging. However, 4 patients who were labeled as stage IV in vivo were then classified in 4 different stages postmortem (Fig. 3). Neither clinical severity nor the time interval between the PET scan and death was useful for predicting the individual postmortem stage from in vivo staging.
The number of patients with a positive signal for 18F-flortaucipir in the cerebellum (n = 29) exceeded the number of patients with a positive result for frontal 18F-flortaucipir binding (n = 10). Although this finding might reflect earlier involvement of the cerebellum in our cohort, regional differences in the density of tau aggregates and predominant cytopathologies could contribute to regional differences in tracer retention (11,13,21)—for example, neuronal and oligodendroglial tau predominates in the cerebellum, whereas astrocytic tau predominates in cortical regions.
Off-target binding for 18F-flortaucipir is well characterized, but this problem alone would still leave open the possibility of quantifying tau pathology in areas without significant monoamine oxidase levels or neuromelanin, such as the cerebellum and medial frontal cerebral cortex (22). However, recent PET-to-autopsy correlational studies suggested that 18F-flortaucipir PET does not reliably correspond to postmortem tau pathology in non-Alzheimer tauopathies (13,23). This finding suggests that 18F-flortaucipir lacks sensitivity in non-Alzheimer tau pathology. This characteristic might explain the underperformance of this tracer in defining an in vivo classification that systematically aligns with postmortem staging. Next-generation tau tracers might prove to be more useful for tracking in vivo PSP pathology progression because of a combination of good affinity for 4-repeat tau and lower off-target binding to monoamine oxidases (i.e., 18F-PI-2620 (24)). However, evidence from PET-to-autopsy studies for these new ligands is needed, together with better segmentation and signal detection from small regions. These features would be particularly important for early-stage pathology detection and the classification of stage I/II of the system of Kovacs et al. (7).
CONCLUSION
We conclude that 18F-flortaucipir PET is not a useful marker of the neuropathologic stage in PSP, despite increased binding and some regional concordance between tau pathology and ligand binding. This analytic approach, seeking to mirror in vivo neuropathology staging with PET-to-autopsy correlational analyses, could be applied to test next-generation tau PET tracers. However, comparisons with postmortem data are also required.
DISCLOSURE
This study was cofunded by the Cambridge University Centre for Parkinson-Plus (RG95450); the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre (BRC-1215-20014), including their financial support for the Cambridge Brain Bank; the PSP Association (MAPT-PSP Award); the Alzheimer’s Research UK East-Network pump priming grant; the Wellcome Trust (grant number: 220258); the Medical Research Council (MR/P01271X/1; G1100464); the Association of British Neurologists; the Patrick Berthoud Charitable Trust (RG99368); the Alzheimer’s Society (443 AS JF 18017); the Evelyn Trust (RG84654); RCUK/UKRI (via a Research Innovation Fellowship awarded by the Medical Research Council to Caroline H. Williams-Gray [MR/R007446/1]); and the Guarantors of Brain (G101149). James B. Rowe serves as an associate editor to Brain and is a nonremunerated trustee of the Guarantors of Brain, Darwin College, and the PSP Association (United Kingdom). He provides consultancy to Asceneuron, Astex, SVHealth and Curasen, and UCB and has received research grants from AZ-Medimmune, Janssen, and Lilly as industry partners in the Dementias Platform U.K. John T. O’Brien has received honoraria for work as Drug and Safety Monitoring Board chair or member for TauRx, Axon, Eisai, and Novo Nordisk; has acted as a consultant for Biogen and Roche; and has received research support from Alliance Medical and Merck. Maria Grazia Spillantini is in the Scientific Advisory Board of the Tau Consortium supported by the Rainwater Charitable Foundation. Caroline H. Williams-Gray has received honoraria from Lundbeck and Profile Pharma Ltd. and consultancy fees from Modus Outcomes and Evidera, Inc./GlaxoSmithKline. Unrelated to this work, Timothy Rittman has received honoraria from Biogen, Oxford Biomedica, and the National Institute for Health and Clinical Excellence (NICE). No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can the novel postmortem pathologic staging of PSP be reproduced in vivo with 18F-flortaucipir PET?
PERINENT FINDINGS: Conditional inference tree analyses were performed on regional 18F-flortaucipir PET binding potential values to define in vivo staging in 42 patients with probable PSP, comparing the results in 9 participants with postmortem confirmation of the diagnosis and stage. 18F-flortaucipir PET did not provide accurate in vivo staging of PSP; in particular, the nominal stage derived from 18F-flortaucipir PET did not correlate with disease severity or relate to staging postmortem.
IMPLICATIONS FOR PATIENT CARE: This analytic approach, seeking to mirror in vivo neuropathology staging with PET-to-autopsy correlational analyses, might be more effective with next-generation tau PET tracers.
ACKNOWLEDGMENTS
We thank our participant volunteers for their participation in this study, and we gratefully acknowledge the participation of all National Institute for Health Research (NIHR) Cambridge BioResource volunteers. We thank the National Institute for Health Research Cambridge Biomedical Research Centre and staff for their contributions. We thank NIHR and NHS Blood and Transplant. We thank the radiographers and technologists at the Wolfson Brain Imaging Centre and Addenbrooke’s Hospital PET/CT Unit for their role in data acquisition. We thank the East Anglia Dementias and Neurodegenerative Diseases Research Network (DeNDRoN) for help with participant recruitment. In addition, we thank Dr. Istvan Boros, Dr. Joong-Hyun Chun, and other WBIC RPU staff for the manufacture of the radioligand. We thank Avid (Lilly) for supplying the precursor for the production of flortaucipir used in this study.
Footnotes
Published online Nov. 18, 2021.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
Immediate Open Access: Creative Commons Attribution 4.0 International License (CC BY) allows users to share and adapt with attribution, excluding materials credited to previous publications. License: https://creativecommons.org/licenses/by/4.0/. Details: http://jnm.snmjournals.org/site/misc/permission.xhtml.
REFERENCES
- Received for publication July 31, 2021.
- Revision received October 8, 2021.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}