


Abstract
Targeted radiopharmaceutical therapy (TRT) using α-particle radiation is a promising approach for treating both large and micrometastatic lesions. We developed prostate-specific membrane antigen (PSMA)–targeted low-molecular-weight agents for 212Pb-based TRT of patients with prostate cancer (PC) by evaluating the matching γ-emitting surrogate, 203Pb. Methods: Five rationally designed low-molecular-weight ligands (L1-L5) were synthesized using the lysine-urea-glutamate scaffold, and PSMA inhibition constants were determined. Tissue biodistribution and SPECT/CT imaging of 203Pb-L1–203Pb-L5 were performed on mice bearing PSMA(+) PC3 PIP and PSMA(−) PC3 flu flank xenografts. The absorbed radiation dose of the corresponding 212Pb-labeled analogs was determined using the biodistribution data. Antitumor efficacy of 212Pb-L2 was evaluated in PSMA(+) PC3 PIP and PSMA(−) PC3 flu tumor models and in the PSMA(+) luciferase-expressing micrometastatic model. 212Pb-L2 was also evaluated for dose-escalated, long-term toxicity. Results: All new ligands were obtained in high yield and purity. PSMA inhibitory activities ranged from 0.10 to 17 nM. 203Pb-L1–203Pb-L5 were synthesized in high radiochemical yield and specific activity. Whole-body clearance of 203Pb-L1–203Pb-L5 was fast. The absorbed dose coefficients (mGy/kBq) of the tumor and kidneys were highest for 203Pb-L5 (31.0, 15.2) and lowest for 203Pb-L2 (8.0, 4.2). The tumor-to-kidney absorbed dose ratio was higher for 203Pb-L3 (3.2) and 203Pb-L4 (3.6) than for the other agents, but with lower tumor-to-blood ratios. PSMA(+) tumor lesions were visualized through SPECT/CT as early as 0.5 h after injection. A proof-of-concept therapy study with a single administration of 212Pb-L2 demonstrated dose-dependent inhibition of tumor growth in the PSMA(+) flank tumor model. 212Pb-L2 also demonstrated an increased survival benefit in the micrometastatic model compared with 177Lu-PSMA-617. Long-term toxicity studies in healthy, immunocompetent CD-1 mice revealed kidney as the dose-limiting organ. Conclusion: 203Pb-L1–203Pb-L5 demonstrated favorable pharmacokinetics for 212Pb-based TRT. The antitumor efficacy of 212Pb-L2 supports the corresponding 203Pb/212Pb theranostic pair for PSMA-based α-particle TRT in advanced PC.
Targeted radiopharmaceutical therapy using α-particles (α-TRTs), which cause deposition of ionizing radiation of high-linear-energy transfer, is accelerating in importance for managing prostate cancer (PC). This acceleration is due in part to the unexpected survival benefit conferred by 223RaCl2 in patients with castration-resistant PC metastatic to bone (1). Also contributing to this acceleration has been the remarkable decrease in tumor burden demonstrated on images of patients who received 225Ac-PSMA-617 (2), which targets prostate-specific membrane antigen (PSMA) in patients with metastatic castration-resistant PC who failed prior standard treatment (3,4). However, salivary and lacrimal gland radiotoxicity may affect the overall survival benefit by reducing quality of life (5). As an alternative to 225Ac (half-life, 10 d), 212Pb, which has a shorter physical half-life (10.6 h), is a promising source of α-emissions that has proved safe and effective in both preclinical models and clinical studies for several indications (6–9). 212Pb is commercially available from a 224Ra generator and has well-described radiochemistry (10). It is a β-emitter but serves as an in vivo nanogenerator of 212Bi (half-life, 1.01 h), which decays with an α-particle in its decay chain. 212Pb has been successfully used as a stand-alone treatment and in combination with chemotherapy using peptides and monoclonal antibodies as targeting vectors (6,7,11). Although PSMA-based TRT using low-molecular-weight agents and monoclonal antibodies is expanding in management of metastatic castration-resistant PC, to date this has primarily used agents that deliver β-emitting payloads (12,13). Few preclinical studies describe detailed evaluation of α-TRT (14–17).
A challenge of α-TRT is that the administered therapeutic activities are generally insufficient to be imaged for patient-specific dosimetry. For 212Pb, preclinical evaluation presents additional challenges due to a high-energy γ-emission from a daughter that requires extra shielding. As a surrogate radionuclide, 203Pb (half-life, 51.9 h, γ = 279 keV) is suitable for γ-well counting and SPECT and has been explored to aid development of 212Pb-based α-TRT (18,19). A first-in-humans study using 203Pb-based PSMA SPECT has recently appeared (20).
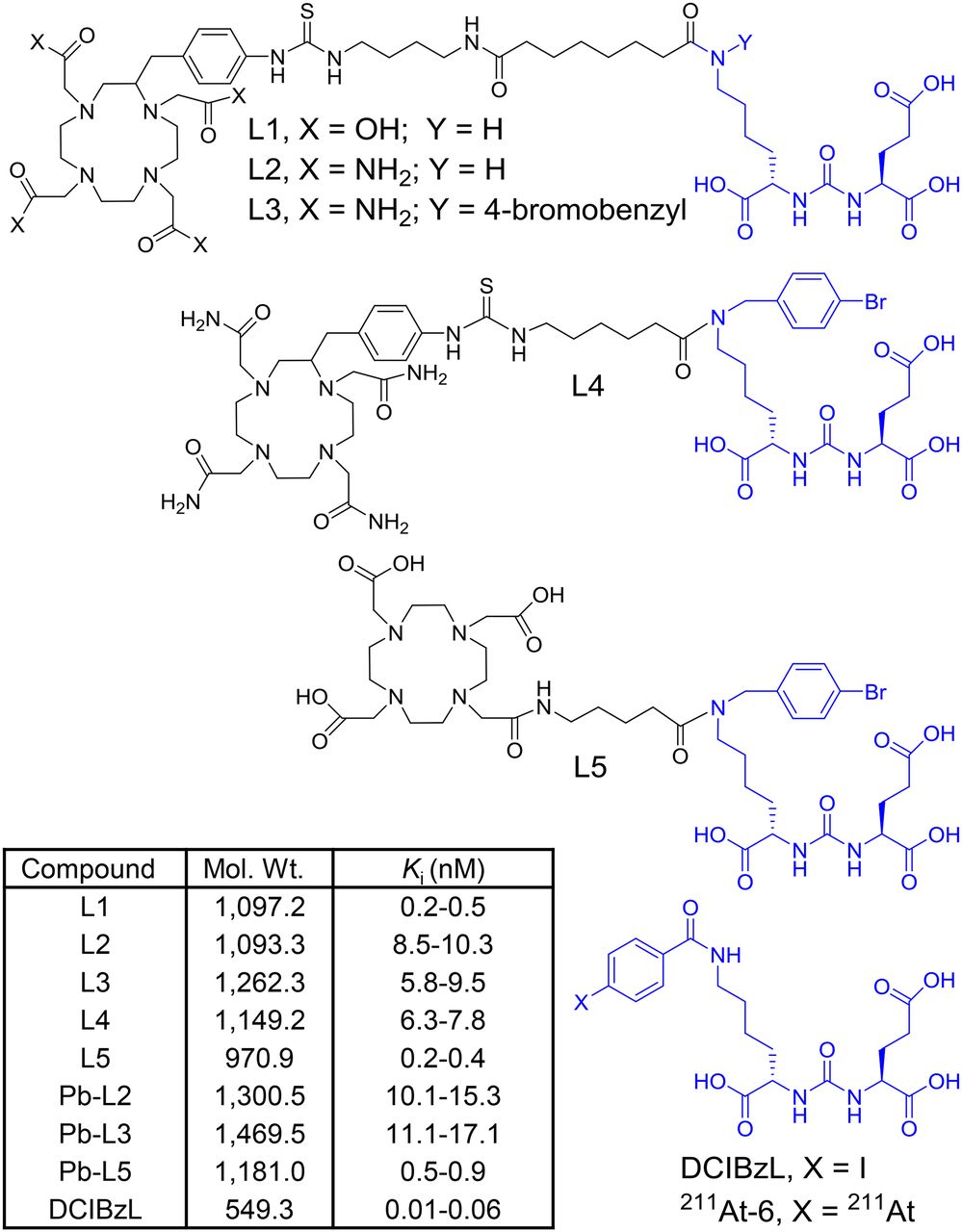
Here we report preclinical evaluation of a series of 203Pb-labeled low-molecular-weight ligands (L1–L5) for PSMA α-TRT. We first reevaluated our previous lead agent, L1 (21), as the 203Pb-labeled analog, and then synthesized 4 new ligands L2–L5 with further alterations to the chelator and inclusion of a 4-bromobenzyl-lysine-urea-glutamate targeting moiety. We used the 4-bromobenzyl derivative of lysine-urea-glutamate as the targeting moiety because of the sustained tumor uptake and high efficacy previously demonstrated by 125I-DCIBzL and its short-half-life α-emitting analog, 211At-6 (half-life, 7.2 h) (15,22) (Fig. 1). The goal of this study was to optimize an α-emitting agent with decreased off-target radiotoxicity relative to 211At-6 for PSMA-based α-TRT.
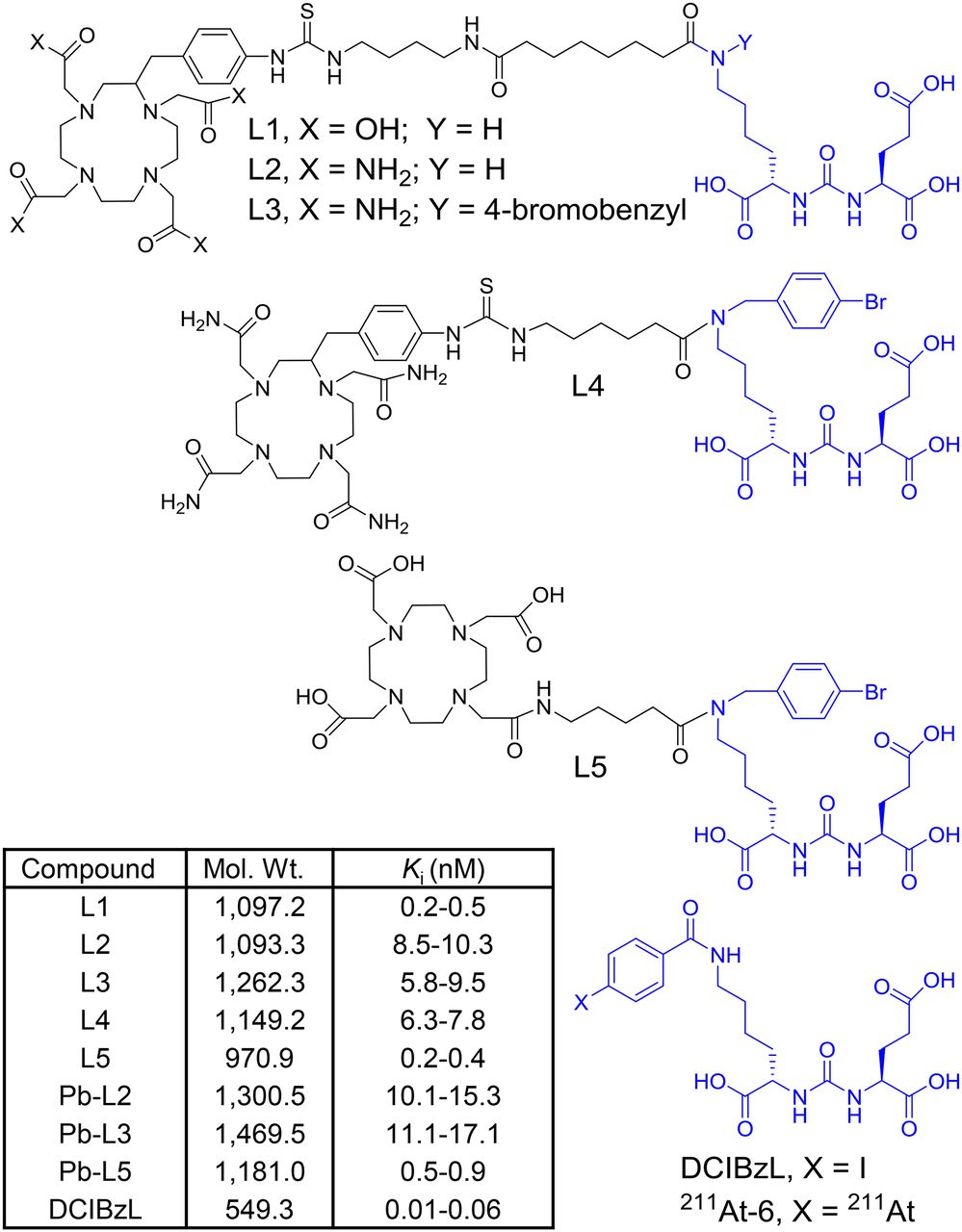
Structures of DCIBzL, 211At-6, and ligands L1–L5, for 203/212Pb-labeled PSMA-targeted α-particle theranostics. Molecular weight and PSMA inhibition constant (Ki) of new compounds are listed in the inset table.
MATERIALS AND METHODS
Reagents, Cell Lines, and Animal Models
203Pb was produced at the NIH Clinical Center cyclotron facility using a 203Tl(d,n)/203Pb reaction and purified from the target as previously described (23). The 212Pb was obtained using a 224Ra/212Pb generator (Oak Ridge National Laboratories). Sublines of the androgen-independent PC3 human PC cell line, originally derived from an advanced androgen-independent bone metastasis, were used (24). Animal studies were in compliance with the regulations of the Johns Hopkins Animal Care and Use Committee. Six- to 8-wk-old male nonobese, diabetic/Shi-scid/IL-2rgnull (NSG; The Jackson Laboratory) mice (Johns Hopkins Animal Resources Core) were implanted subcutaneously with PSMA(+) PC3 PIP (1.5 × 106) cells and PSMA(−) PC3 flu cells (1 × 106) in the forward right and left flanks, respectively.
Chemistry
Ligands L1 (21) and L5 (25) and intermediates 3 (24) and 4 (25) were synthesized following our recent reports. Detailed descriptions for L2, L3 (26), L4, and L5 are provided in the supplemental data (available at http://jnm.snmjournals.org). The PSMA binding affinity of the new compounds was determined using a fluorescence-based competitive binding assay reported by our laboratory (15).
Radiolabeling
An acidic solution of 203PbCl2 (∼25.9 MBq in 100 μL) was neutralized with 6 μL of 5 M NH4OAc to obtain a pH of approximately 4.5–5.5. A solution (40 μL) of L1–L5 (1 mg/800 μL of 0.1N H4OAc) was added, and the reaction mixture was incubated at 60°C–65°C for 45 min. An identical procedure was followed for radiosynthesis of each 203Pb-labeled analog. Radiolabeling was nearly quantitative in each case. 212Pb-L2 was synthesized following a literature method (27) at the National Cancer Institute and transported to Johns Hopkins for treatment studies.
Biodistribution
Mice bearing PSMA(+) PC3 PIP and PSMA(−) PC3 flu xenografts were injected via the tail vein with approximately 1.85 MBq of 203Pb-L1-203Pb-L5 (n = 4). Competitive inhibition studies were performed using ZJ43 (28), a known low-molecular-weight PSMA inhibitor, added to the 203Pb-L2–203Pb-L5 formulation, and biodistribution studies were performed at 2 h (n = 4).
SPECT/CT Imaging
SPECT/CT imaging of 203Pb-L1, 203Pb-L2, 203Pb-L3, and 203Pb-L4 was performed on an X-SPECT device (GammaMedica) following a reported method (24). Data were reconstructed and fused using commercial software from the vendor. Data were analyzed using AMIRA software (Thermo Fisher Scientific).
Dosimetry
Time–activity curves were generated from the murine biodistribution data of the 203Pb-analogs. Normal tissue and tumor absorbed-dose coefficients (ADCs) were estimated for the 212Pb-labeled analog after accounting for the α-radiation deposited locally using the mathematic formalism established by MIRD (29). Only the α-emission was considered in the calculations and was assumed to be deposited locally (φ = 1). Selected human ADCs were estimated using a mouse-to-human conversion formula for time-integrated activities, which were then input into OLINDA/EXM (30).
The preclinical biodistribution data (percentage injected dose [%ID] per gram of tissue) were translated into human whole-organ biodistribution data (%ID/organ) based on the ratio of organ activity concentration to whole-body mass being equal in both species.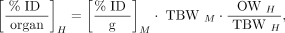 Eq. 1
Eq. 1
where M is mouse, H is human, TBW is total body weight (25 g for a mouse and 73.7 kg for an adult male human), and OW is the average male organ weight, in kilograms. The time-integrated activity coefficients were calculated for the human adult male organs and used as input into OLINDA/EXM, version 1.0, to calculate the clinical ADCs. For the tumor calculations, the OLINDA/EXM version 1 sphere model was used for a 1 g sphere (30).
Radiopharmaceutical Therapy with 212Pb-L2
Therapy in the Xenograft and Micrometastatic Models
Mice were injected subcutaneously in the upper flank with PSMA(+) PC3 PIP or PSMA(−) PC3 flu cells. Treatments were administered when tumor volume was 60–100 mm3. Animals (n = 5/group) received a single dose of 1.5 and 3.7 MBq of 212Pb-L2 intravenously via tail-vein injection or were untreated. Tumors were then measured 2–3 times per week until they reached a volume that was 10-fold the initial volume. The probability of reaching 10 times the initial tumor volume was characterized using Kaplan–Meier curves, and a comparison was performed using the log-rank test. For the PSMA(+) micrometastatic model, mice were injected intravenously with 1 × 106 PC3-ML-Luc-PSMA cells, as previously reported by us (15). At 24 h after injection of the tumor cells, mice (n = 5/group) were injected intravenously with 0, 0.7, 1.5, and 3.7 MBq of 212Pb-L2 and 37 MBq of 177Lu-PSMA-617. Metastatic tumor progression and survival were monitored by in vivo bioluminescence imaging (IVIS Spectrum; Perkin-Elmer).
Determination of Maximum Tolerated Dose (MTD)
The MTD was defined as the highest dose at which no animal died or lost more than 20% of its pretreatment weight. Non–tumor-bearing CD-1 mice (Charles River, n = 5/group) received intravenous injections of 212Pb-L2 and were then weighed and inspected twice per week for at least 12 mo. Urinalysis was performed monthly. On sacrifice, animals were evaluated at the Johns Hopkins Phenotyping Core, which obtained a serum metabolic panel, blood counts, and full necropsy.
Statistical Analysis
Statistical analysis was performed using a 2-tailed t test (GraphPad). P values were considered significant at a level of 0.05.
RESULTS
Synthesis and Radiolabeling
An abbreviated structure–activity relationship study was performed by modifying the chelating agent, linker, and targeting scaffold to develop an optimized agent for α-TRT (Fig. 1). Ligands L1 and L2 were synthesized following our previous report (Supplemental Fig. 1A) (21). Although DOTA-monoamide was successfully used for a 212Pb-labeled peptide (6), considering the unusual stability of Pb-(1,4,7,10-tetra-(2-carbamoyl methyl)-cyclododecane (TCMC) compounds in an acidic environment (11,31), L2–L4 were designed to contain a TCMC chelating agent. L3–L5 were synthesized following a similar route using the 4-bromobenzyl derivative of the Glu-urea-Lys scaffold (Supplemental Figs. 2B and 2C). 203Pb-labeled compounds (203Pb-L1–203Pb-L5) and 212Pb-L2 were synthesized in greater than 95% yield and were separated from the corresponding nonradiolabeled precursor by high-performance liquid chromatography to obtain a pure product with a specific activity of 0.7–1.9 MBq/nmol. The stability of the radiolabeled compounds was determined by incubation in phosphate-buffered saline and in 0.1% human serum albumin in phosphate-buffered saline (×1) at 37°C up to 72 h and was greater than 95%.
In Vivo Evaluation
Tissue Biodistribution
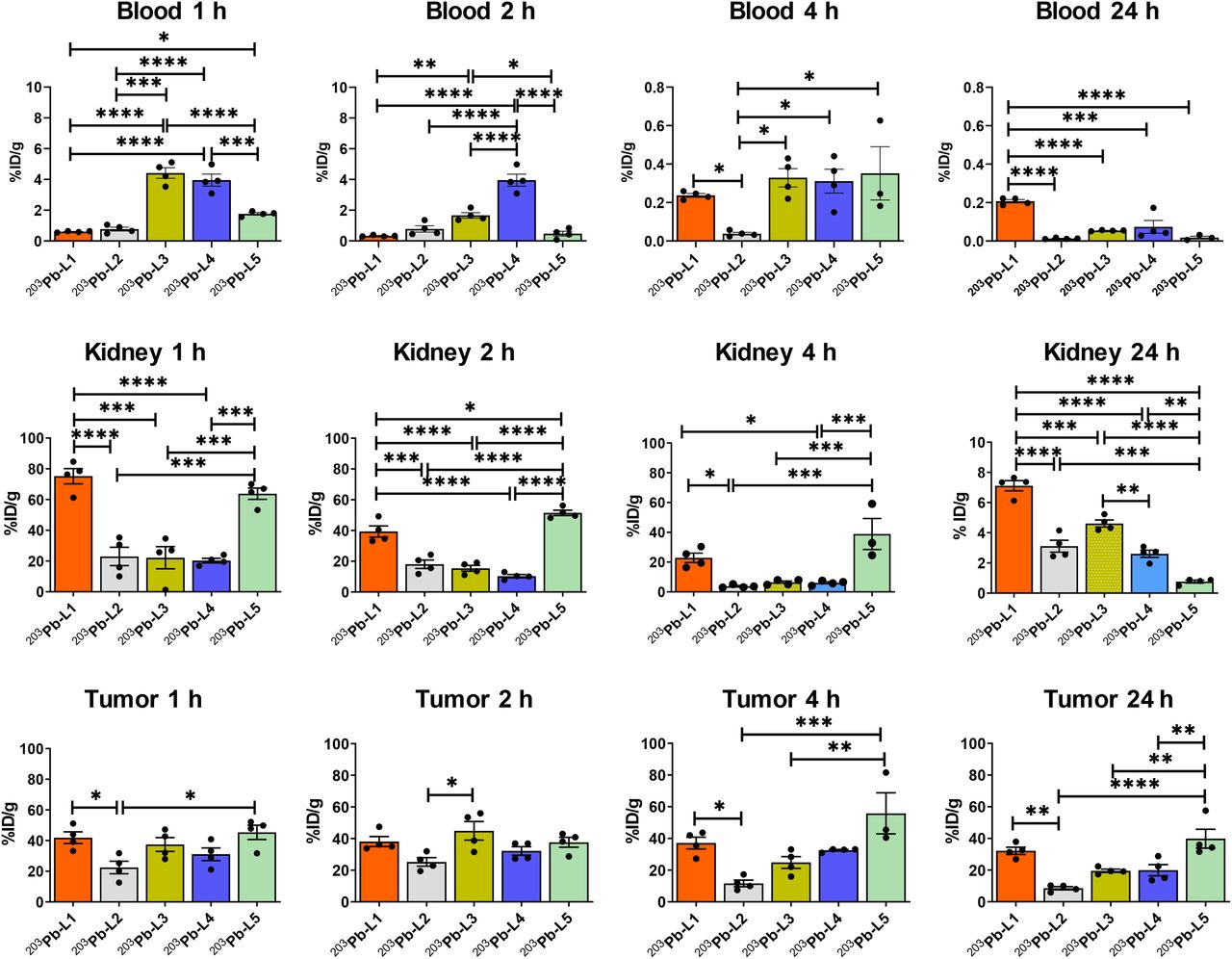
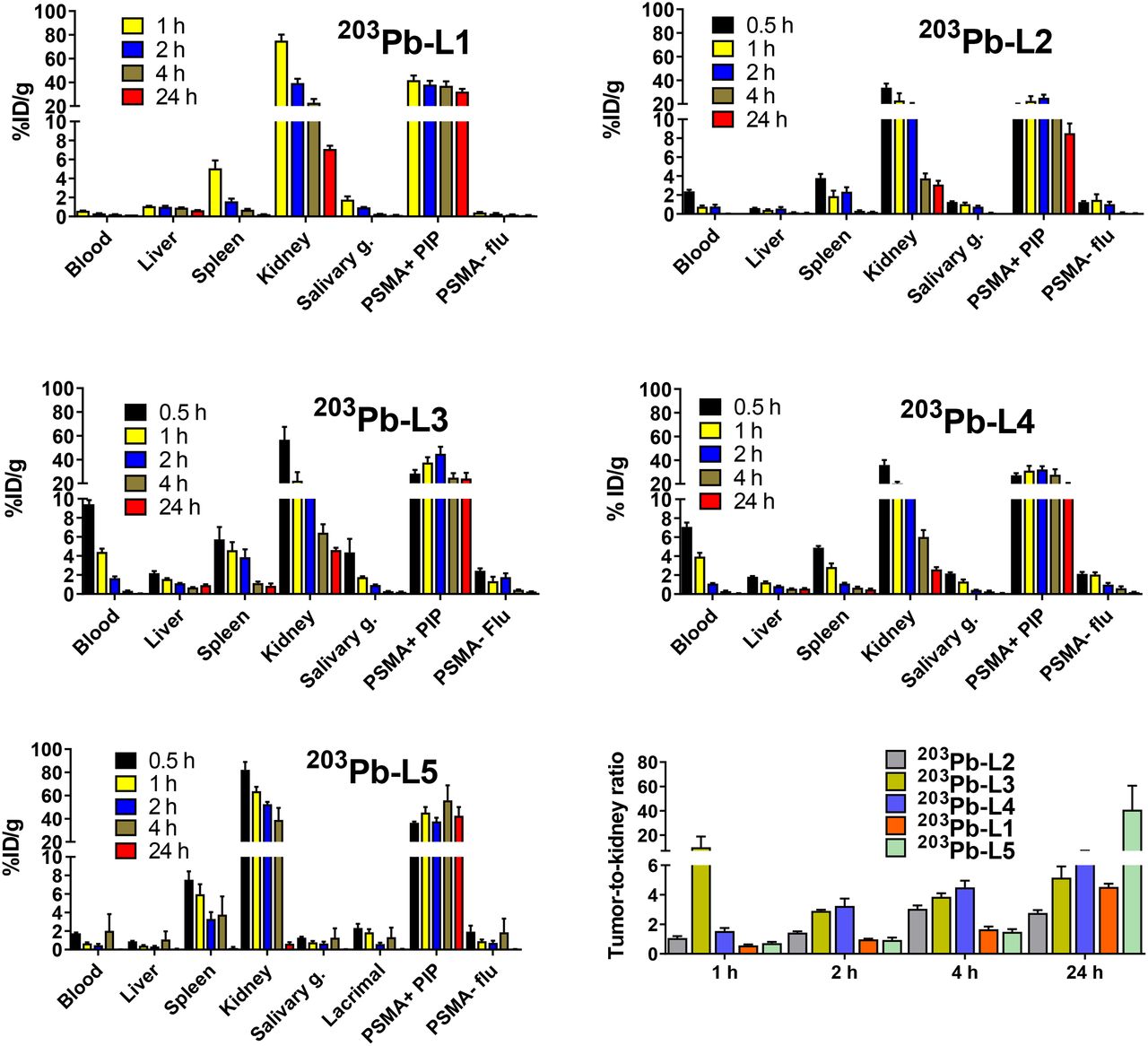
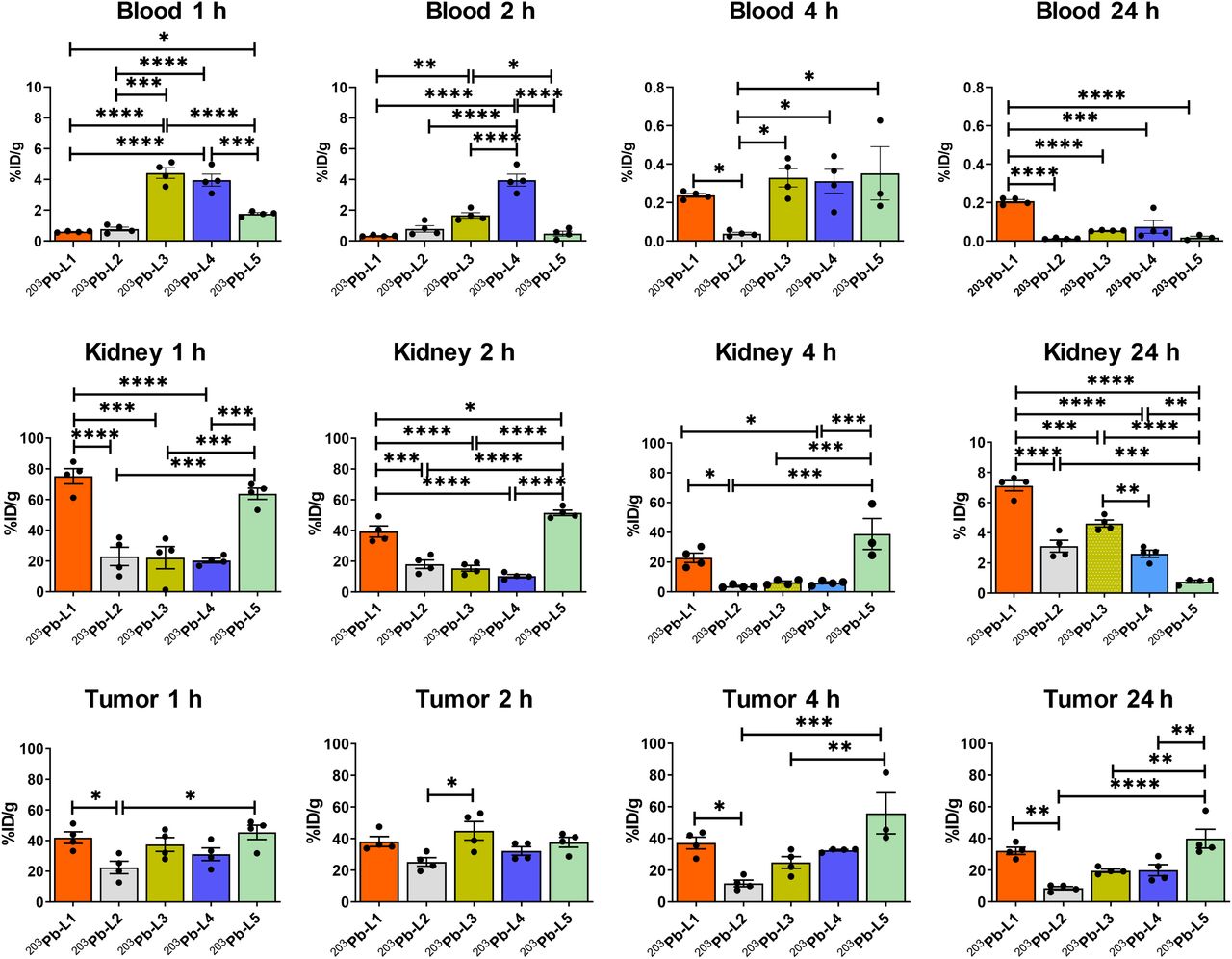
Biodistribution data (expressed in %ID/g) of 203Pb-L1–203Pb-L5 are shown in Figure 2 and Supplemental Tables 1–5. 203Pb-L1 exhibited high uptake in the PSMA(+) PC3 PIP tumor as early as 1 h, remained high at 4 h, and decreased at 24 h after injection. Unlike 203Pb-L1, 203Pb-L2 demonstrated the highest uptake in the PSMA(+) tumor at 2 h, followed by gradual clearance during 4–24 h after injection. 203Pb-L2 displayed fast clearance from all normal tissues including kidneys and PSMA(+) tumor. Observing the significant change in biodistribution, especially within the PSMA(+) PC3 PIP tumor and the kidneys by simply changing the chelating agent as we previously experienced (32), we further investigated radioligands 203Pb-L3 and 203Pb-L4 bearing the same chelating agent, TCMC-Bn-NCS, and 203Pb-L5 bearing the DOTA-monoamide chelating agent. Figure 3 summarizes the head-to-head comparison of the PSMA(+) tumor and selected tissues of the tested agents. 203Pb-L2 demonstrated significantly lower tumor uptake up to 2 h than 203Pb-L1, 203Pb-L3, and 203Pb-L5 (P < 0.01). At 4 h, 203Pb-L2 displayed significantly lower tumor uptake than any other compound (203Pb-L1 and 203Pb-L4, P < 0.001; 203Pb-L3 and 203Pb-L5, P < 0.05). Additionally, at 24 h after injection, tumor uptake of 203Pb-L2 was significantly lower than that of 203Pb-L1, 203Pb-L3, and 203Pb-L5. There was no significant difference in PSMA(+) tumor uptake between 203Pb-L3 and 203Pb-L4 during the 0.5- to 24-h time-points. Both 203Pb-L1 and 203Pb-L5 showed significantly higher tumor retention than 203Pb-L3 at 24 h.
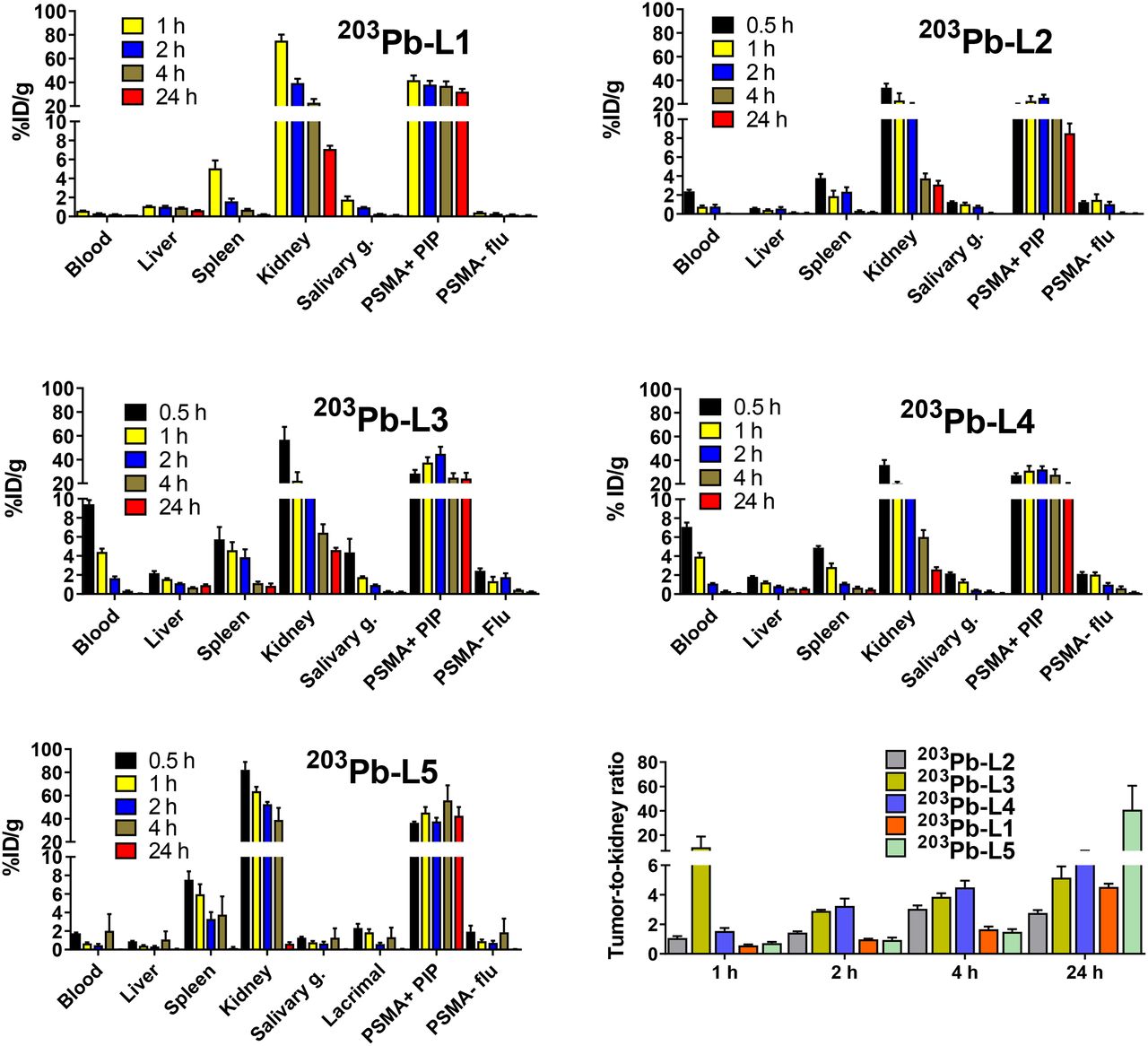
Tissue biodistribution in mice bearing PSMA(+) PC3 PIP and PSMA(−) PC3 flu tumors on either flank (n = 4) and tumor–to–normal-organ ratios.
Time-dependent uptake of 203Pb-L1–203Pb-L5 in selected tissues. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.0001.
203Pb-L2–203Pb-L4 with the TCMC chelating agent displayed significantly lower renal uptake during 1–4 h after injection than 203Pb-L1 and 203Pb-L5. At 24 h, renal uptake was significantly lower for 203Pb-L2–203Pb-L4 than for 203Pb-L1 (P < 0.001). 203Pb-L5 displayed significantly higher renal uptake at 2 h and remained high compared with 203Pb-L1 during 4–24 h. There was no significant difference in renal uptake between 203Pb-L2, 203Pb-L3, and 203Pb-L4 up to 4 h and a small but significant difference at 24 h only between 203Pb-L2 and 203Pb-L3. Blocking (PSMA binding specificity) studies were performed for 203Pb-L2–203Pb-L5 by coadministration of 50–100 nmol of the known PSMA inhibitor, ZJ43 (28), showing significant blockade in the PSMA(+) PC3 PIP tumor for all agents (Supplemental Fig. 2). Significant renal blockade was observed for all agents, further indicating specificity for PSMA.
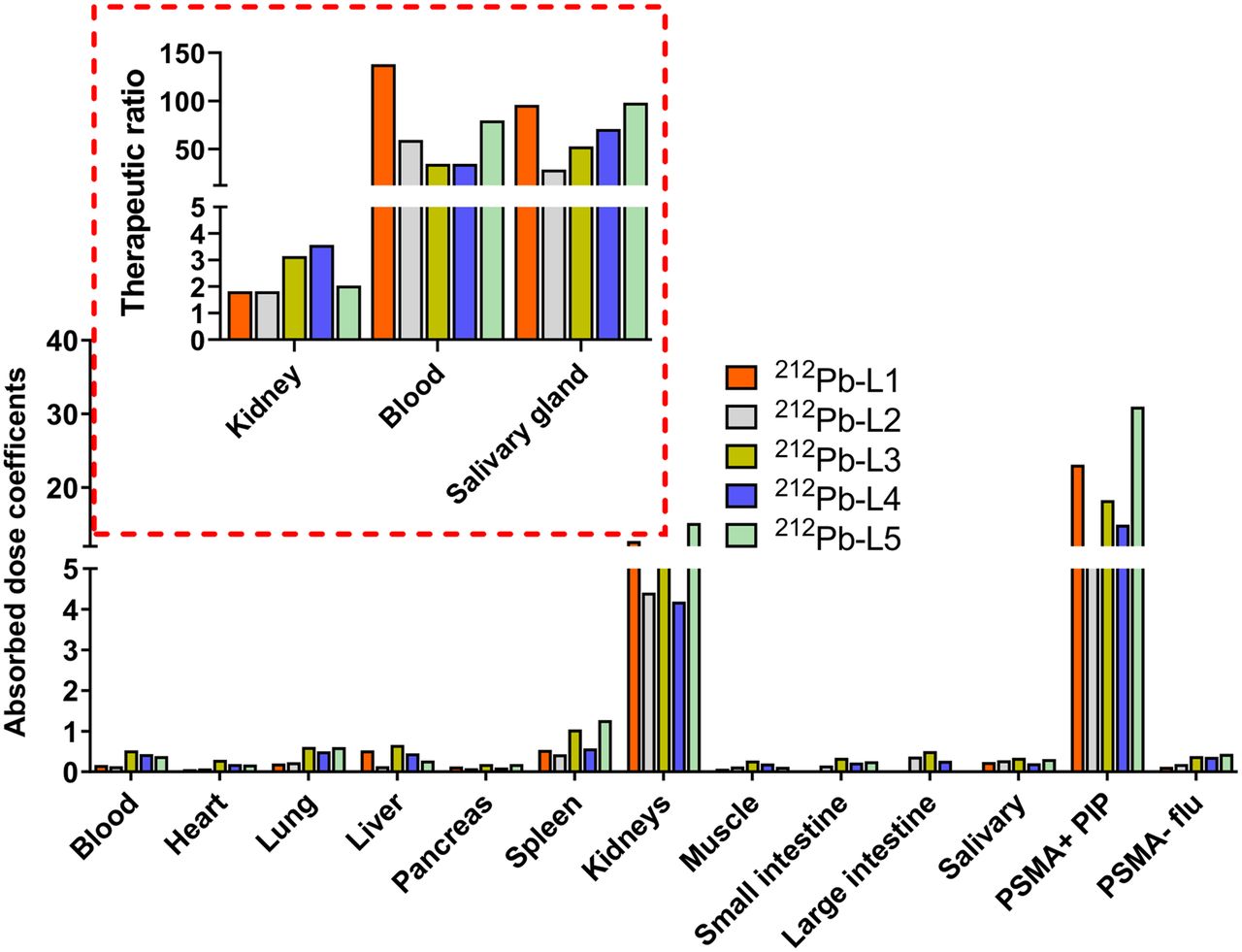
Organ-Absorbed Doses
Figure 4 and Supplemental Table 6 provide a selected list of the murine ADC for 212Pb-analogs. Tumors received ADCs of 23.1, 8.0, 18.3, 15.0, and 31.0 mGy/kBq for 212Pb-L1, 212Pb-L2, 212Pb-L3, 212Pb-L4, and 212Pb-L5, respectively. Kidneys received the highest ADC and followed a similar trend to that of the PSMA(+) tumors, with 23.1, 4.4, 5.8, 4.1, and 15.2 mGy/kBq for 212Pb-L1, 212Pb-L2, 212Pb-L3, 212Pb-L4, and 212Pb-L5, respectively. The other potential dose-limiting organ was blood, which demonstrated a nearly 2-fold higher ADC for 212Pb-L3 (0.5 mGy/kBq) and 212Pb-L4 (0.4 mGy/kBq) than for 212Pb-L1 (0.2 mGy/kBq) and 212Pb-L2 (0.1 mGy/kBq). Absorbed doses for other tissues were low, including heart, lung, liver, spleen, and muscle. Salivary gland ADCs were low, within the range of 0.28–0.35 mGy/kBq for all agents. Therapeutic PSMA(+) tumor–to–normal-organ ratios were calculated for kidney, blood, and salivary glands (Fig. 5 inset). Therapeutic ratios with respect to kidney demonstrated the following trend: 212Pb-L3 > 212Pb-L4 > 212Pb-L5 > 212Pb-L1 ∼ 212Pb-L2. With respect to blood, the trend was 212Pb-L1 > 212Pb-L5 > 212Pb-L4 > 212Pb-L3 ∼ 212Pb-L2. Therapeutic ratios with respect to salivary glands were in the range of blood, indicating kidney as the dose-limiting organ. Estimated human ADCs from OLINDA/EXM, based on mouse-to-human time-integrated activity conversion, are listed in Supplemental Table 7.

Estimated ADCs (mGy/kBq). Inset shows therapeutic ratio, calculated as ADCs of tumor-to-blood (Blood), tumor-to-kidney (Kidney), and tumor-to-salivary glands (Salivary gland).
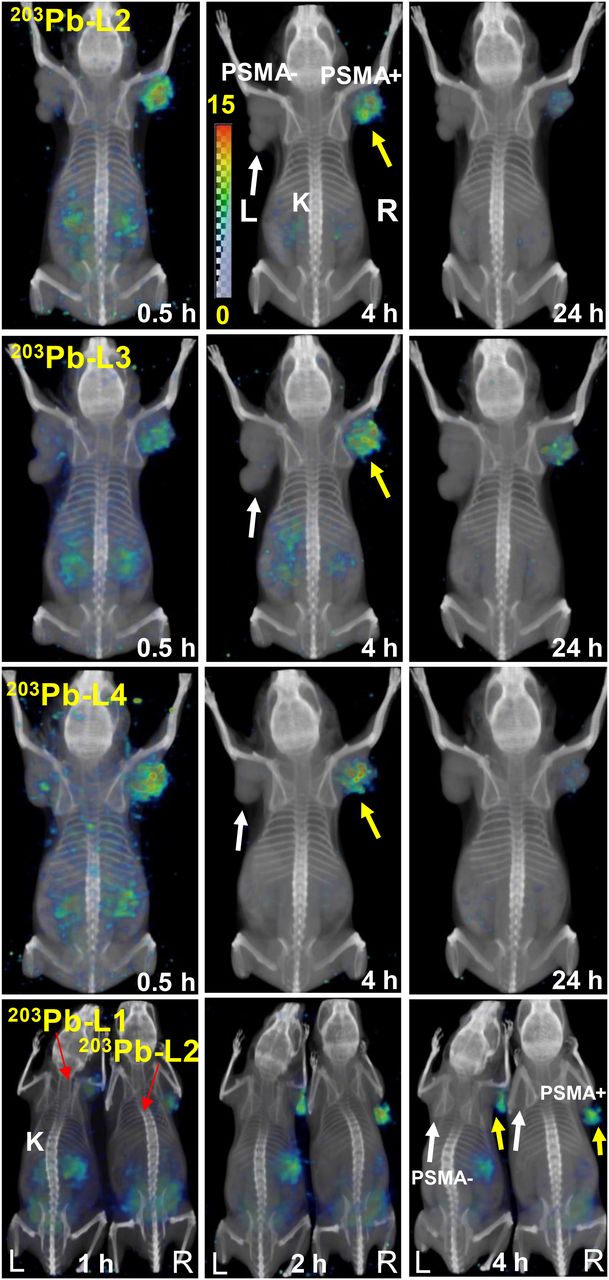
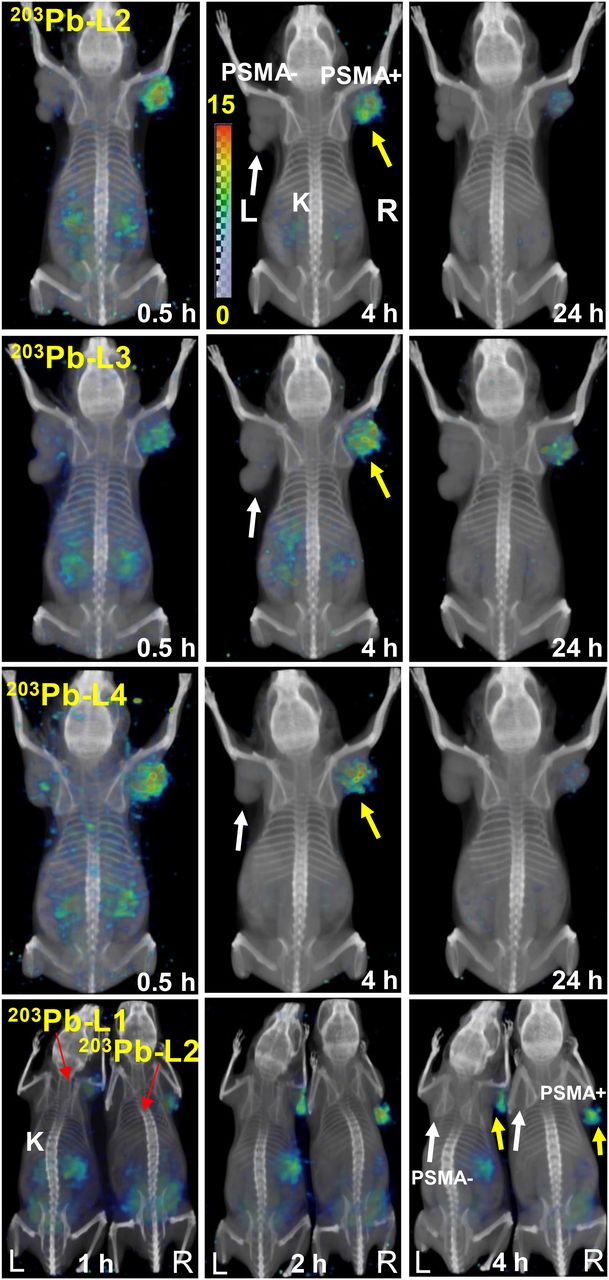
Whole-body volume-rendered SPECT/CT in mice bearing PSMA(+) PC3 PIP (yellow arrows) and PSMA(−) PC3 flu (white arrows) tumors. Mice were injected intravenously with ∼26 MBq, showing uptake only in PSMA(+) PC3 PIP tumor and kidneys. K = kidney.
In Vivo Imaging
SPECT/CT imaging was performed for 203Pb-L1–Pb-L4 for a visual demonstration of in vivo pharmacokinetics (Fig. 5). SPECT/CT images during 0.5–24 h after administration confirmed high uptake in the PSMA(+) PC3 PIP tumors but not in the PSMA(−) PC3 flu tumors. Also consistent with the biodistribution data, 203Pb-L2, 203Pb-L3, and 203Pb-L4 displayed very low renal uptake compared with 203Pb-L1 at 2 h after injection. Fast blood clearance of all agents was also evident from the imaging study. Compared with 203Pb-L4 (short linker), 203Pb-L3, which bears a long linker, displayed high spleen uptake up to 4 h.
Radiopharmaceutical Therapy
Antitumor Effect in the Flank Tumor Model
The treatment effects of 212Pb-L2 on the tumor growth rate and body weight of the mice are shown in Figures 6A–6B. A single administration of 1.5 or 3.7 MBq showed significant tumor growth delay only in PSMA(+) tumors (P = 0.003), compared with the other groups; however, slow tumor regrowth was observed after 8 wk. The median time to reach a 10-fold increase from the initial tumor volume (tumor volume/initial volume ≤ 10) was 25 and 39 d for the treatment groups bearing PSMA(+) tumors administered 1.5 and 3.7 MBq, respectively (Fig. 6C). For control groups, untreated PSMA(+) and PSMA(−) tumors reached a 10-fold increase from the initial tumor volume at 13 and 15 d, respectively. The median time for a 10-fold tumor increase in the groups with PSMA(−) tumors treated with 1.5 and 3.7 MBq were 7.5 and 16 d.
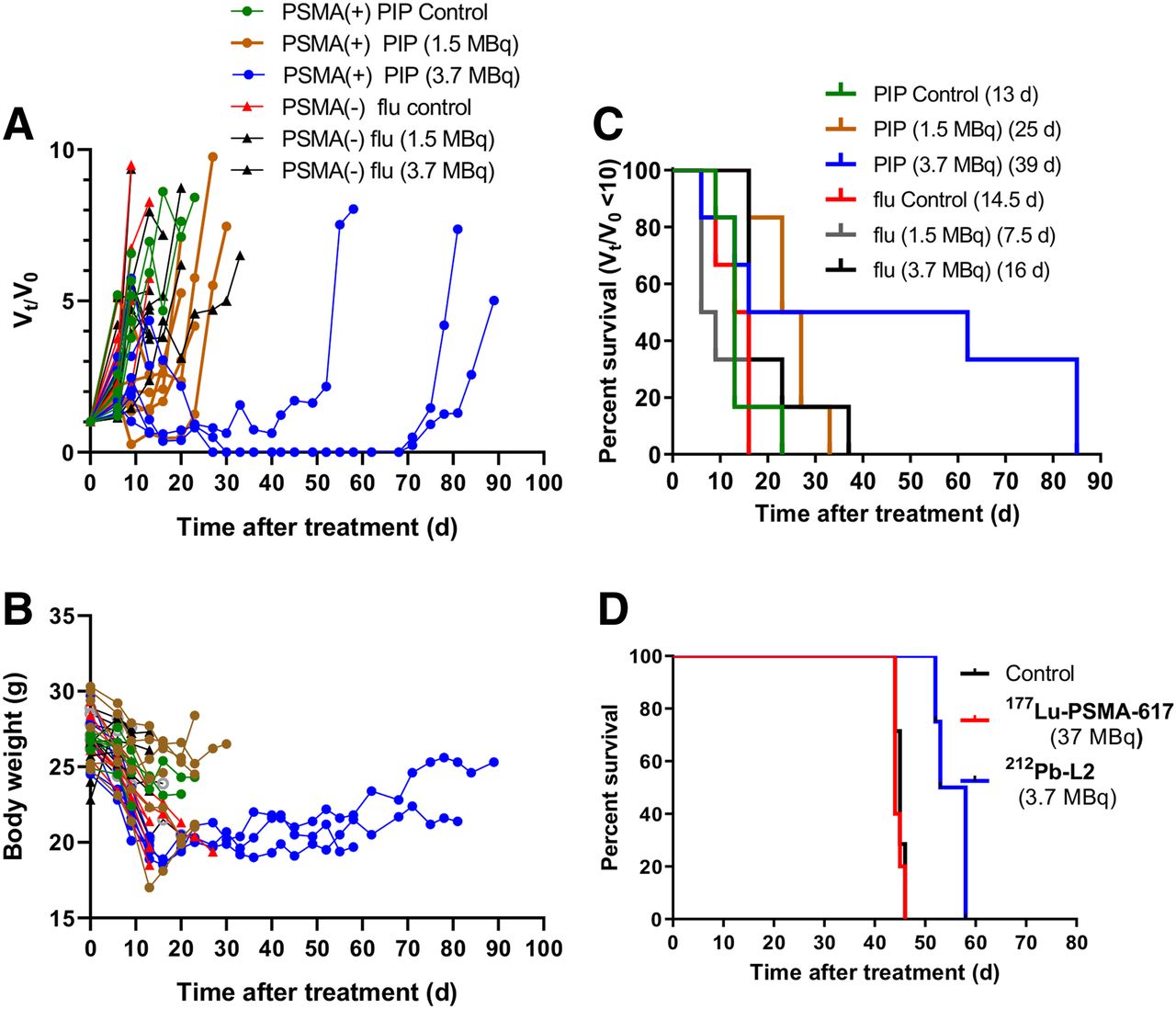
Treatment effect of 212Pb-L2. (A) Ratio of tumor volume (tumor volume/initial volume) changes on treatment with single administration (n = 5). Each line represents 1 mouse. (B) Changes in body weight of the corresponding treatment group. Dose and median time for (tumor volume/initial volume) = 10 are in parentheses. (C) Kaplan–Meier curves illustrating time to grow 10-fold in tumor volume after treatment with a single administration of 212Pb-L2 or control. (D) Kaplan–Meier curves showing significant improvement in survival after treatment in the micrometastatic model compared with control and 177Lu-PSMA-617.
Antitumor Effect in the PSMA(+) Micrometastatic Model
For the PSMA(+) micrometastatic tumor model, doses were administered 24 h after tumor cell inoculation. At that time, tumors were considered to be clusters of relatively few cells, which should be favorable for the short range of α-particles, compared with long-range β−-TRT. The efficacy of α-particle–emitting 212Pb-L2 (single administration of 0.7, 1.5, and 3.7 MBq) was compared with an untreated group and a group treated with β-emitting 177Lu-PSMA-617 (37 MBq) (Fig. 6D). No survival benefit was seen for the group treated with 177Lu-PSMA-617 compared with the control group (median survival time, 46 and 47 d, respectively); in contrast, the median survival time for the mice administered 212Pb-L2 (3.7 MBq) was 58 d, demonstrating moderate but significant improvement (P = 0.002).
In Vivo Toxicity and MTD
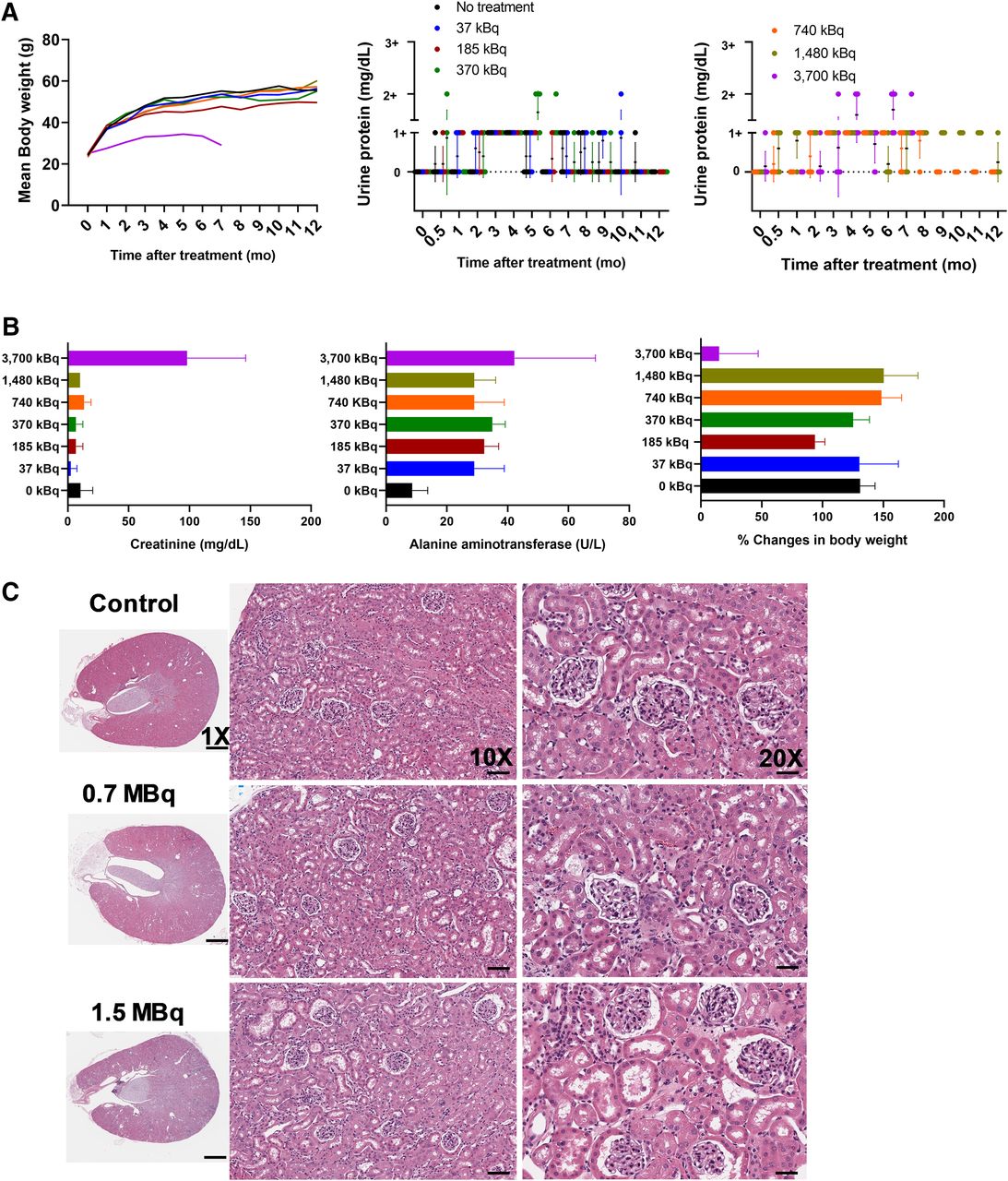
Mean body weight and urinalysis data after a single administration of 0.04- to 3.7-MBq doses are presented in Figure 7A. The MTD of 212Pb-L2 in immunocompetent CD-1 mice was 1.5 MBq. Necropsy at 12 mo after treatment for lower doses (0.74 and 1.5 MBq) showed acceptable changes in hematologic parameters, including blood urea nitrogen (24–44 mg/dL) and creatinine (0.3 mg/dL) for kidney function and alanine aminotransferase and aspartate aminotransferase for liver function (Fig. 7B; Supplemental Tables 8–11). Relative kidney mass was comparable for both groups. Histopathologic evaluation revealed moderate nephrotoxicity and tubule epithelial karyomegaly in the treated compared with control animals (Fig. 7C), also chronic nephropathy with typical degenerative and regenerative changes that are expected in obese mice in chronic studies (33). No histopathologic radiotoxicity was noted in other organs, including bone marrow. Clinically evident toxicity of mice treated with 3.7 MBq was identified at 7 mo, including body weight loss, proteinuria, anemia, and azotemia in several animals. Those results support the dosimetry that designated kidney as the dose-limiting organ. The projection to human data is simplistic but indicates that activities on the order of 1–3 GBq could be delivered to humans, assuming MTD constraints of 23 Gy to the kidneys and 2 Gy to the bone marrow (Supplemental Table 7).

Analysis of radiotoxicity parameters after single administration of 212Pb-L2 in healthy CD-1 mice (n = 5) for 1–12 mo. (A) Mean body weight and urine protein level measured by dipstick showing dose-dependent proteinuria occurring in the 3.7-MBq treatment group. A different batch of mice (n = 7) for treatment group was administered 3.7 MBq. Each dot represents the urine protein value for each mouse (trace, 0–10 mg/dL; 1+, 30 mg/dL; 2+,100 mg/dL). (B) Creatinine concentration and alanine aminotransferase in serum, and percentage body weight change at 13 mo after injection. All measurements for the 3.7-MBq treatment group were done at 7 mo after injection. (C) Hematoxylin and eosin staining of kidneys from nontreated mouse and mouse treated with 0.7 and 1.5 MBq of 212Pb-L2 after 12 mo showing mild changes in cortical tubules (scale bar, 50 μm; ×20).
DISCUSSION
Although encouraging, PSMA-targeted 177Lu-based β−-TRT is not effective in about 30% of patients and is considered unsuitable for patients with diffuse red marrow infiltration (12). 225Ac-based α-TRT has been pursued to overcome resistance to β−-TRT as salvage therapy for treatment-refractory metastatic castration-resistant PC (2). Those clinical investigations have revealed that α-emitters are effective in controlling large lesions in addition to their predicted role in elimination of microscopic disease because of their short range of energy deposition and a noted bystander or abscopal effect. Several α-emitting isotopes, such as 213Bi, 211At, 212Pb, and 225Ac, have been studied clinically, and all are being investigated preclinically by us and others in the context of PSMA-based TRT (15,16,19,20).
There are several radiobiologic effects that contribute to the superior efficacy of α-emitters relative to β−-emitters, one of which is by activating several unique molecular pathways (34). That mechanism of radiotoxicity is independent of tissue oxygenation, dose rate, and cellular resistance to γ- or β−-irradiation and chemotherapy (35). Accordingly, normal tissues might also be expected to receive those higher toxic doses, causing severe side effects for the α- compared with β−-emitters. A careful evaluation of the absorbed doses from radiosensitive vital organs based on long-term toxicity studies as described in this report may provide reliable dose prediction for an initial phase I dose escalation trial.
The major acute toxicity from clinical PSMA-based α-TRT is related to salivary and lacrimal gland dysfunction (5). Although renal toxicity has so far proved minimal for PSMA β−- and α-TRT (2,12), late nephrotoxicity remains a concern, as an insubstantial number of patients have been evaluated many years out from therapy. For example, over the long term, chronic nephrotoxicity was reported as a major side effect for patients treated with 177Lu-/90Y-octreotate (36–38). The α-emitters, because of their short-range radiation, may actually yield a lower absorbed dose to the radiosensitive glomeruli; however, they are much more potent with respect to promoting damage to the renal tubules (39). 225Ac in particular, along with its 3 α-emitting daughters, is expected to have substantial radiotoxicity due to the redistribution of daughters to the normal organs after each α-decay. It is known that free bismuth is accumulated by the renal cortex (39), which is of concern because of the radioactive bismuth daughters of 225Ac.
212Pb offers an alternative for PSMA-based α-TRT because of its short half-life and its availability through a commercial generator. 212Pb-based α-TRT using low-molecular-weight agents has not been studied extensively (6). The preclinical work described here leveraged several key features of the Lys-Glu-urea scaffold to optimize 212Pb-based α-TRT targeting PSMA. An abbreviated structure–activity relationship study allowed us to modulate off-target toxicity while maintaining higher tumor-absorbed radiation dose. Because of the short half-life of 212Pb, a high dose is expected to be delivered to the kidneys within the first few hours; consequently, we paid careful attention to the renal dose. Additionally, we anticipate a lower salivary gland absorbed dose for the 212Pb-labeled compounds than for 225Ac-labeled compounds because of the shorter half-life and less complicated dosimetry of 212Pb. We recognize that direct comparison of salivary gland absorbed dose for 212Pb- and 225Ac-labeled analogs based on biodistribution data from 203Pb-labeled compounds may be speculative since the studies were performed only out to 24 h after injection and the compounds harboring each radioisotope have their own pharmacokinetic properties.
Biodistribution data revealed that the agents with DOTA and DOTA-monoamide chelating agents (203Pb-L1 and 203Pb-L5) tended to display higher renal retention than the positively charged agents that carry TCMC as the chelator (203Pb-L2–203Pb-L4). Similarly, higher tumor uptake and retention were observed with 203Pb-L1 and 203Pb-L5 than with TCMC-chelated agents after 24 h. Among the TCMC-chelated compounds, 203Pb-L2 without a 4-bromobenzyl moiety from the targeting lysine-urea-glutamate displayed significantly faster clearance from the PSMA(+) tumor and normal tissues at all time points. Although 203Pb-L3 and 203Pb-L4 demonstrated tumor uptake comparable to 203Pb-L1 and 203Pb-L5, they displayed significantly lower renal uptake at 4 h. However, the high tumor-to-kidney ratios of 203Pb-L3 and 203Pb-L4 were offset somewhat by their relatively high blood activity levels at early time-points compared with 203Pb-L1 and 203Pb-L5. Accordingly, 212Pb-L2 was selected for the proof-of-concept α-TRT.
212Pb-L2 significantly delayed growth of the PSMA(+) tumors. The median survival of animals receiving the agent was comparable to that for 211At-6 (15). Significantly, 212Pb-L2 demonstrated an approximately 6-fold lower kidney absorbed dose than our previous short-half-life α-emitting agent, 211At-6 (15). Consequently, the MTD observed for a single administered dose in healthy, immunocompetent CD-1 mice increased from 37 kBq for 211At-6 to 1.5 MBq for 212Pb-L2. An administered dose of 3.7 MBq of 212Pb-L2 showed characteristic features of late radiation nephropathy at 7 mo after treatment. In contrast, a dose of up to 1.5 MBq induced only discrete, nonspecific changes in the kidney while still enabling a measure of tumor growth control. To address the late nephropathy issue, we anticipate that a fractionated dose regimen would be more appropriate for this short-half-life radioligand. Alternatively, a partial kidney-blocking strategy using DCIBzL could be useful for 212Pb-based α-TRT.
212Pb-L2 also proved more effective in treating micrometastases than did β−-emitting 177Lu-PSMA-617 in our PSMA(+) micrometastatic tumor model (15). That finding was most likely due to the superior capability of the high-linear-energy radiation of 212Pb in this type of model and is consistent with previous reports using similar low-molecular-weight peptides and antibodies (40–42). Theoretically, the total energy for each 212Pb disintegration is 6–8 MeV, compared with a mean β-radiation energy of 0.4 MeV for each 177Lu disintegration—a 15-fold difference. The administered dose ratio for 212Pb:177Lu was 1:10, favored for 212Pb-L2. A higher dose may be allowed for 212Pb-L2 when considering a possible loss of activity after α-emission, and the half-life of 212Pb-L2 (0.4 d) is much shorter than that of 177Lu (6.7 d).
The high MTDs projected for humans as compared with other α-emitters can be explained by only 1 α-emission per 212Pb decay, versus 4 for 225Ac and 223Ra; the shorter half-life; and the faster normal-organ biologic clearance. This explanation is also consistent with the murine MTD of 1.85–3.7 MBq, which is in the range of the fractional 223Ra activity administered to humans. Nevertheless, such projections are highly uncertain, and any human application should be performed in increasing increments from values well below the calculated MTD.
Although 212Pb-L2 displayed a short circulation time within blood and low renal uptake relative to other compounds in this series, long-term renal toxicity for the higher doses is a concern that is admittedly related to the physical characteristics of 212Pb decay rather than to the in vivo stability of the 212Pb-TCMC or 212Pb-DOTA interactions. β-particle emission of 212Pb is associated with γ-ray emission pathways that compete with internal conversion with 30% efficiency. Internal conversion decay destabilizes the resulting bismuth complexes, promoting rupture of the bichelator chemical bonds and resulting in release of 212Bi, which is known to accumulate mainly in the renal proximal tubules (11). Therefore, the safety and efficacy of these new 212Pb-based compounds could be further optimized with ex vivo murine studies and small-scale (macro-to-micro) dosimetry, with biokinetic modeling applied to clinical scenarios (43). An additional clinical consideration is that the 212Pb decay chain includes several high-energy photons (1.6 MeV [1.5%]; 727 keV [6.6%]; 785 keV [1.1%]; 861 keV [4.5%]) that would possibly require hospitalization for radiation safety reasons.
CONCLUSION
We have evaluated in preclinical models the theranostic radionuclide pair 203Pb/212Pb in a focused series of compounds for PSMA-based α-TRT. 212Pb-L2 demonstrated PSMA-specific tumor growth delay in both large and micrometastatic tumor models. The kidney was identified as the dose-limiting organ from the long-term toxicity study. Future studies are directed toward evaluation of the safety and efficacy of 212Pb-L3–212Pb-L5 studied at the MTD, in comparison with the corresponding long-lived α-emitters 225Ac-L3–225Ac-L5, as we work toward clinical translation with our optimized, lead compound.
DISCLOSURE
This work was supported by the Patrick C. Walsh Prostate Cancer Research Fund, P30CA006973, CA151838, CA134675, CA184228, CA183031, EB024495, and the Commonwealth Foundation. Drs. Banerjee, Minn, Mease, and Pomper are coinventors on one or more U.S. patents covering compounds discussed in this article and as such are entitled to a portion of any licensing fees and royalties generated by this technology. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict-of-interest policies. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Do PSMA-targeted α-emitting 212Pb-labeled low-molecular-weight radioligands display the required safety and efficacy in preclinical studies for potential clinical translation as an alternative to long-half-life 225Ac-based therapy with reduced off-target effects?
PERTINENT FINDINGS: Our report has addressed the question by examining strategic preclinical research. We generated 5 212Pb-labeled PSMA-targeted compounds and chose a lead among them, 212Pb-L2, which demonstrated tumor growth control in both flank and micrometastatic models with the lowest off-target effects in this series. We determined the MTD of 212Pb-L2 in healthy, immunocompetent mice to be 1.5 MBq to inform a future phase I clinical trial.
IMPLICATIONS FOR PATIENT CARE: 212Pb-labeled α-emitters expand the possibilities for PSMA-targeted management of PC. The promising preclinical data, availability of commercial generators to produce 212Pb, and relatively straightforward dosimetry, compared with other α-emitters, make Pb-labeled theranostic agents, including 203/212Pb-L2, attractive alternatives to existing TRT for PC.
Footnotes
Published online Jun. 28, 2019.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication April 4, 2019.
- Accepted for publication June 18, 2019.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Dosimetry in Radiopharmaceutical Therapy
- An Improved 211At-Labeled Agent for PSMA-Targeted {alpha}-Therapy
- Harnessing {alpha}-Emitting Radionuclides for Therapy: Radiolabeling Method Review
- Preclinical Evaluation of 213Bi- and 225Ac-Labeled Low-Molecular-Weight Compounds for Radiopharmaceutical Therapy of Prostate Cancer
- Preclinical Applications of Multi-Platform Imaging in Animal Models of Cancer