


Abstract
The adenosine triphosphate-binding cassette transporters P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) are 2 efflux transporters at the blood–brain barrier (BBB) that effectively restrict brain distribution of dual ABCB1/ABCG2 substrate drugs, such as tyrosine kinase inhibitors. Pharmacologic inhibition of ABCB1/ABCG2 may improve the efficacy of dual-substrate drugs for treatment of brain tumors, but no marketed ABCB1/ABCG2 inhibitors are currently available. In the present study, we examined the potential of supratherapeutic-dose oral erlotinib to inhibit ABCB1/ABCG2 activity at the human BBB. Methods: Healthy men underwent 2 consecutive PET scans with 11C-erlotinib: a baseline scan and a second scan either with concurrent intravenous infusion of the ABCB1 inhibitor tariquidar (3.75 mg/min, n = 5) or after oral intake of single ascending doses of erlotinib (300 mg, n = 7; 650 mg, n = 8; or 1,000 mg, n = 2). Results: Although tariquidar administration had no effect on 11C-erlotinib brain distribution, oral erlotinib led, at the 650-mg dose, to significant increases in volume of distribution (23% ± 13%, P = 0.008), influx rate constant of radioactivity from plasma into brain (58% ± 26%, P = 0.008), and area under the brain time–activity curve (78% ± 17%, P = 0.008), presumably because of combined partial saturation of ABCG2 and ABCB1 activity. Inclusion of further subjects into the 1,000-mg dose group was precluded by adverse skin events (rash). Conclusion: Supratherapeutic-dose erlotinib may be used to enhance brain delivery of ABCB1/ABCG2 substrate anticancer drugs, but its clinical applicability for continuous ABCB1/ABCG2 inhibition at the BBB may be limited by safety concerns.
The blood–brain barrier (BBB) constitutes a major obstacle to the pharmacologic treatment of primary and secondary brain tumors (1,2). The adenosine triphosphate–binding cassette transporters P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) were shown to work together in limiting the passage of many anticancer drugs from blood across the BBB into brain parenchyma (3,4). Many anticancer drugs, including virtually all currently available members of the important class of tyrosine kinase inhibitors, are dual ABCB1/ABCG2 substrates, leading to low BBB penetration and limited efficacy against brain tumors and metastases (3,4). There is evidence that newly formed blood vessels supplying brain tumors are leaky, leading to higher drug concentrations in tumors than in healthy brain tissue (1,2). Nonetheless, it has been shown that disruption of the tumor BBB is restricted mostly to the central necrotic parts of brain tumors, whereas tumor cells in the invasive margin remain protected by an intact BBB (1,2). Moreover, despite a disruption of the tumor BBB, ABCB1 and ABCG2 may remain sufficiently active to effectively limit brain uptake of many substrate drugs (5,6).
The pharmacologic treatment of brain tumors may greatly benefit from measures to inhibit ABCB1/ABCG2 transport activity. Some multidrug-resistance reversal agents have been repurposed to inhibit ABCB1 and ABCG2 at the human BBB. Promising results have been obtained with tariquidar, which led to up to 5-fold increases in brain distribution of the ABCB1 substrates (R)-11C-verapamil and 11C-N-desmethyl-loperamide in healthy human volunteers (7,8). However, at clinically feasible doses, tariquidar inhibits only ABCB1, so that brain uptake of dual ABCB1/ABCG2 substrates is only marginally increased (9). The dual ABCB1/ABCG2 inhibitor elacridar has shown promise in increasing brain distribution of dual ABCB1/ABCG2 substrates in preclinical species (10,11). However, elacridar was ineffective in humans because of its low oral bioavailability, leading to plasma concentrations far below those required to inhibit ABCB1/ABCG2 at the human BBB (12). In addition, elacridar is a more potent inhibitor of ABCB1 than of ABCG2 (12), whereas ABCG2 is the predominant transporter at the human BBB, with approximately 1.3-fold higher expression levels than those of ABCB1 (13). Both elacridar and tariquidar are nonmarketed, experimental compounds that can effectively inhibit BBB transporters only at high intravenous doses, hindering their broader clinical use to enhance brain delivery of anticancer drugs. There is consequently an unmet need for a clinically available, marketed oral ABCB1/ABCG2 inhibitor. Numerous in vitro studies have investigated the potential of marketed tyrosine kinase inhibitors to overcome ABCB1/ABCG2-mediated multidrug resistance of tumor cells (14–16). However, to date no clinical data are available to prove that ABCB1/ABCG2 inhibition can be achieved at the human BBB with these drugs.
Erlotinib is a first-generation tyrosine kinase inhibitor that targets the epidermal growth factor receptor and is approved for treatment of locally advanced or metastatic non–small cell lung cancer and metastatic pancreatic cancer. Erlotinib is a substrate of ABCB1 and ABCG2 with limited brain distribution (17) and was also shown to inhibit ABCG2 and ABCB1 at higher concentrations (14,18). Our recent preclinical data indicated that high-dose intravenous erlotinib administration leads to partial saturation of ABCG2 and ABCB1 transport activity at the BBB, resulting in a nonlinear dose-dependent increase in erlotinib brain distribution (10,11). These data are of considerable interest for a clinical translation, as pulsatile (weekly) supratherapeutic-dose oral erlotinib treatment (up to dosages of 2,550 mg) was shown to be relatively well tolerated in cancer patients and to lead to improved response rates of non–small cell lung cancer brain metastases as compared with standard clinical dosing (19).
The aim of this study was to provide proof of the concept that erlotinib can achieve ABCB1/ABCG2 inhibition at the human BBB. We used PET imaging with 11C-erlotinib in healthy subjects to study the effect of single ascending oral doses of erlotinib on 11C-erlotinib brain penetration.
MATERIALS AND METHODS
The study was registered with European Union Drug Regulating Authorities Clinical Trials (EudraCT) number 2015-001593-18, approved by the local Ethics Committee, and conducted in accordance with the Declaration of Helsinki and its amendments. Written consent was obtained from all subjects. Only medication-free male volunteers, confirmed as healthy by medical history, routine physical examination, and blood and urine laboratory examination, were eligible for the study.
Genotyping
Venous blood was drawn during the screening examination from all study participants for assessment of common ABCG2 and ABCB1 single-nucleotide polymorphisms using previously described procedures (9). For ABCG2 the c.421C>A variant (rs2231142), and for ABCB1 the c.1236C>T (rs1128503), c.2677G>T (rs2032582), and c.3435C>T (rs1045642) single-nucleotide polymorphisms, were determined.
Imaging and Blood Sampling Procedures
Subjects underwent MRI of the brain (T1-weighted Magnetom Skyra 3.0-T MRI; Siemens Medical Solutions). Two dynamic 60-min 11C-erlotinib PET scans were conducted with a frame sequence of 1 × 15, 3 × 5, 3 × 10, 2 × 30, 3 × 60, 2 × 150, 2 × 300, and 4 × 600 s, either on the same day for the tariquidar group or on 2 separate days for the erlotinib group, on an Advance scanner (GE Healthcare). 11C-erlotinib, which was synthesized as described elsewhere (20), was injected as an intravenous bolus over 20 s (injected radioactivity, 369 ± 22 MBq for scan 1 and 372 ± 17 MBq for scan 2, corresponding to 3.5 ± 2.7 μg of unlabeled erlotinib for scan 1 and 4.1 ± 3.9 μg of unlabeled erlotinib for scan 2). In parallel with PET imaging, serial blood samples were drawn from the radial artery. In the tariquidar group (n = 5), the second PET scan was performed with a concurrent intravenous infusion of tariquidar (AzaTrius Pharmaceuticals) (3.75 mg/min), which had been initiated 1 h before the start of the PET scan, as described elsewhere (9). In the erlotinib group (n = 17), the second PET scan was performed at approximately 3 h after oral intake of 300, 650, or 1,000 mg of erlotinib (Tarceva; Roche Pharma [50- and 150-mg tablets]) after a standardized high-fat meal. Ten blood samples were collected at baseline and hourly for 8 h and at approximately 21 h after erlotinib intake. Plasma obtained from centrifuged blood samples was kept at −80°C until analysis of erlotinib concentrations, which was performed as previously described (21).
Blood and Metabolite Analysis
Radioactivity concentrations in blood and plasma aliquots were measured in a γ-counter, which was cross-calibrated with the PET camera. Plasma samples collected at 3.5, 5, 10, 20, 30, 40, and 60 min after radiotracer injection were analyzed for radiolabeled metabolites of 11C-erlotinib using a previously described solid-phase extraction procedure (21,22). In 6 subjects, metabolite data were available for only the 20- and 40-min time points. Because of the low percentage of radiolabeled metabolites in plasma (<10%), total radioactivity counts were considered for construction of an arterial input function, similar to a previous study in which 11C-erlotinib PET data were modeled in human lung tumors (23). Plasma protein binding of 11C-erlotinib was determined by incubating plasma samples obtained before each PET scan with 11C-erlotinib followed by ultrafiltration as described previously (21).
PET Data Analysis and Modeling
A whole-brain gray-matter region of interest was defined on individual MR images acquired for all study participants using the Hammersmith n30r83 3-dimensional maximum-probability atlas of the human brain (24) and coregistered to average PET images. In addition, the pituitary gland was manually outlined in 8 subjects. Time–activity curves were extracted from the dynamic PET images. Compartment models (1-tissue–2-rate-constant and 2-tissue–4-rate-constant) were fitted to the brain and arterial plasma time–activity curves from 0 to 60 min after radiotracer injection as described previously (25). The fractional arterial blood volume in brain was included as a fitting parameter. The primary outcome parameters of 11C-erlotinib brain distribution were total volume of distribution (VT), which equals the brain-to-plasma concentration ratio at steady state, and the influx rate constant of radioactivity from plasma into brain (K1). To obtain a model-independent estimate of VT, Logan graphical analysis was performed (26). As a measure of erlotinib brain exposure, the area under the brain time–activity curves was calculated as an exploratory outcome parameter.
Pharmacokinetic and Statistical Analysis
Pharmacokinetic parameters of unlabeled erlotinib in plasma were determined with the Kinetica 2000 software package, version 3.0 (InnaPhase). All data are given as mean ± SD. Differences in outcome parameters between scans 1 and 2 were tested using the Wilcoxon matched-pairs signed-rank test (Prism 7.04 software; GraphPad Software). To assess correlations, the Spearman correlation coefficient (r) was calculated. The level of statistical significance was set to a P value of less than 0.05.
RESULTS
Study participants underwent 2 PET scans with 11C-erlotinib: a first baseline scan in which a microdose of erlotinib was administered, and a second scan that was performed either during intravenous infusion of tariquidar (3.75 mg/min) or at 3 h after the intake of single ascending oral doses of erlotinib (300, 650, or 1,000 mg). Our initial study protocol contained only the 300-mg erlotinib dose but was later amended to include the 650- and 1,000-mg doses. Supplemental Table 1 summarizes all adverse events that occurred during the study (supplemental materials are available at http://jnm.snmjournals.org). Erlotinib intake led to skin rash in 9 of 17 subjects. These adverse events were classified as mild or moderate, were resolved within 5 wk, and occurred mainly in the higher-dose groups. Inclusion of further subjects into the 1,000-mg dose group was stopped for safety reasons.
Most radioactivity in plasma comprised unchanged 11C-erlotinib in both scans. Erlotinib intake slightly but significantly increased the percentage of unchanged 11C-erlotinib in plasma (20 min: scan 1, 97.3% ± 1.0%; scan 2 [mean of all doses], 98.3% ± 0.5% [P = 0.001]; 40 min: scan 1, 95.6% ± 2.1%; scan 2 [mean of all doses], 97.8% ± 0.8% [P = 0.001]; Supplemental Table 2). The percentage of plasma protein binding of 11C-erlotinib did not significantly differ between the scans without and with erlotinib intake (scan 1, 94.7% ± 2.4%; scan 2 [mean of all doses], 94.1% ± 2.7%; n = 11). The plasma-to-blood radioactivity ratio (mean of 7 blood samples taken between 3.5 and 60 min after radiotracer injection) did not significantly differ between scans 1 and 2 (scan 1, 0.89 ± 0.10; scan 2, 0.90 ± 0.05).
Concentrations of unlabeled erlotinib in plasma were measured in all study subjects. Maximum plasma concentration and area under the curve increased less than dose proportionally with increasing erlotinib doses (Supplemental Table 3). Across all dose groups, the mean erlotinib plasma concentration at the time of the PET scan (average of values at 3 and 4 h after oral intake) was 5.8 ± 1.9 μmol/L (range, 2.6–9.2 μmol/L), corresponding to an unbound erlotinib concentration of 0.35 ± 0.16 μmol/L (range, 0.09–0.73 μmol/L).
As a parameter of 11C-erlotinib brain distribution, we determined VT using either a 2-tissue–4-rate-constant model or Logan graphical analysis, which provided similar VT estimates. The 1-tissue–2-rate-constant model provided poorer fits of the PET data than the 2-tissue–4-rate-constant model and was therefore not further considered (Supplemental Fig. 1). Correction of plasma radioactivity for the low percentages of radiolabeled metabolites yielded parameter estimates similar to a non–metabolite-corrected plasma input function (Supplemental Fig. 2). For the tariquidar group, VT did not significantly differ between scans 1 and 2 (Supplemental Table 4; Fig. 1). Subjects in the tariquidar group were grouped according to their c.421C>A genotype. In scan 2, VT calculated with Logan graphical analysis (VT,Logan) was increased by 11% ± 1% in the c.421C>C group (n = 4) and by 38% in the c.421C>A subject (Supplemental Table 4, Fig. 1).

Whole-brain gray matter VT,Logan in individual subjects for baseline scan and scan during intravenous infusion of tariquidar. Subjects with c.421C>C (n = 4) and c.421C>A (n = 1) genotype are shown as black circles and red triangles, respectively. P value in parentheses refers to c.421C>C subjects only.
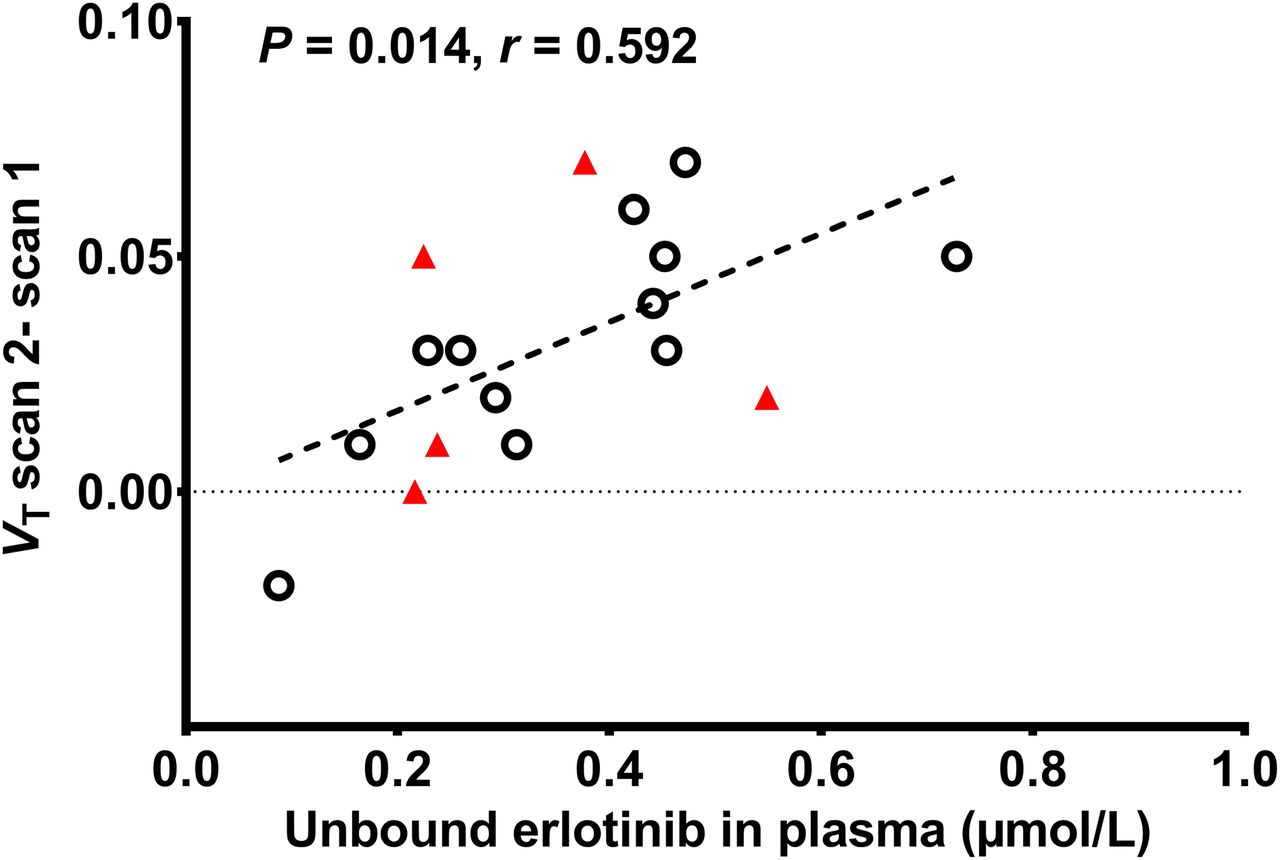
In Figure 2 representative transaxial PET images, and in Figure 3 mean time–activity curves in plasma and brain, are shown for the baseline scan and for the scans after intake of different erlotinib doses. Plasma concentrations of total radioactivity rose significantly after erlotinib intake as compared with baseline scans (area under the plasma time–activity curve [SUV × min]: scan 1, 59 ± 13, and scan 2 (mean of all dose groups), 90 ± 19; percentage change in scan 2, +52% ± 17%; P = 0.0003). Modeling outcome parameters for all erlotinib dose groups are summarized in Table 1, and the VT,Logan values for the individual subjects of the different dose groups in scans 1 and 2 are shown in Figure 4. For the 300-mg dose group, none of the outcome parameters except for area under the brain time–activity curve (+67% ± 46%, P = 0.016) were significantly changed from the baseline group. For the 650-mg dose group, VT, K1, and area under the brain time–activity curve were significantly increased by 23% ± 13% (P = 0.008), 58% ± 26% (P = 0.008), and 78% ± 17% (P = 0.008), respectively, as compared with individual baseline values. For the 1,000-mg dose group, VT, K1, and area under the brain time–activity curve were increased by 27% ± 4%, 83% ± 24%, and 94% ± 23%, respectively, but statistical testing was not possible because the group was too small (Table 1). In Supplemental Table 5, the ABCG2 and ABCB1 genotypes of individual subjects are stated and related to the individual percentage changes in VT,Logan in scan 2. There was a significant positive correlation (P = 0.014, r = 0.592) between the increase in whole-brain VT,Logan in scan 2 and unbound erlotinib plasma concentrations (Fig. 5).

Representative transaxial PET summation images (15–60 min) for scans at baseline (left scan of each pair) and after oral intake of 300, 650 or 1,000 mg of erlotinib (right scan of each pair).

Mean time–activity curves (SUV ± SD) in arterial plasma (A) and whole-brain gray matter (B) for scans at baseline and after oral intake of 300 mg (n = 7), 650 mg (n = 8), or 1,000 mg (n = 2) of erlotinib. Insets show early data after radiotracer injection.
11C-Erlotinib Outcome Parameters for Baseline Scan and Scan after Oral Dosing of Erlotinib

Whole-brain gray matter VT,Logan in individual subjects for scans at baseline and after oral intake of 300, 650, or 1,000 mg of erlotinib. Subjects with c.421C>C (n = 12) and c.421C>A (n = 5) genotype are shown as black circles and red triangles, respectively. For 1,000-mg group, statistical testing was not feasible because group was too small.

Relationship between scan 2 increase in 11C-erlotinib whole-brain gray matter VT,Logan (VT,Logan scan 2 − scan 1) and unbound erlotinib plasma concentration (μmol/L) during PET scan (average of values determined at 3 and 4 h after erlotinib intake) and corresponding linear fit (r = Spearman correlation coefficient). Subjects with c.421C>C (n = 12) and c.421C>A (n = 5) genotype are shown as black circles and red triangles, respectively.
The pituitary gland, a brain region that has fenestrated capillaries and thus is not protected by the BBB, was also outlined on the PET images (Supplemental Fig. 3). VT,Logan in the pituitary gland did not significantly differ between baseline scans and scans after oral intake of erlotinib (scan 1, 0.79 ± 0.11; scan 2, 0.74 ± 0.17). Whole-brain VT,Logan was 5.5 ± 0.9-fold and 4.4 ± 1.0-fold lower than pituitary gland VT,Logan in scans 1 and 2, respectively.
DISCUSSION
ABCB1 and ABCG2 work in concert at the human BBB to limit brain distribution of dual ABCB1/ABCG2 substrates (3,4). Only when both ABCB1 and ABCG2 are inhibited can significant increases in brain distribution of dual-substrate drugs be achieved. We found in previous studies that intravenous tariquidar infusion led to only small increases in brain distribution of the dual ABCB1/ABCG2 substrates 11C-elacridar and 11C-tariquidar, whereas substantial increases in brain distribution of the ABCB1 substrate (R)-11C-verapamil were achieved (8,9). In the present study, we used 11C-erlotinib as a clinically relevant dual ABCB1/ABCG2 substrate. Enhanced brain distribution of erlotinib would be highly beneficial for the treatment or prevention of brain metastases, which frequently occur in non–small cell lung cancer patients but show a poor response to systemic erlotinib treatment. At the same time, erlotinib can be considered a model tyrosine kinase inhibitor, as most currently marketed tyrosine kinase inhibitors are dual ABCB1/ABCG2 substrates with limited BBB penetration (3,4).
In line with our previous findings, we showed that the same tariquidar infusion protocol as used in our previous studies (8,9) led to negligible increases in brain distribution of 11C-erlotinib (Fig. 1). The only exception was 1 heterozygous carrier of the ABCG2 single-nucleotide polymorphism c.421C>A, who showed a 38% increase in 11C-erlotinib VT. This finding is in accordance with our previous observation that single-nucleotide polymorphism carriers have reduced ABCG2 transport activity at the BBB, leading to higher increases in brain distribution of dual ABCB1/ABCG2 substrates after ABCB1 inhibition with tariquidar (9).
In nonhuman primates, as a relevant model of the human BBB, continuous intravenous infusion of erlotinib (10 mg/kg/h) led to a 70% increase in brain VT of 11C-erlotinib as compared with PET scans without erlotinib infusion (11). This increase in brain distribution of 11C-erlotinib was most likely caused by partial saturation of ABCG2- and ABCB1-mediated efflux transport of erlotinib at the BBB. In line with the nonhuman primate data, 11C-erlotinib VT was increased by 27% in the highest erlotinib dose group (1,000 mg) (Table 1). 11C-erlotinib brain exposure (area under the brain time–activity curve), which may be more relevant for therapeutic effect than VT, was increased by 94%. The mean erlotinib plasma concentration at the time of the PET scan was 5.8 μmol/L, whereas in the nonhuman primate study the erlotinib plasma concentration was approximately 16 μmol/L. In vitro studies have shown that erlotinib is a more potent inhibitor of ABCG2 than of ABCB1, with half-maximum inhibitory concentrations of 0.13 and 2.0 μmol/L, respectively (18). Erlotinib is approximately 95% bound to plasma proteins, and the percentage of plasma protein binding of 11C-erlotinib did not significantly differ between the baseline scan and the scan after erlotinib administration. This finding supports the presumption that the increase in 11C-erlotinib brain exposure after erlotinib administration was not due to changes in the plasma-unbound fraction of 11C-erlotinib. Theoretically, erlotinib administration could have increased cerebral blood flow, which may have contributed to the observed increase in 11C-erlotinib brain distribution. However, this possibility could not be assessed in our study because we did not measure cerebral blood flow (i.e., with 15O-H2O). Unbound erlotinib plasma concentrations at the time of the PET scan ranged from 0.09 to 0.73 μmol/L, which was similar to or above the in vitro half-maximum inhibitory concentration of erlotinib for ABCG2 inhibition (0.13 μmol/L) and should therefore have led to substantial inhibition of ABCG2 transport activity at the BBB. In addition, it can be expected that partial inhibition of ABCB1 transport activity occurred. The higher ABCG2 than ABCB1 inhibitory potency of erlotinib is advantageous, as ABCG2 is the predominant efflux transporter at the human BBB (13). The largest individual increase in 11C-erlotinib brain VT observed in our study was 50% (Supplemental Table 5). To estimate maximum possible brain uptake of 11C-erlotinib in the absence of ABCB1/ABCG2 activity, we assessed 11C-erlotinib distribution to the pituitary gland, a brain region that is not protected by the BBB because it possesses fenestrated capillaries (Supplemental Fig. 3) (9). The VT values estimated for the pituitary gland were approximately 5-fold higher than those for whole-brain gray matter. This result was in reasonably good agreement with our previous nonhuman primate study, in which a maximum 3.5-fold increase in 11C-erlotinib brain distribution was achieved after elacridar infusion (12 mg/kg/h), presumably because of complete ABCB1/ABCG2 inhibition (11). This indicated that the increases in 11C-erlotinib brain distribution measured in our PET study were situated at the lower end of the concentration-effect curve. Consequently, higher plasma concentrations of erlotinib will be needed to achieve higher increases in erlotinib brain exposure. We used oral doses of up to 1,000 mg, which was below the maximum oral dose that had been used in cancer patients receiving pulsatile (weekly), supratherapeutic-dose erlotinib (2,550 mg) (19). Administration of higher erlotinib doses to the healthy volunteers included in our study was precluded by the known adverse skin events of erlotinib. Erlotinib plasma pharmacokinetics were quite variable, as was in line with previous reports (27). Moreover, erlotinib plasma exposure did not increase proportionally with administered erlotinib doses; that is, area under the curve and maximum plasma concentration increases were lower than expected with increasing doses (Supplemental Table 3). The exact reasons for this observation are unknown but may be related to solubility-rate–limited absorption, leading to lower oral bioavailability of erlotinib at higher doses. To overcome the variability in erlotinib plasma pharmacokinetics and achieve higher erlotinib plasma exposure, intravenous administration of erlotinib may be an option (28). An intravenous administration mode, however, may not be feasible in routine patient care and may be applicable only as a research tool in a controlled experimental setting.
The observed moderate increases in 11C-erlotinib brain distribution after supratherapeutic-dose erlotinib administration contrasted with results from a previous study, in which administration of oral elacridar failed to increase brain uptake of 11C-erlotinib in cancer patients (12). This finding was attributed to elacridar’s having a low oral bioavailability, leading to plasma concentrations insufficiently high to effectively inhibit ABCG2 at the human BBB. Elacridar plasma concentrations in the study by Verheijen et al. were in the range of 271–619 ng/mL (12), whereas Tournier et al. showed in their nonhuman primate study, in which elacridar was dosed intravenously, that elacridar plasma concentrations associated with ABCB1/ABCG2 inhibition at the nonhuman primate BBB were in the range of 10,000 ng/mL (11). An intravenous formulation of elacridar for human use is not currently available, and its development is severely hampered by the low aqueous solubility of elacridar (29). Another study, by Saleem et al., found no effect of oral therapeutic-dose lapatinib on ABCB1/ABCG2-mediated efflux transport of 11C-lapatinib at the BBB of breast cancer patients with brain metastases (30), suggesting that lapatinib is a less potent ABCB1/ABCG2 inhibitor than erlotinib.
Because of the higher ABCG2 than ABCB1 inhibitory potency of erlotinib, higher increases in brain exposure may be achieved for preferential ABCG2 substrates with low affinity for ABCB1 (e.g., sorafenib, momelotinib, or temozolomide) (3,31), which needs to be confirmed in future studies. The efficacy of supratherapeutic-dose erlotinib as an enhancer of brain distribution of ABCG2/ABCB1 substrate drugs may differ in brain tumor patients with a partly disrupted tumor BBB. However, because of safety concerns, the use of supratherapeutic-dose erlotinib will most likely be limited to a once-weekly dosing regimen, which will not allow for continuous ABCB1/ABCG2 inhibition at the BBB.
We observed a significantly increased plasma exposure to radioactivity after oral intake of erlotinib, as compared with the baseline scan. The mechanism behind this phenomenon has been elucidated before and was shown to be caused by a dose-dependent decrease in liver distribution of 11C-erlotinib, most likely because of saturation of the activity of organic anion-transporting polypeptide 2B1 (SLCO2B1) in hepatocytes (21). Therefore, erlotinib may lead to changes in the peripheral pharmacokinetics of other ABCB1/ABCG2 substrate drugs also, and this possibility needs to be considered when erlotinib is used as an ABCG2/ABCB1 inhibitor.
We genotyped all our study participants for common ABCG2 and ABCB1 single-nucleotide polymorphisms (Supplemental Table 5). We failed to detect an effect of these single-nucleotide polymorphisms on the percentage increase in 11C-erlotinib brain distribution after erlotinib administration (Figs. 4 and 5). This finding may be due to the relatively small sample size of our study or to the lack of an effect of ABCB1 single-nucleotide polymorphisms on ABCB1 activity at the human BBB (32).
CONCLUSION
We provided proof of the concept that supratherapeutic-dose erlotinib administration leads to a nonlinear increase in 11C-erlotinib brain distribution, presumably because of partial saturation of ABCG2/ABCB1 activity at the BBB, whereas ABCB1 inhibition alone with tariquidar failed to increase brain uptake of 11C-erlotinib. This result underlines the relatively greater importance of ABCG2 than of ABCB1 in limiting brain distribution of dual ABCB1/ABCG2 substrates at the human BBB. Additionally, it suggests that supratherapeutic-dose erlotinib may be used to enhance brain delivery of other anticancer drugs, in particular those preferentially transported by ABCG2. However, a clinical application for continuous ABCB1/ABCG2 inhibition at the BBB may be hampered by safety concerns.
DISCLOSURE
This work was supported by the Austrian Science Fund (FWF) (grants F 3513-B20 and KLI 480-B30) and by the Lower Austria Corporation for Research and Education (NFB) (grant LS15-003). No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Harald Ibeschitz, Ingrid Leitinger, and the other staff members of the PET Center at the Division of Nuclear Medicine for cooperating with this study; Johann Stanek (Department of Clinical Pharmacology, Medical University of Vienna, Austria, and the Center for Health and Bioresources, Austrian Institute of Technology GmbH, Seibersdorf, Austria) for technical support with radiosynthesis and metabolite analysis; and Elisabeth Ponweiser and Manuela Repl (MedUni Wien Biobank, Medical University of Vienna) for technical assistance.
Footnotes
Published online Sep. 20, 2018.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication June 20, 2018.
- Accepted for publication September 10, 2018.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- ABCB1 and ABCG2 Regulation at the Blood-Brain Barrier: Potential New Targets to Improve Brain Drug Delivery
- ABCG2- and ABCB1 Inhibition Using Supratherapeutic Doses of Erlotinib: Clinical Implications in the Treatment of Central Nervous System Metastases
- Pharmacokinetic Imaging with Radiolabeled Molecularly Targeted Anticancer Drugs