


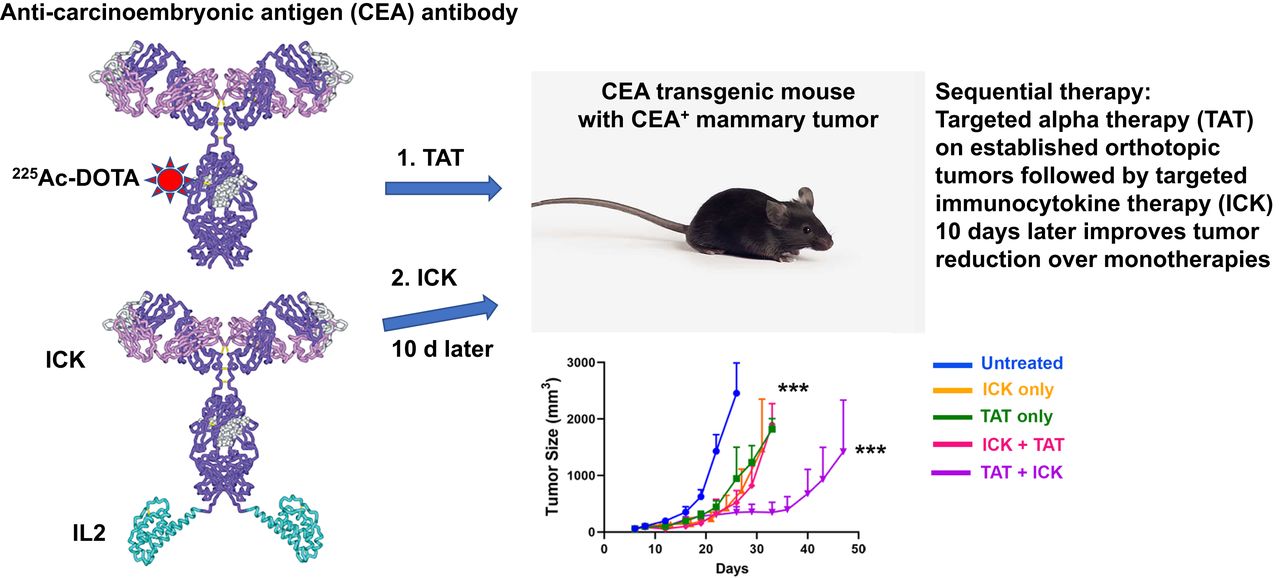
Visual Abstract

Abstract
Targeted α-therapy (TAT) delivers high-linear-transfer-energy α-particles to tumors with the potential to generate tumor immune responses that may be augmented by antigen-targeted immunotherapy. Methods: This concept was evaluated in immunocompetent carcinoembryonic antigen (CEA) transgenic mice bearing CEA-positive mammary or colon tumors. Tumors were targeted with humanized anti-CEA antibody M5A labeled with 225Ac for its 10-d half-life and emission of 4 α-particles, as well as being targeted with the immunocytokine M5A–interleukin 2. Results: A dose response (3.7, 7.4, and 11.1 kBq) to TAT only, for orthotopic CEA-positive mammary tumors, was observed, with a tumor growth delay of 30 d and an increase in median survival from 20 to 36 d at the highest dose. Immunocytokine (4 times daily) monotherapy gave a tumor growth delay of 20 d that was not improved by addition of 7.4 kBq of TAT 5 d after the start of immunocytokine. However, TAT (7.4 kBq) followed by immunocytokine 10 d later led to a tumor growth delay of 38 d, with an increase in median survival to 45 d. Similar results were seen for TAT followed by immunocytokine at 5 versus 10 d. When a similar study was performed with subcutaneously implanted CEA-positive MC38 colon tumors, TAT (7.4 kBq) monotherapy gave an increase in median survival from 29 to 42 d. The addition of immunocytokine 10 d after 7.4 kBq of TAT increased median survival to 57 d. Immunophenotyping showed increased tumor-infiltrating interferon-γ–positive, CD8-positive T cells and an increased ratio of these cells to Foxp3-positive, CD4-positive regulatory T cells with sequential therapy. Immunohistochemistry confirmed there was an increase in tumor-infiltrating CD8-positive T cells in the sequential therapy group, strongly suggesting that immunocytokine augmented TAT can lead to an immune response that improves tumor therapy. Conclusion: Low-dose (7.4 kBq) TAT followed by a 4-dose immunocytokine regimen 5 or 10 d later gave superior tumor reductions and survival curves compared with either monotherapy in breast and colon cancer tumor models. Reversing the order of therapy to immunocytokine followed by TAT 5 d later was equivalent to either monotherapy in the breast cancer model.
- radionuclide therapy
- carcinoembryonic antigen
- breast cancer
- colon cancer
- targeted alpha therapy
- targeted immunotherapy
Carcinoembryonic antigen (CEA) is a pancarcinoma antigen highly expressed in colon (1) and breast cancers. Radiolabeled CEA antibodies have been used to image a variety of CEA-expressing tumors (2–4), confirming their tumor-targeting specificity. Since most anti-CEA antibodies are not cytotoxic, they require conjugation to drugs or radionuclides for therapeutic applications. In this respect, radioimmunotherapy with anti-CEA antibodies radiolabeled with the β-emitters 131I (5–7) or 90Y (8,9) have met with some success in the clinic. In the case of immunotherapy, anti-CEA bifunctional antibodies have shown promising preclinical results (10,11). In addition, there are also several clinical trials (NCT04513431, NCT04348643, NCT02349724) investigating chimeric antigen receptor (CAR) T-cell therapy targeting CEA—trials that have met with mixed results (12). Thus, there is an unmet need for improvement in CEA-targeted therapies.
Sequential targeted radiotherapy followed by targeted immunotherapy is a promising approach in that it may stimulate an immune response. In this respect, we have recently shown that stereotactic body radiotherapy (SBRT) plus a CEA-targeted immunocytokine gave tumor reduction superior to that from either monotherapy (13). Those studies were performed with humanized anti-CEA antibody M5A (14) and an M5A–interleukin 2 fusion protein (13). We now extend those studies to sequential targeted α-therapy (TAT) plus immunocytokine. The choice of α- over β-based radionuclide therapy is based on the fact that α-emitters deliver more energy to the tumor and tumor vasculature because of their high linear-energy transfer (15), with a potential increase in tumor cytotoxicity due to stimulation of an immune response. In addition, their low tissue penetration is expected to reduce the hematologic toxicity of the systemic radiolabeled antibody, one of the major off-target effects of β-emitter–based radioimmunotherapy (16). For this study, we chose the α-emitter 225Ac for its long half-life (10 d) and emission of 4 α-particles (17). We have previously investigated the use of 225Ac-based TAT in the treatment of ovarian cancer (18) and multiple myeloma (19), finding that in one study, 225Ac-radionuclide TAT was superior to 177Lu-radionuclide radioimmunotherapy (19).
We hypothesized that TAT was more likely to stimulate a tumor immune response when followed by targeted immunotherapy. To test this hypothesis, it was necessary to perform these studies on immune-competent mice that expressed the target antigen of interest in normal tissues. For this reason, we used CEA transgenic mice in which the entire human CEA gene was expressed, conferring tissue-specific CEA expression that mimics that found in humans (20). We have previously shown that an all-murine anti-CEA immunocytokine significantly reduced CEA-positive tumor growth in this CEA transgenic model (21) and, more recently, that an all-murine anti-CEA CAR T-cell therapy plus the humanized immunocytokine reduced tumor growth in the same model (22). We now show that sequential TAT followed by immunocytokine therapy significantly improves tumor responses in both breast and colon tumors compared with either monotherapy in the CEA transgenic model.
MATERIALS AND METHODS
M5A, DOTA-M5A, immunocytokine, and the cell lines E0771/CEA and MC38/CEA were previously described (13).
Radiolabeling
DOTA-M5A (50 μg) was incubated with 225Ac at a labeling ratio of 1.85 kBq/μg for 45 min at 43°C, chased with 1 mM diethylenetriaminepentaacetic acid and purified by size-exclusion high-performance liquid chromatography.
Immunohistochemistry, Blood Analysis, and Flow Cytometry
Details of immunohistochemistry staining blood analysis and flow cytometry are provided in the supplemental materials (available at http://jnm.snmjournals.org).
Animal Studies
Animals were handled in accordance with institutional animal care and use committee protocol 91037, approved by the City of Hope Institutional Animal Care and Use Committee. CEA transgenic mice were previously described (20). E0771/CEA cells (105 in Matrigel [Corning]:phosphate-buffered saline 1:1) were injected into the mammary fat pad, and MC38/CEA (106) were injected subcutaneously.
Statistical Analysis
Two-way ANOVA was used to analyze tumor growth curves, and the log-rank Mantel–Cox test was used to analyze survival curves, using Prism, version 8.3.0 (GraphPad Software). Survival was the time at which the tumor reached 1,500 mm3. Treated groups were compared with untreated, unless otherwise stated. Differences were considered significant if P values were 0.05 or less.
RESULTS
TAT of E0771/CEA Mammary Tumors
Murine mammary cancer cells E0771 transfected with CEA (13) were injected into the mammary fat pad of CEA transgenic mice (20) to establish mammary tumors in an immunocompetent model that expressed the human CEA gene. Since we have previously shown that these tumors do not respond to naked humanized anti-CEA antibody M5A (13), we tested their response to TAT using 225Ac-DOTA-M5A (Fig. 1A). Increasing doses from 3.7 to 11.1 kBq showed a significant dose response compared with untreated controls (Fig. 1B; Supplemental Table 1), with the highest dose leading to a delay in tumor growth of about 30 d and a significant increase in median survival from about 20 to 36 d compared with untreated controls (Fig. 1C; Supplemental Table 1). Whole-body toxicity as measured by weight loss (≥20% loss) was not observed at all doses (Fig. 1D). In addition, there was no evidence of acute liver or kidney toxicity as measured by enzymes at the end of the study (Supplemental Table 2).

(A) Treatment schema and color codes for TAT 225Ac-DOTA-M5A TAT doses in orthotopic breast cancer model. (B) Tumor growth curves. (C) Kaplan–Meier survival plot. (D) Weight loss. Groups contained 8 mice, with 2 mice removed at days 21 and 22 for blood analysis. **P < 0.01. ***P < 0.001.
Flow cytometry analysis of the blood on day 21 indicated a significant decrease in CD8 T cells and B cells for the 2 highest doses of TAT, with no effect on CD11b myeloid cells (Supplemental Fig. 1A). The highest dose of TAT significantly reduced tumor infiltration of both CD4 and CD8 T cells (Supplemental Fig. 1B), and there was an increase in tumor-infiltrating neutrophils by 7.4 and 11.1 kBq of TAT (Supplemental Fig. 1C). These results suggest the possibility that TAT had a major effect on the immune response to the tumor, but it was unclear if the effect was immunosuppressive or immunostimulatory.
Comparison of Immunocytokine Plus TAT Versus TAT Plus Immunocytokine in E0771/CEA Mammary Tumors
The order of sequential therapy was tested since it was likely that TAT could kill tumor-resident CD8 cells that would otherwise respond to immunocytokine given first (Fig. 2A). The immunocytokine therapy schedule of 4 daily doses of 1 mg/kg starting 8 d after tumor inoculation into the mammary fat pad, adopted from our previous study (13), significantly delayed tumor growth by about 20 d, compared with untreated tumors (Fig. 2B; Supplemental Table 1). Interestingly, the delay in tumor growth for immunocytokine therapy alone was equivalent to the 7.4 kBq of TAT only (Fig. 2B). When immunocytokine was given before TAT (Fig. 2B), the results were similar to either monotherapy. Thus, the addition of TAT to immunocytokine given first did not boost the antitumor response.

(A) Treatment schema for immunocytokine first followed by TAT (bottom) or TAT first followed by immunocytokine (top) in breast cancer model. Groups contained 8 mice, with 2 mice removed at days 21 and 22 for blood analysis. (B and C) Tumor growth curves (B) and Kaplan–Meier survival plot (C) for immunocytokine-first vs. TAT-first sequential therapy. P values are vs. untreated controls. **P < 0.01. ***P < 0.001. ICK = immunocytokine.
For TAT first followed by immunocytokine, we chose 10 d later for the start of immunocytokine, based on the 2- to 4-d half-life of the circulating antibody in the blood (23) and the 10-d half-life of 225Ac. When TAT was followed by immunocytokine 10 d later, tumor growth was reduced to 38 d (Fig. 2B), with an increase in median survival to 45 d compared with about 30 d for either monotherapy (Fig. 2C; Supplemental Tables 1 and 3). In addition, sequential therapy did not result in significant loss of whole-body weight (Supplemental Fig. 2A) or liver and kidney toxicity (Supplemental Table 2). White blood cell analysis of TAT followed by immunocytokine showed a 50% reduction early after therapy (21 d) that recovered at the late time point (30–50 d) (Supplemental Fig. 2B). A breakdown of white blood cells into component cells revealed that early effects in this group were mostly on lymphocytes and neutrophils, with recovery in the TAT-plus-immunocytokine group by the later time point (Supplemental Figs. 2C and 2D). There was no effect on red blood cells in any group (Supplemental Fig. 2E), and there was about a 50% reduction in platelets immediately after therapy that recovered at the end of the study (Supplemental Fig. 2F). Overall, the toxicities of the sequential therapies were transient and minimal.
TAT alone or TAT plus immunocytokine affected the cellular viability of treated tumors at 21 d as shown by flow cytometry (Supplemental Fig. 3A). Although the percentage of tumor-infiltrating CD8 or CD4 T cells was reduced by TAT only and increased by immunocytokine monotherapy, the changes were not statistically significant. However, when analyzed for interferon-γ (IFNγ) production, there was a significant increase in tumor-infiltrating IFNγ-positive, CD8-positive T cells at 21 d (Supplemental Fig. 3B), of which IFNγ-positive, PD-1–positive tumor-infiltrating CD8-positive T cells also increased, followed by a significant decrease in IFNγ-negative, PD-1–positive exhausted T cells (Supplemental Fig. 3C). In addition, the ratio of CD8-positive, IFNγ-positive to CD4-positive regulatory T cells (Tregs) increased in both the immunocytokine-only and the sequential therapy groups (Supplemental Fig. 3D), suggesting that the Tregs play a more important role in tumor response than does the percentage of PD1-positive, CD8-positive T cells. This analysis confirmed that TAT followed by immunocytokine was superior to immunocytokine followed by TAT, suggesting that TAT adversely affected immunocytokine given first in sequential therapy.
Timing of Immunocytokine After TAT in E0771/CEA Mammary Tumors
Since TAT can adversely affect immunocytokine therapy when immunocytokine is given first but not when immunocytokine was given 10 d after TAT, we tested the possibility of administering immunocytokine 5 d after TAT (Fig. 3A). Five days was chosen as a time point when circulating M5A is at about 25% of initial levels (24) and total 225Ac would be at about 50% of its initial levels. Although the tumor reduction and survival curves showed slight differences between immunocytokine at 5 versus 10 d after TAT, the results overall were statistically identical to untreated controls and monotherapies (Figs. 3B–3C; Supplemental Table 1). A comparison of median survivals for all the mammary tumor studies shows increased survivals for the +5-d and +10-d groups out to 44–45 d (Supplemental Table 3). Flow analysis of infiltrating leukocytes in the tumor were also similar between the 2 sequential therapy groups (Supplemental Figs. 4A–4C). Notably, the percentage of CD4-positive, Foxp3-positive Tregs was reduced (Supplemental Fig. 4C), and the percentage of IFNγ-positive, CD8-positive cells to Tregs was higher in the +10-d and +5-d immunocytokine groups (Supplemental Fig. 4D). We conclude that delaying immunocytokine after TAT may prove to be beneficial as early as 5 d after TAT. This result suggests that immunocytokine may have its greatest effects on tumors that are damaged by prior radiation therapy, whether SBRT or TAT.

(A) Treatment schema for TAT followed by immunocytokine 5 d later (top) or 10 d later (bottom) in breast cancer model. Groups contained 8 mice, with 2 mice removed at days 21 and 22 for blood analysis. (B and C) Tumor growth curves (B) and Kaplan–Meier survival plot (C). P values are vs. untreated controls. **P < 0.01. ***P < 0.001. ICK = immunocytokine.
The timing of immunocytokine after TAT had minimal effect on whole-body toxicity (Supplemental Fig. 5) or liver and kidney toxicity (Supplemental Table 2). Hematologic analysis was similar between the 2 studies, showing a reduction in lymphocytes with TAT only at the early time point and recovery in the TAT-plus-immunocytokine group at the late time point (Supplemental Figs. 5A–5D). There were no effects on red blood cells (Supplemental Fig. 5E), and there was a transient effect on platelets (Supplemental Fig. 5F).
TAT of CEA-Positive MC38/CEA Colon Tumors
To confirm the efficacy of TAT in a second tumor model, CEA-transfected murine colon carcinoma MC38 cells were engrafted subcutaneously in CEA transgenic mice. In a dose response study of 3.7–11.1 kBq, there was little difference in tumor reduction or survival between control and treated tumors at the lowest dose (Figs. 4A and 4B; Supplemental Table 1), but at the middle (7.4 kBq) and highest (11.1 kBq) doses, there was a significant reduction in tumor growth and increase in median survival from 29 d (no treatment) to 50 d for the highest dose (Supplemental Table 4). In the colon cancer model, we evaluated the effects of increased TAT by changing the dose in 2 ways, first by raising the maximum dose to 14.8 kBq, and second by administering the 7.4-kBq dose twice, once at 13 d after tumor inoculation and again 10 d later. The tumor growth curves for individual mice shown in Figure 4C reveal an interesting spread in response to TAT, suggesting that minor differences in tumor sizes or microenvironment affect the responses to TAT that are not apparent in the control tumors. However, both the tumor growth (Fig. 4C; Supplemental Table 1) and the survival curves (Fig. 4D; Supplemental Tables 1 and 4) demonstrate that the single 14.8-kBq dose was superior to the fractionated 2 × 7.4-kBq dose. No significant whole-body (Supplemental Fig. 6A), liver or kidney (Supplemental Table 5), or chronic hematologic (Supplemental Figs. 6B–6F) toxicities were noted in this TAT monotherapy model.

(A and B) Tumor growth curves (A) and Kaplan–Meier survival plot (B) for TAT dose response in colon cancer model (6 per group, with 2 removed at day 27). (C and D) Tumor growth curves (C) and Kaplan–Meier survival plot (D) for 2 × 7.4 kBq vs. 14.8 kBq of TAT (n = 5–6). P values are vs. untreated controls. **P < 0.01. ***P < 0.001.
Sequential TAT Plus Immunocytokine Therapy of MC38/CEA Colon Tumors
To directly compare the 2 tumor model responses to sequential therapy, the identical TAT dose of 7.4 kBq was chosen with a delay of 10 d for the start of immunocytokine (Fig. 5A). The tumor growth curves for TAT or immunocytokine monotherapy were identical until 35 d, after which immunocytokine monotherapy showed tumor regrowth (Fig. 5B; Supplemental Table 1). Even though some of the mice treated with TAT had prolonged tumor growth inhibition and survival, the sequential therapy showed an improved tumor growth reduction out to about 45 d, after which tumor regrowth became obvious (Fig. 5B; Supplemental Table 1). Median survival for sequential therapy was 57 versus 29 d for untreated controls (Fig. 5C; Supplemental Tables 1 and 4). No whole-body (Supplemental Fig. 7A) or liver or kidney (Supplemental Table 5) toxicities were noted in the TAT-plus-immunocytokine group; however, early lymphocyte and platelet numbers and percentages were reduced but recovered at the late time point (Supplemental Figs. 7B–7F).

Treatment schema and TAT doses (A), tumor growth curves (B), and Kaplan–Meier survival plot (C) for TAT first followed by immunocytokine therapy in colon cancer model (7 per group). P values are vs. untreated controls. **P < 0.01. ***P < 0.001. ICK = immunocytokine.
Surprisingly, there was a significant increase in tumor-infiltrating CD4-positive and CD8-positive T cells only in mice treated with immunocytokine as analyzed by flow cytometry at 27 d (Supplemental Fig. 8A). However, both IFNγ-positive, CD4-positive T cells and CD8-positive T cells were significantly increased by sequential therapy (Supplemental Fig. 8B). Since these findings did not explain the significant improvement in tumor growth inhibition and survival in the TAT-plus-immunocytokine group, an additional study was performed in which tumors were collected and analyzed on days 1, 5, and 8 after the last dose of immunocytokine in the sequential therapy group. The results of this study showed a gradual increase in CD8-positive T-cell infiltration into tumors (Supplemental Fig. 8C). The increase in tumor-infiltrating IFNγ-positive, CD8-positive T cells on days 5 and 8 after the last dose of immunocytokine in the sequential therapy group was especially evident (Supplemental Fig. 8D). As in the other sequential therapy model, the change in the ratio of IFNγ-positive, CD8-positive T cells to regulatory T cells on days 5 and 8 after the last dose of immunocytokine in the sequential therapy group was significant (Supplemental Fig. 8E).
We conclude that both tumor models show a similar augmented response to TAT followed by immunocytokine, in which the increase in cytotoxic infiltrating T cells and decrease in tumor Tregs are due to the addition of immunocytokine, suggesting an immunologic mechanism.
Immunohistochemistry Analysis of Therapies
A limited number of tumors were harvested 21 or 27 d (for breast or colon cancer models, respectively) after tumor injection to study tumor morphology, vascularity, CEA expression, and lymphocyte infiltration. In the orthotopic mammary tumor model, vascularity as measured by CD31 staining was most affected by sequential therapy, as evidenced by increased staining and vessel size, especially at the tumor periphery (Supplemental Figs. 9A–9D). In the colon cancer model, the vascularity of untreated tumors showed even CD31 staining across the entire tumor that was greatly disrupted in all therapy groups (Supplemental Figs. 9E–9H).
CD8 numbers were lowest in the TAT-alone breast cancer group, with similar expression in the untreated control and immunocytokine groups, and were highest in the TAT-plus-immunocytokine group (Supplemental Figs. 10A–10D). CEA expression was largely limited to the tumor periphery in untreated controls and was markedly decreased toward the tumor center (Supplemental Fig. 10E), indicating an in vivo effect on CEA expression in this tumor model. Interestingly, TAT greatly reduced CEA expression only at the tumor periphery while preserving expression toward the tumor center (Supplemental Fig. 10F), whereas the opposite was true for immunocytokine-only therapy (Supplemental Fig. 10G). CEA expression in the sequential therapy tumors was similar to TAT only (Supplemental Fig. 10H), suggesting that TAT was most efficient in killing CEA-positive cells at the tumor periphery, a result that may be explained by the low tissue penetration of α-particles. In addition, breast tumors were stained for macrophages with the antibody F4/80 (Supplemental Fig. 11). In this series, F4/80 staining was most intense at the tumor periphery and was relatively unchanged for immunocytokine-only therapy. However, TAT alone or TAT plus immunocytokine greatly increased myeloid staining throughout the tumor, suggesting that TAT mobilized myeloid infiltration.
For CD8 staining of colon cancer tumors, untreated controls had large numbers of resident CD8 cells (Supplemental Fig. 12A), which were greatly reduced by TAT only (Supplemental Fig. 12B). The profile in immunocytokine-only therapy was intermediate, with clusters of CD8 cells observed in regions of the tumor (Supplemental Fig. 12C), suggesting a redistribution or elimination of CD8 subtypes. Sequential therapy was similar to TAT only (Supplemental Fig. 12D). CEA staining was uniformly intense throughout the tumor in untreated controls (Supplemental Fig. 12E), but with islands of low staining in TAT-only tumors (Supplemental Fig. 8F). Conversely, controls treated with immunocytokine only stained lightly for CEA, with islands of CEA-negative cells (Supplemental Fig. 12G). The sequential therapy tumors showed intense CEA staining at the periphery, with a centralized area of less intense staining (Supplemental Fig. 12H). Myeloid cell staining with F4/80 exhibited a profile different from the mammary tumors, with intense sporadic staining throughout the untreated controls changing to peripheral tumor staining in the treated groups (Supplemental Fig. 13).
DISCUSSION
Both tumor models responded similarly to targeted monotherapies, in which higher doses caused transient white blood cell and platelet reduction. Although no early kidney toxicity was observed, we did not test for delayed kidney toxicity, a potential problem with 225Ac-based therapy (17). In the case of the colon cancer model, an even higher dose (14.7 kBq), divided into a single-treatment or a split-treatment regimen, showed that the single higher dose increased the median survival to 65 d, versus 51 d for the split dose.
The order of sequential therapy in the breast cancer model showed that immunocytokine first followed by TAT was no better than either monotherapy, but TAT followed by immunocytokine increased survival from 30 to 45 d. Since immunocytokine therapy increases CD8 infiltration into both tumor models, these cells may be killed by subsequent TAT, which is cytotoxic to all cells in the tumor and T cells in particular. This may explain the controversial results of tumor-targeted radiation therapy, which was found to either suppress or stimulate the immune response (25,26). Although antibody-targeted radiation therapy can deliver significant tumor doses (27,28), they are accompanied by hematologic doses that are unavoidable because of circulating antibody. However, hematologic doses can be reduced by the short range of α-particles used in TAT (17). In a previous study with the same breast cancer model, tumor regrowth with SBRT plus immunocytokine occurred at 23 d, versus 15 d with SBRT alone (13). Tumor regrowth with TAT plus immunocytokine occurred at 40 d, versus 20 d for TAT alone. Thus, TAT plus immunocytokine may have advantages over SBRT plus immunocytokine. Clinical studies with SBRT plus anti-PD-1 or anti-PD-L1 immunotherapy demonstrate modest improvements in tumor response and suggest that the toxicities associated with these immunotherapies is limiting (29). The use of targeted immunotherapy with agents such as immunocytokine may improve outcomes.
Immunohistochemistry staining of tumors showed that tumors responded spatially differently for vascular effects, immune cell infiltration, and target antigen expression. Notably, TAT was more effective than immunocytokine in destroying or modifying tumor vasculature. CEA expression was most affected by TAT. However, effective therapy was observed in both models, suggesting that loss of CEA expression was not the major factor. Thus, in a single targeted treatment regimen, the initial antigen expression played the dominant role.
Myeloid cell infiltration was pronounced for TAT only or for TAT plus immunocytokine versus immunocytokine only in both models, but TAT caused little, if any, increase in CD8 cell infiltration, whereas immunocytokine therapy had a greater effect on CD8 infiltration, whether alone or followed by TAT in both models. A role for myeloid cell infiltration after TAT was also noted by Dabagian et al. (30), who ascribed this effect to immunosuppression. Thus, immunotherapy may increase the effectiveness of TAT by reducing myeloid infiltration. In a study by Dabagian et al. (30), anti-PD-1 immunotherapy was more effective than TAT, making comparisons to our study difficult.
CONCLUSION
In 2 tumor models, we showed comparable tumor reduction by TAT or immunocytokine monotherapy, which, when performed sequentially with TAT followed by immunocytokine, produced improved therapy. Both monotherapy and sequential therapies have minimal whole-body, hematologic, liver, and kidney toxicities. Increased infiltration of IFNγ-positive CD8 lymphocytes and an increased ratio of IFNγ-positive CD8 T cells to Foxp3-positive CD4 T cells is a mechanistic observation for immunocytokine-only or sequential TAT-plus-immunocytokine therapy in both tumor models.
DISCLOSURE
This research was partially supported by NIH cancer center support grant P30CA033572. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Is sequential TAT plus targeted immunocytokine therapy more effective than either monotherapy, and what is the best order for sequential targeted therapies?
PERTINENT FINDINGS: In an immunocompetent animal model, TAT followed by immunocytokine was more effective than either monotherapy, and TAT before immunocytokine performed better than immunocytokine followed by TAT. A major effect of immunocytokine was to improve the effector CD8 T-cell–to–Treg ratio.
IMPLICATIONS FOR PATIENT CARE: The effect of targeted radiotherapy on the immune system is controversial, making it hard to predict whether and when targeted immunotherapy should be added to the treatment regimen. Our study suggests that TAT followed by immunocytokine may be effective because of the limited tissue range of α-particles, which cause less hematologic immune suppression.
Footnotes
Published online Jun. 30, 2022.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication March 22, 2022.
- Revision received May 25, 2022.
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.