


Abstract
The purinergic receptor subtype 7 (P2X7R) represents a novel molecular target for imaging neuroinflammation via PET. GSK1482160, a potent P2X7R antagonist, has high receptor affinity, high blood–brain barrier penetration, and the ability to be radiolabeled with 11C. We report the initial physical and biologic characterization of this novel ligand. Methods: 11C-GSK1482160 was synthesized according to published methods. Cell density studies were performed on human embryonic kidney cell lines expressing human P2X7R (HEK293-hP2X7R) and underwent Western blotting, an immunofluorescence assay, and radioimmunohistochemistry analysis using P2X7R polyclonal antibodies. Receptor density and binding potential were determined by saturation and association–disassociation kinetics, respectively. Peak immune response to lipopolysaccharide treatment in mice was determined in time course studies and analyzed via Iba1 and P2X7R Western blotting and Iba1 immunohistochemistry. Whole-animal biodistribution studies were performed on saline- or lipopolysaccharide-treated mice at 15, 30, and 60 min after radiotracer administration. Dynamic in vivo PET/CT was performed on the mice at 72 h after administration of saline, lipopolysaccharide, or lipopolysaccharide + blocking, and 2-compartment, 5-parameter tracer kinetic modeling of brain regions was performed. Results: P2X7R changed linearly with concentrations or cell numbers. For high-specific-activity 11C-GSK1482160, receptor density and Kd were 1.15 ± 0.12 nM and 3.03 ± 0.10 pmol/mg, respectively, in HEK293-hP2X7R membranes. Association constant kon, dissociation constant koff, and binding potential (kon/koff) in HEK293-hP2X7R cells were 0.2312 ± 0.01542 min−1⋅nM−1, 0.2547 ± 0.0155 min−1, and 1.0277 ± 0.207, respectively. Whole-brain Iba1 expression in lipopolysaccharide-treated mice peaked by 72 h on immunohistochemistry, and Western blot analysis of P2X7R for saline- and lipopolysaccharide-treated brain sections showed a respective 1.8- and 1.7-fold increase in signal enhancement at 72 h. Biodistribution of 11C-GSK1482160 in saline- and lipopolysaccharide-treated mice at 72 h was statistically significant across all tissues studied. In vivo dynamic 11C-GSK1482160 PET/CT of mice at 72 h after administration of saline, lipopolysaccharide, or lipopolysaccharide + blocking showed a 3.2-fold increase and 97% blocking by 30 min. The total distribution volumes for multiple cortical regions and the hippocampus showed statistically significant increases and were blocked by an excess of authentic standard GSK1482160. Conclusion: The current study provides compelling data that support the suitability of 11C-GSK1482160 as a radioligand targeting P2X7R, a biomarker of neuroinflammation.
One essential step in the progression of brain diseases is neuroinflammation (1), for which proinflammatory cytokines play a central role (2). Recent work has shown that the pathogenesis of neuroinflammation is mediated in part by the release of adenosine or uridine derivatives from the damaged site, involving a family of ionotropic purinergic receptors (P2XRs) with elevated expression (3). Purinergic receptors are adenosine-triphosphate–gated nonselective ion channels that are expressed on the cell surface membranes of hematopoietic cells and include, but are not limited to, macrophages, monocytes, and microglia (4,5). In particular, receptor subtype 7 (P2X7R) has been implicated in microglial proliferation, phagocytosis of the injured site (6,7), and release of proinflammatory cytokines (4,8–10). This mechanism is common among a wide array of neurodegenerative inflammatory diseases, which include Alzheimer disease, Parkinson disease, Huntington disease, frontotemporal dementia, atrophic lateral sclerosis, multiple sclerosis, and traumatic brain injury (11–13).
P2X7R represents a novel molecular target for imaging neuroinflammation via PET. GSK1482160, a potent P2X7R antagonist, has recently been labeled with 11C at our institution (14) as a potential biomarker of neuroinflammation. This article reports the initial characterization of this novel radioligand via in vitro and in vivo preclinical studies.
MATERIALS AND METHODS
Radiosynthesis
11C-GSK1482160 was synthesized according to methods described by Gao et al. (14). The radiopharmaceutical was obtained with high radiochemical purity (93% ± 3%), chemical purity (>95%), and specific activity (571.23 ± 130.8 GBq/μmol), which are consistent with prior reports from our institution (15) and others (16) where similar synthetic N-11C methylation schemes of the amide precursors have been used.
Cell Culture and Membrane Preparation
HEK293 cells expressing human recombinant P2X7 receptor (hP2X7R) or human P2X4 receptor were obtained from B’SYS GmbH and cultured according to the supplier’s procedure. Cells were grown to 80% confluency and then rinsed with phosphate-buffered saline (PBS), detached with trypsin, and harvested. Cell pellets were obtained by centrifugation at 200g for 5 min at 4°C. Collected cell pellets were frozen at −80°C until membrane preparation. For the membrane preparation, pellets from 10 T225 flasks were homogenized in 50 mM Tris-HCl, 5 mM ethylenediaminetetraacetic acid, and 140 mM NaCl at pH 7.4 and 4°C. The homogenate was then centrifuged at 48,000g for 20 min at 4°C, gently rinsed with deionized water, and then resuspended in 50 mM Tris-HCl at pH 7.4 and 4°C. This homogenate was then centrifuged as before, with the resulting pellet being homogenized in 50 mM Tris-HCl at pH 7.4. Total protein concentration was determined via the Bradford protein assay (Bio-Rad). Aliquots were stored in cryovials at −80°C until the day of assay.
Validation of Cell Line Receptor Expression
To validate the hP2X7R expression in HEK293 cells, low passage (4–6) cells were plated, cultured, harvested, and pelleted as described above. An aliquot of cells was counted via hemocytometer to obtain the desired density. Cell pellets were resuspended in a cryomold with 1 mL of Tissue-Tek optimal-cutting-temperature compound (Sakura Finetek), serial-diluted with optimal-cutting-temperature compound to yield 500, 250, 125, and 62.5 million cells/mL, and frozen at −20°C. Each mold was cryosectioned at 20 μm, dry-mounted to gelatin-coated slides, and fixed for 15 min in 4% paraformaldehyde in PBS. HEK293-hP2X7R cells were also used for confirmatory assays, including immunofluorescence (n = 3) and radioimmunohistochemistry (n = 3). Blocking was performed for 45 min in PBS with 5% bovine serum albumen and 0.1% Triton-X100 (The Dow Chemical Company), followed by incubation overnight at 4°C in P2X7R primary antibody (APR-008; Alomone) in PBS with 1% bovine serum albumen and 0.1% Triton-X100 at a 1:200 dilution. There were three 5-min washes in PBS with 0.5% Triton-X100. Secondary antibodies were 1:800 IRDye 680LT goat antirabbit IgG (H + L) for the immunofluorescence assay, and 1:1,000 125I-protein A (NEX146025UC; PerkinElmer) for the radioimmunohistochemistry, both incubated 1 h at room temperature and washed as described above. For detection, immunofluorescence assay slides were imaged on an Odyssey near-infrared imager (LiCor) at 680 nm, whereas radioimmunohistochemistry slides were exposed to a TR2025 phosphor screen (GE Healthcare) overnight and read on a Typhoon FLA-7000IP (GE Healthcare). To determine the protein level expression of hP2X7R via Western blotting, a separate aliquot of cells was further serial-diluted, yielding 12,500, 6,250, 3,125, 1,562, and 781 cells per sample. HEK293-hP2X7R cells were digested in radioimmunoprecipitation assay buffer and run on Novex 10% tris-glycine gels (Invitrogen). The resulting transfer on nitrocellulose was probed via Western blotting techniques using the Millipore SNAP i.d. protein detection system with Tris-buffered saline blocking buffer (LiCor). Primary antibody against P2X7R (APR-008; Alomone) was used at 1:150 followed by IRDye 680LT goat antirabbit IgG (H + L) secondary antibody at 1:6,000 (926-68021; LiCor). Western blots (n = 2) were imaged on an Odyssey imager at 680 nm. In all cases, band intensities were quantified with Image Studio Lite (version 5.2.5; LiCor).
11C-Standards
For each experiment, the highest concentration of radioactivity used was diluted twice in assay buffer, followed by eleven 2-fold dilutions. Twenty microliters of each dilution were then spotted onto a GF/B UniFilter-96 plate (Perkin-Elmer) and allowed to air-dry until the end of the experiment. An additional 20 μL of each dilution were added to a scintillation vial containing 7 mL of Optiphase Hisafe 3 (Perkin-Elmer) and counted on an LS6000 scintillation counter (Beckman). Aliquots of the working concentrations of radioactivity used on each day were counted in the same manner.
Radioligand Depletion Experiments
Ligand depletion experiments were performed to measure the bound ligand concentration (n = 3; 4 technical replicates per dose level). Four replicates of membrane preparation (0.004–4.5 mg of protein/mL of assay medium) were incubated with 0.3 nM and 5 nM 11C-GSK1482160 in a final volume of 0.2 mL in assay buffer (50 mM Tris-HCl, pH 7.4, 0.1% bovine serum albumen) for 30 min at 22°C. To measure the total radioactivity added to the experiment, 20 μL of stock ligand (amount added to each experimental well) were spotted onto a GF/B UniFilter plate and then allowed to air-dry. For termination of the binding reaction, the samples were filtered onto GF/B UniFilter plates that had been presoaked in 0.5% polyethyleneimine for 30 min using a UniFilter-96 cell harvester. The plates were washed 5 times with ice-cold saline, dried under a vacuum, and exposed to a TR2025 phosphor screen for 20–60 min. Phosphor screens were then read on a Typhoon FLA-7000IP along with 11C-calibration standards.
Radioligand Association and Dissociation Experiments
For the association study, membrane protein (0.054 mg of protein/mL of assay medium) was incubated with 3 nM 11C-GSK1482160 at 22°C. The binding was started at different time points in reverse order and terminated simultaneously in all samples at time zero by filtration as described above. For the dissociation study, incubation of samples was terminated after 30 min of association by adding 10 μM authentic standard GSK1482160 solution in dimethyl sulfoxide and saline (14). The competition reaction was assessed at different time points by rapid filtration and exposed to a phosphor screen as described in “Radioligand Depletion Experiments” (n = 3; 8 technical replicates).
Radioligand Saturation Experiments
Membrane protein (0.054 mg of protein/mL of assay medium) was incubated with increasing concentrations of 11C-GSK1482160 (0.15–15,000 pM) at 22°C for 20 min. Nonspecific binding was obtained by incubation of adjacent samples with 10 μM authentic standard GSK1482160. The assay was terminated by rapid filtration and exposure to a phosphor screen as described in “Radioligand Depletion Experiments” (n = 3; 4 technical replicates per treatment).
Lipopolysaccharide Time Course
To demonstrate microglial activation and determine the peak of the immune response, a lipopolysaccharide time course study was conducted in which C57BL6 mice (n = 4/treatment/time) were injected intraperitoneally with saline or a 5 mg/kg dose of lipopolysaccharide (O111:B4; Sigma-Aldrich) dissolved in PBS and returned to their home cages for 24, 48, 72, and 96 h (saline and lipopolysaccharide). At the time of harvesting, groups of mice were deeply anesthetized using isoflurane and sacrificed via rapid decapitation. The brains were rapidly removed and rinsed in chilled PBS, and the frontal cortex was dissected, frozen on dry ice, and stored at −80°C until use. For Western blotting, protein was extracted from approximately 100 mg of tissue that was weighed out and placed in a microcentrifuge tube with 100 μL of N-PER neuronal protein extraction reagent (Pierce Biotechnology) on ice. The tissue was homogenized on ice for approximately 20 strokes with a pellet pestle, incubated for 10 min on ice, and centrifuged at 10,000g for 10 min at 4°C. The supernatant was collected into fresh microcentrifuge tubes, protein content was determined via the Bradford assay, and aliquots were stored at −80°C until analysis. For Western blotting, tissue lysates were run on Novex gels (Invitrogen) at 5 μg/lane. For Iba1, 10%–20% Tris-glycine gels were used, with subsequent blotting performed on a SNAP i.d. system, according to the manufacturer’s protocol, at a 1:1,000 dilution in Anti-Iba1 (EPR16588) primary antibody (ab178846; Abcam). For P2X7R, 10% tris-glycine gels and Alomone antibody were used as described in “Cell Line Receptor Expression Validation.” Both blots were probed secondarily with IRDye 680LT goat antirabbit IgG (H + L) secondary antibody at a 1:6,000 dilution (926-68021) before imaging on an Odyssey imager at 680 nm. The blots were stripped and reprobed for β-actin (ab8226; Abcam) at a 1:1,000 dilution in Tris-buffered saline buffer with 1:6,000 IRDye 800CW goat antimouse IgG (H + L) secondary antibody (926-32210l LiCor) before imaging on an Odyssey imager at 800 nm. In all cases, band intensities were quantified with Image Studio Lite. On the basis of the results of the time course, a subset of brains was postfixed overnight in 4% paraformaldehyde and then dehydrated in gradient sucrose solution (20%–30%) at 4°C. At completion, the brains were rinsed in PBS, blotted dry, mounted into cryomolds with Tissue-Tek optimal-cutting-temperature compound, frozen, and stored at −80°C until cryotomy. For immunohistochemistry analysis, the brains from mice treated with saline or 72 h after lipopolysaccharide administration were frozen in optimal-cutting-temperature compound and cut into 50-μm-thick slices. Immunohistochemistry was performed on floating sections after a 30-min treatment in 0.3% H2O2 in methanol to remove endogenous peroxides. Primary incubation was performed overnight at 4°C in 5% nonfat dry milk in 50 mM Tris + 1.5% NaCl and 0.1% Triton X-100, pH 7.2. Iba1 antibody (19-19741; Wako) was used at a 1:500 dilution. There was a single 5-min wash in 50 mM Tris + 1.5% NaCl and 0.1% Triton X-100, pH 7.2. A Vectastain Elite avidin–biotin complex horseradish peroxidase kit (catalog no. PK-6101 [Vector Labs]; peroxidase, rabbit IgG) was used for the secondary and avidin–biotin steps, with the secondary antibody solution being diluted, per manufacturer protocol, in 5% nonfat dry milk in 50 mM Tris + 1.5% NaCl and 0.1% Triton X-100 buffer, pH 7.2. Colorimetric development was via a 3,3-diaminobenzidine horseradish peroxidase kit (SK-4100; Vector Labs), and after rinses in deionized water, sections were dry-mounted on Superfrost Plus (model 12-550-15; Fisher) charged glass slides, cleared through a progression of increasing ethanol solutions (80%–100%) followed by immersion in xylene, cover-slipped, and imaged on an Aperio scanner (Leica Biosystems). Images were analyzed via manual segmentation of the cortex and hippocampal regions using Analyze, version 12.0 (AnalyzeDirect), a voxel-based normalized entropy algorithm (n = 3; 8 technical replicates). In the same cohort as used for immunohistochemistry, a subset of mice (3 from the saline group and 4 from the lipopolysaccharide group) were used to determine the protein levels of P2X7R and β-actin via Western blot analysis as described in “Cell Line Receptor Expression Validation.”
Radiotracer Biodistribution
Tissue biodistribution of 11C-GSK1482160 was measured in C57BL6 mice administered either saline or lipopolysaccharide (5 mg/kg) and allowed to incubate for 72 h. At 15, 30, and 60 min after tail-vein tracer administration of 155 ± 10 MBq/kg of 11C-GSK1482160, tissues were harvested. At the time of harvesting, the animals were deeply anesthetized using isoflurane and cardiac puncture was performed to collect terminal blood samples. The animals were rapidly decapitated, and the following organs were removed for analysis: brain, heart, lung, liver, spleen, kidneys, intestines (small and large), and skeletal muscle. All samples were placed into vials, weighed, counted via a γ-counter (model 8000; Beckman), and decay-corrected to the time of injection. Total tissue activity for each subject, tissue or organ, and time point was expressed according to the following: Eq. 1where At, Wt, Wv, Dt, Dr, i, j, and t are the tissue activity, total weight, vial weight, total dose, residual dose, i’th subject, j’th tissue or organ, and i’th time point, respectively.
Eq. 1where At, Wt, Wv, Dt, Dr, i, j, and t are the tissue activity, total weight, vial weight, total dose, residual dose, i’th subject, j’th tissue or organ, and i’th time point, respectively.
PET and CT Imaging
To determine tracer kinetics under microglial resting–surveilling and activated states, adult C57BL6 mice were intraperitoneally administered either saline (n = 3) or a 5 mg/kg dose of lipopolysaccharide (O111:B4) (n = 4) and returned to their home cage for 72 h. At the time of imaging, the mice were anesthetized using 3%–4% isoflurane balanced with medical-grade oxygen at a flow rate of 2 L/h. Once anesthetized, the animals were placed on a custom carbon-fiber bed and maintained at 1.5%–2% isoflurane, and access was secured via a 27-gauge butterfly catheter into the lateral tail vein. Dynamic list-mode PET images were acquired using the IndyPET3 scanner (17). List-mode images were reconstructed into 80 × 100 mm volumes using filtered backprojection with the following frame durations: 12 × 5 s, 6 × 10 s, 6 × 20 s, 6 × 60 s, and 9 × 300 s. In all cases, images were corrected for decay, random coincidence events, and dead-time loss (18). At the completion of the PET procedure, the animal beds were transferred to an EVS R9 small-animal CT scanner (Enhanced Vision Systems Corp.) and images were acquired using 80 kVp, 1,000 mA, 200 ms/degree, and 1.08°/step and were reconstructed into forty 65-mm volumes using a vendor-supplied filtered backprojection algorithm. To determine the specific binding, blocking studies were performed on lipopolysaccharide-treated mice at 72 h as described above. Approximately 10 min before 11C-GSK1482160 administration, the mice were anesthetized, venous access was secured, and a 1 mg/kg dose of authentic standard GSK1482160 was administered intravenously.
Image Analysis
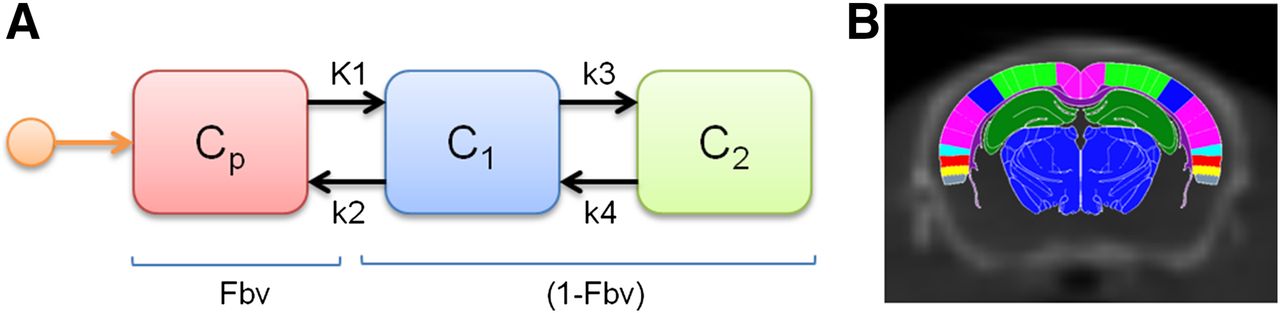
PET and CT images were registered using Analyze (19). To quantify regional activity in mouse brains with time, PET/CT images were then coregistered to a Paxinos–Franklin (20) mouse brain atlas, and time–activity curves were extracted and kinetically modeled using a 2-compartment, 5-parameter tracer kinetic model (Fig. 1A) with custom modeling tools developed by Paul R. Territo for the following brain regions: retrosplenial dysgranular cortex, cingulate cortex, primary motor cortex, secondary motor cortex, primary somatosensory cortex, secondary somatosensory cortex, dysgranular insular cortex, and hippocampus (cornu ammonis 1–3). In addition, image-based arterial input functions for each mouse were obtained via manual segmentation of the left ventricle, with care being taken to avoid the septal and free walls of the myocardium. Regions of interest were then extracted from the PET time series and were decay-corrected, and a tracer kinetic model describing 11C-GSK1482160 metabolite-uncorrected kinetics in left ventricular blood and brain tissue using a 2-tissue-compartment, 5-parameter model (Fig. 1) was applied: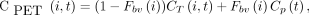 Eq. 2where CPET is the measured 11C-GSK1482160 concentration in the voxel of interest, CT is the tissue 11C-GSK1482160 concentration, Cp is the arterial plasma 11C-GSK1482160 concentration using a hematocrit level of 0.42 (21), Fbv is the fractional blood volume, i is the i’th brain region, and t is the t’th time point. After fitting of the 2-tissue-compartment model, the total distribution volume of the P2X7R is computed as
Eq. 2where CPET is the measured 11C-GSK1482160 concentration in the voxel of interest, CT is the tissue 11C-GSK1482160 concentration, Cp is the arterial plasma 11C-GSK1482160 concentration using a hematocrit level of 0.42 (21), Fbv is the fractional blood volume, i is the i’th brain region, and t is the t’th time point. After fitting of the 2-tissue-compartment model, the total distribution volume of the P2X7R is computed as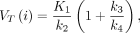 Eq. 3where VT is the total distribution volume of the P2X7R; K1, k2, k3, and k4 are the estimated model rate constants; and i is the i’th brain region.
Eq. 3where VT is the total distribution volume of the P2X7R; K1, k2, k3, and k4 are the estimated model rate constants; and i is the i’th brain region.
Statistical Analysis
Statistical analysis of the immunofluorescence assay and radioimmunohistochemistry dose response curves for the cell line receptor validation studies was performed using Prism, version 6.02 (GraphPad Software), with 1-way ANOVA in which pairwise analysis was according to the Tukey multiple-comparison tests. Kinetic modeling for association–disassociation kinetics was performed using a nonlinear fit of association and then dissociation, whereas saturation kinetics were modeled using a 1-site saturation model with Prism. Statistical analysis of Iba1 immunohistochemistry staining and P2X7R Western blot analyses was performed in Excel, version 12.0 (Microsoft Corp.) using an unpaired 1-tailed Student t test with unequal variances. For tissue biodistribution studies and VT, data were analyzed with 2-way ANOVA using Prism. The tracer kinetics of mouse brain regions were modeled using a custom tool (i.e., eNumerate) developed and validated by our lab. In all cases, data are presented as mean ± SEM, with statistical significance taken at the P ≤ 0.05 level.
RESULTS
Cell Line Receptor Expression Validation
Dry-mounted and immunofluorescently stained sections of HEK293 cell blocks against P2X7R revealed a statistically significant (P < 0.0001, 1-way ANOVA, n = 3; 4 technical replicates) increase in signal levels with cell density (Fig. 2). Pairwise analysis of immunofluorescence assay intensity with density revealed statistically significant differences (P < 0.05, Tukey multipairwise test, n = 3; 4 technical replicates) for all density levels studied. Given that immunofluorescence assay is reliant on fluorescence emission, which is subject to inner-filter effects (both primary and secondary) and light scatter, these measures are considered semiquantitative at best. To overcome these limitations, we used a new method (radioimmunohistochemistry) that takes advantage of the receptor specificity afforded by immunohistochemistry and the sensitivity of autoradiographic imaging. Analysis of P2X7R radioimmunohistochemistry in dry-mounted cell-block sections revealed a statistically significant (P < 0.0001, 1-way ANOVA, n = 3; 4 technical replicates) increase in tissue activity with cell density. Pairwise analysis of radioimmunohistochemistry dose levels showed that virtually all dose levels were statistically significantly different at P < 0.05 (Tukey multipairwise test, n = 3; 4 technical replicates) for all cell density levels studied except 62.5 versus 125 and 250 versus 500 million cells. To further quantitatively probe the expression level as a function of cell number, Western blot analysis of P2X7R was performed in serial-diluted samples from 12,500 to 781 cells and revealed a clear dose dependence of P2X7R on cell number.
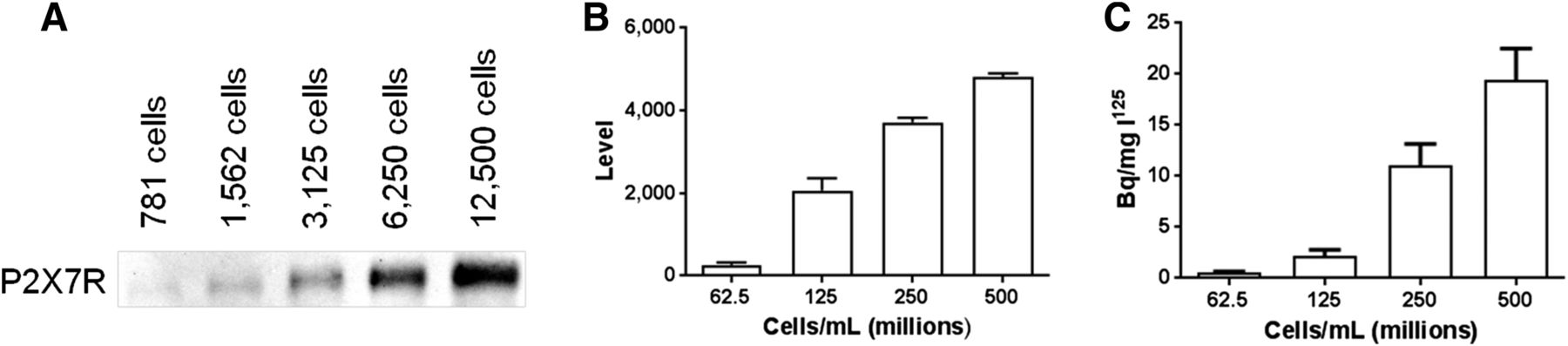
P2X7R expression in HEK293 cell line. (A) WB using anti-P2X7R antibody showing band of expected Kd size increasing in intensity with increasing number of cells. (B) Immunofluorescence assay on cell blocks showing increased signal with increasing cell number. (C) Radioimmunohistochemistry confirming increased signal with increased cell number. Data are mean ± SEM.
Radioligand Association–Disassociation and Saturation Kinetics Experiments
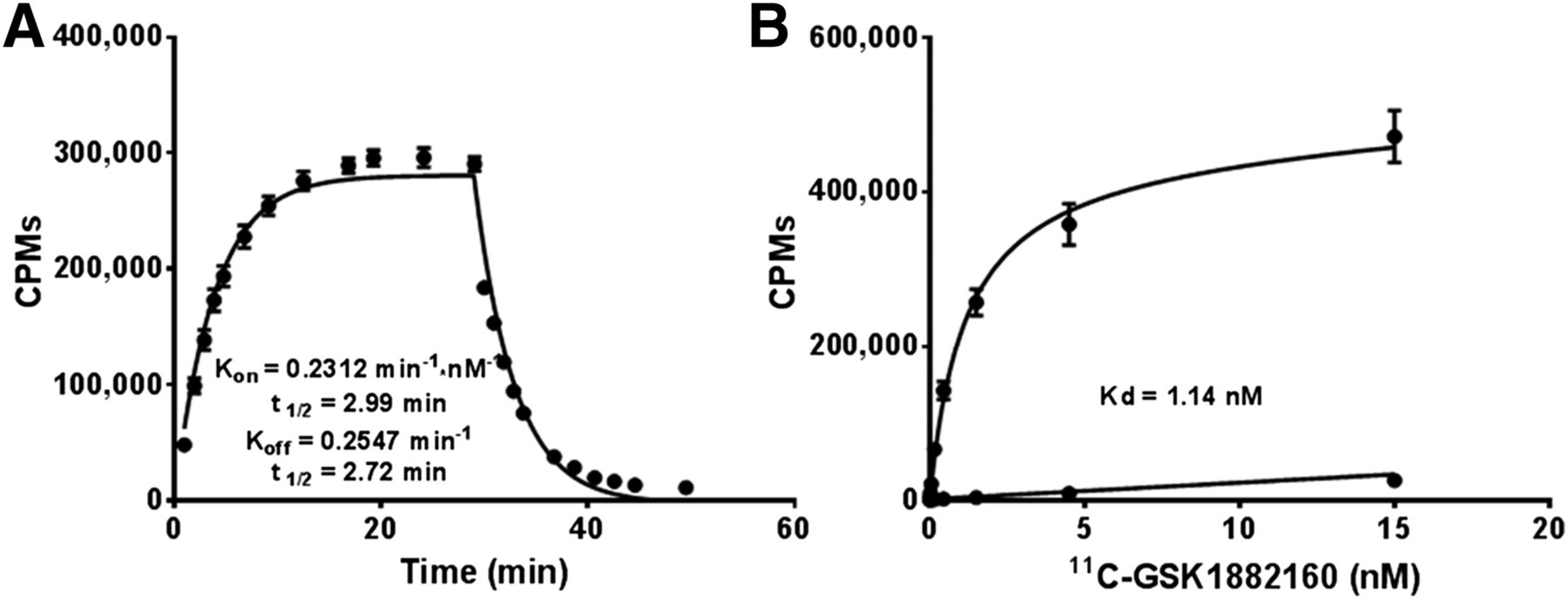
Estimates of the association constant kon, dissociation constant koff, and binding potential (kon/koff) were 0.2312 ± 0.01542 min−1⋅nM−1, 0.2547 ± 0.0155 min−1, and 1.0277 ± 0.207, respectively (Fig. 3A). Provided this, the ratio of total-to-nonspecific binding was determined over a broad range of 11C-GSK1482160 concentrations (0.15–15,000 pM) in the absence and presence of 10 μM authentic standard GSK1482160, and Kd and receptor density values of 1.15 ± 0.12 nM and 3.03 ± 0.10 pmol/mg membrane protein were obtained, respectively (Fig. 3B). Nonspecific binding in the presence of an excess of authentic standard GSK1482160 was 5 orders of magnitude below that seen without competitive inhibition. To demonstrate the specificity of 11C-GSK1482160 to hP2X7R, total binding was determined in hP2X4R membrane preparations and showed no affinity to this receptor construct.

(A) Association–disassociation kinetics of 11C-GSK1482160 in membrane preparations from HEK293 cells expressing hP2X7R. Kd = 1.10. (B) Saturation binding curve for 11C-GSK1482160 in absence and presence of excess of parent. Data are mean ± SEM.
Lipopolysaccharide Time Course
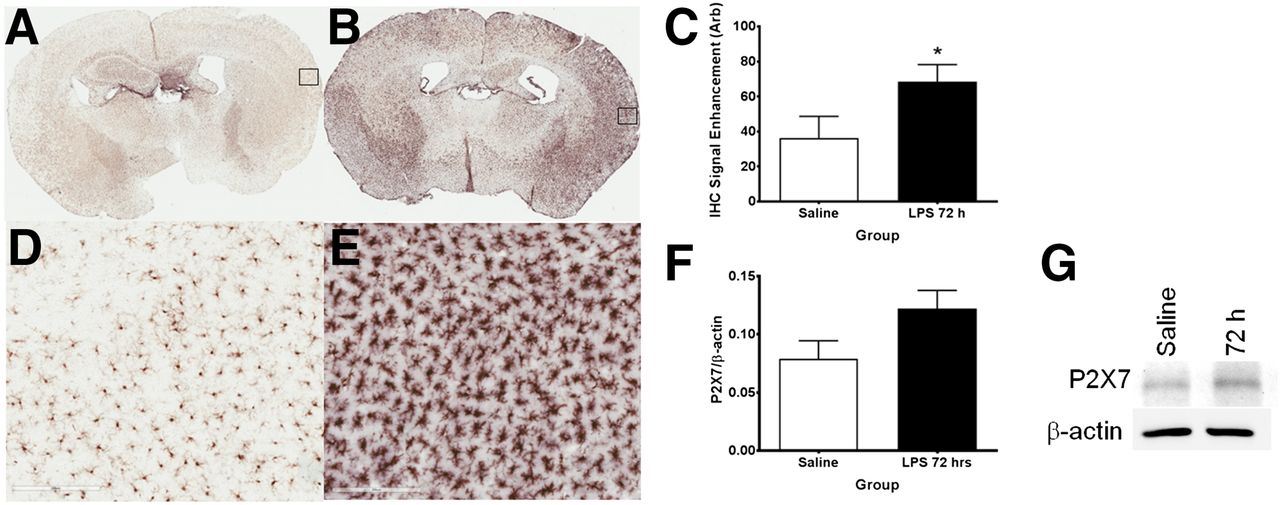
Expression levels of Iba1 showed a progressive rise in band density with increasing time, with the highest expression level occurring at 72 h after administration (Fig. 4A). By contrast, β-actin, a cellular housekeeping gene product, showed no elevation across the time points studied. Ratios of Iba1–to–β-actin band intensities, which control for sample loading, showed similar responses in Iba1, with peak expression occurring at 72 h (Fig. 4B) and being 2.4-fold higher than that for saline. Figure 5 illustrates coronal brain sections at 72 h after treatment with saline (Fig. 5A) and lipopolysaccharide (Fig. 5B); the density and morphology of the microglia have transitioned from a resting–surveying state to a condensed activated state. This transition can be seen with greater detail in Figures 5D and 5E, respectively. Quantitative analysis of the immunohistochemistry staining indicated a statistically significant (P < 0.05, Student t test, n = 3) increase (∼1.8-fold) in average signal enhancement of Iba1 staining. To determine the P2X7R and β-actin expression, a parallel cohort of mice was injected with saline or lipopolysaccharide, sacrificed, and their frontal cortex homogenates analyzed via Western blotting at 72 h. As with Iba1 expression, P2X7R levels increased in the lipopolysaccharide-treated group, whereas β-actin remained constant. Quantitation of P2X7R–to–β-actin band-intensity ratios showed a 1.7-fold increase in receptor expression with treatment (P = 0.054, Student t test, 3 saline-treated and 4 lipopolysaccharide-treated).

Time course for increased microglial activation after systemic lipopolysaccharide-induced inflammation. (A) Western blot of Iba1 protein expression in mouse cortex after lipopolysaccharide injection. (B) Ratio of Iba1 expression level to β-actin loading controls. Data are mean ± SEM; n = 3/time point.
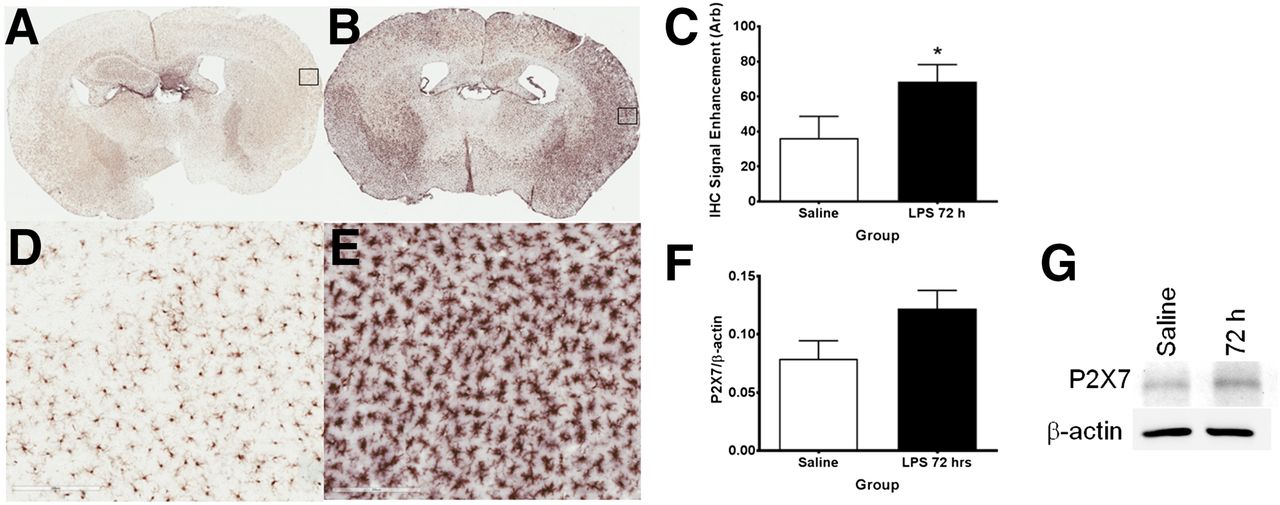
Iba1 immunohistochemistry showing increased microglial activation in lipopolysaccharide-treated mice. (A) Coronal brain section of mouse injected with saline. (B) Coronal brain sections of mouse injected with lipopolysaccharide after 72 h. (D and E) Respective ×20 magnification of insets in A and B. (C) Iba1 staining intensity in saline- vs. lipopolysaccharide-treated mice. (F) Ratio of P2X7R expression level to β-actin in saline- vs. lipopolysaccharide-treated mice. (G) Western blot of P2X7R protein expression in cortex of lipopolysaccharide-treated mice after 72 h. Data are mean ± SEM. *P < 0.05, unpaired t test.
Radiotracer Biodistribution
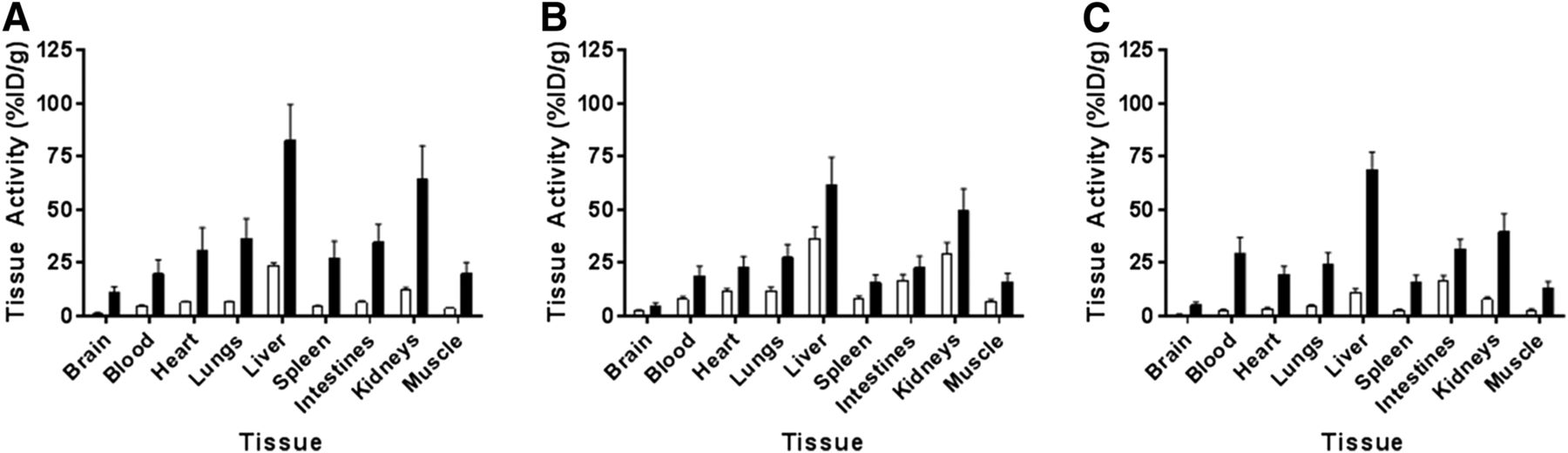
Compared with saline treatment, lipopolysaccharide treatment resulted in a statistically significant (P < 0.001, 2-way ANOVA, n = 3/time and treatment) 2.9- to 5.7-fold increase in total activity across all organs studied (Figs. 6A–6C).

Biodistribution of 11C-GSK1482160 in saline- (white bars) and lipopolysaccharide-treated (5 mg/kg) (black bars) mice 72 h after administration. Samples were acquired at 15 min (A), 30 min (B), and 60 min (C) after 11C-GSK1482160 administration. Two-way ANOVA showed that lipopolysaccharide treatment significantly increased tracer biodistribution (P < 0.0001). Data are mean ± SEM; n = 3/time point.
PET and CT Imaging
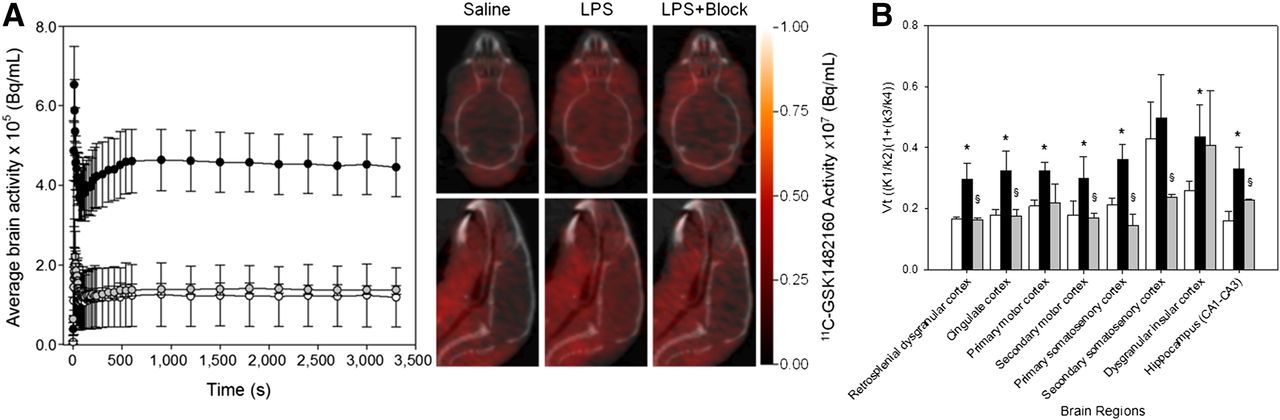
The average whole-brain time–activity curves for saline-treated mice (n = 3) and lipopolysaccharide-treated mice (n = 4) show the characteristic rise in brain tracer levels within the first 5–10 s after administration of 11C-GSK1482160 and stable tissue levels within approximately 10 min. As expected, lipopolysaccharide resulted in a 3.6-fold increase in total brain activity over the time course (Fig. 7A), and this increase was 97% displaceable when blocked with a 1 mg/kg dose of authentic standard GSK1482160 over the same time course. In all cases, the model was fit to a high degree (R2 = 0.915 ± 0.037, n = 10) across all subjects and treatments. Estimates of VT for the retrosplenial dysgranular cortex, cingulate cortex, primary motor cortex, primary somatosensory cortex, dysgranular insular cortex, and hippocampus showed statistically significant (P < 0.001, 2-way ANOVA, 3 saline-treated and 4 lipopolysaccharide-treated) increases in regional binding (Fig. 7B). To determine the degree of specific binding, blocking studies were performed at 72 h in lipopolysaccharide-treated mice by intravenously administering a 1 mg/kg dose of authentic standard GSK1482160 10 min before 11C-GSK1482160 administration. As observed with whole-brain analysis, VT significantly decreased (P < 0.001, 2-way ANOVA, 4 lipopolysaccharide-treated and 3 lipopolysaccharide + blocking–treated) for all brain regions studied except the dysgranular insular cortex.
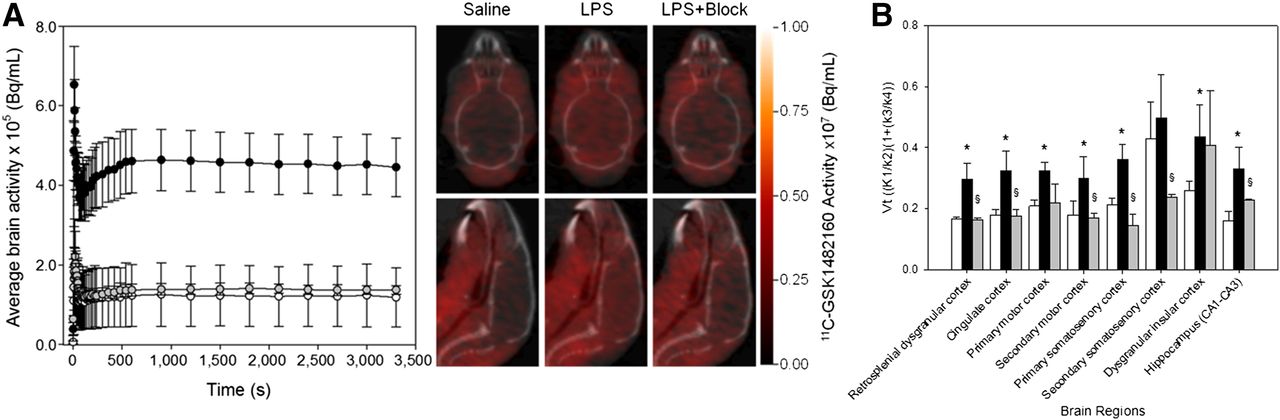
(A) Graph of dynamic 11C-GSK1482160 average brain PET activity in saline-treated mice (white circles, n = 3), lipopolysaccharide-treated mice (black circles, n = 4), and lipopolysaccharide + block–treated mice (gray circles, n = 3) 72 h after intraperitoneal administration, along with representative static transverse (top) and sagittal (bottom) PET/CT images. For blocking, 1 mg/kg dose of authentic standard GSK1482160 was administered intravenously approximately 10 min before tracer. (B) VT reported as measure of ligand binding for saline (white bars), lipopolysaccharide (black bars), and lipopolysaccharide + block (gray bars). Data are mean ± SEM. *Statistically significant difference from saline, P < 0.05. §Statistically significant difference from lipopolysaccharide, P < 0.05.
DISCUSSION
Here, we have presented a comprehensive analysis of the molecular and physiologic characteristics of 11C-GSK1482160, a novel radioligand for imaging of P2X7R and neuroinflammation.
HEK293 cells expressing hP2X7R were stable and dose-dependent over a broad range of concentrations, as revealed by immunofluorescence, radioimmunohistochemistry, and Western blot analyses (Fig. 1). Inherently, the immunofluorescence and radioimmunohistochemistry methods provide spatial information and receptor densities but do so at the cost of sensitivity. As expected, protein levels via Western blot analysis were significantly more sensitive but lacked the spatial information encoding of the immunofluorescence and radioimmunohistochemistry methods. The combined data clearly show that the expression levels of hP2X7R in this system are stable and receptor content–dependent over a broad range and are suitable for studying receptor kinetics in membrane preparations.
Given the reliability of the HEK293-hP2X7R cell line, membrane preparations were generated and used for association–disassociation and saturation kinetic analyses. The association–disassociation kinetics showed a high degree of fit across the replicates studied and yielded consistent kon rates (0.2312 ± 0.01542 min−1⋅nM−1) and koff rates (0.2547 ± 0.0155 min−1), with binding potential (kon/koff) estimates indicating approximately 100% receptor occupancy by 30 min. These data are consistent with literature reports of a functional analog of GSK1482160 that showed rapid onset of ligand binding and equilibrium within 30–128 min (22). Saturation kinetic experiments were conducted over a broad range of concentrations and in the presence of an excess of authentic standard GSK1482160 and produced a Kd (1.15 ± 0.12 nM) that displays favorable binding kinetics. Similar results were obtained from association–disassociation studies (koff/kon), which also yielded a comparable Kd (e.g., 1.10 nM) despite being derived from different preparations and methods. These combined data are consistent with previous reports for antagonists from this drug class in HEK293-P2X7R membranes, which had a Kd and receptor density of 1.4 nM and 5.4 pmol/mg, respectively (22). Importantly, 11C-GSK1482160 specificity to hP2X7R was shown to be selective and specific for this receptor subtype.
To determine the time course of peripheral cytokine release, microglial activation, and receptor expression in C57BL6 mice, lipopolysaccharide time course studies were conducted. Previous literature reports using the same mouse strain, dose level, and route of administration were reported to peak within 30 min (23) when monitored by circulating cytokines and 12 h (24) to 24 h (25) after lipopolysaccharide administration for microglial activation as determined via Iba1 expression. In the current study, brain-derived protein levels of Iba1 (normalized to β-actin) in response to intraperitoneal lipopolysaccharide administration revealed a progressive rise in Iba1 content through 72 h (Fig. 4B), which is significantly longer than previously reported (23–25). It is unclear what the differences between these studies may be, but recent work on the role of gut microflora in germ-free mice suggests that elevated IL10 levels may play a role in murine immune response to lipopolysaccharide (26) and that prior exposure via gut microflora may play a substantial role in a heightened immune response. To determine the morphology and spatial distribution of microglial activation, immunohistochemistry analysis against Iba1 was performed in the brains of saline- and lipopolysaccharide-treated mice. As shown in Figures 5A–5D, administration of lipopolysaccharide at 72 h resulted in a frank increase in Iba1 staining over saline, which is consistent with prior reports at earlier time points (24,25). Interestingly, the magnitude of the response as measured by image analysis of signal enhancement was consistent with Western blot analysis obtained from the lipopolysaccharide time courses at 72 h (Fig. 4B). Importantly, the expression level of P2X7R/β-actin in this same system tracks that of Iba1/β-actin, with a 1.7-fold increase over saline (Figs. 5F–5G). These data clearly illustrate not only that lipopolysaccharide activates the microglia, as determined by Iba1/β-actin levels, but that P2X7R/β-actin is also upregulated during this same interval, consistent with the theory of microglial activation and the role of P2X7R during this process (6).
To assess the tissue distribution of 11C-GSK1482160 under baseline and lipopolysaccharide-activated states, biodistribution studies were performed at 72 h after lipopolysaccharide administration. As seen in Figure 6, administration of 11C-GSK1482160 resulted in a range of uptake levels (0.8–23.8 %ID/g) depending on tissue type and time point studied. As expected, lipopolysaccharide resulted in a significant increase in tissue uptake for all tissues and organs studied, with liver showing the largest uptake overall across time points. These data are consistent with prior reports implicating liver as a primary route of lipopolysaccharide clearance during bacterial sepsis (27,28). Moreover, hepatic myeloid interstitial dendritic cell ectonucleoside triphosphate diphosphohydrolase-1 (CD39) hyporesponsiveness has been reported in mice (29). Combined, the aforementioned processes would result in an accumulation of lipopolysaccharide, activation of hepatic Toll-like receptor 4, and suppression of extracellular adenosine-triphosphate hydrolysis (via CD39 hyporesponsiveness) and thus stimulate upregulation of P2X7R expression. Although total uptake was higher in liver than in the other organs collected, uptake in the brain was approximately 8-fold higher than in saline-treated animals (Fig. 6). These data, along with previous reports that GSK1482160 has excellent blood–brain barrier penetration (30), suggest that binding in activated microglia could be quantified if dynamically imaged via PET.
To evaluate the in vivo tracer kinetics under resting–surveilling and activated states, dynamic whole-body 11C-GSK1482160 PET/CT imaging was performed on mice at 72 h after saline or lipopolysaccharide administration. As shown in Figure 7, the administration of lipopolysaccharide resulted in a marked increase (3.6-fold) in whole-brain 11C-GSK1482160 uptake, which was consistent with both the Iba1/β-actin increase and the P2X7R/β-actin increase observed (Fig. 5). The administration of an excess of authentic standard GSK1482160 in lipopolysaccharide-treated mice resulted in a marked decrease in tracer uptake across all time points, clearly showing that whole-brain uptake was 97% receptor-dependent. Tracer kinetic modeling of the individual time courses revealed a high degree of fit across all subjects studied and all conditions studied. To evaluate the appropriateness of the model, koff rate constants from both membrane preparations and PET kinetic modeling (i.e., k4∼koff) were compared, and excellent agreement was found between in vitro (0.2547 ± 0.0155 min−1) and in vivo (0.3296 ± 0.0774 min−1, n = 10) measures, suggesting that the model was appropriate for describing the system.
Although the aforementioned tracer kinetic modeling was performed on metabolite-uncorrected data, which have been shown to affect interpretation of many PET radiopharmaceuticals (31), work on GSK1482160 in liver microsomes, liver S9 fraction, and hepatocytes in multiple species has been shown to be metabolically stable (32). Moreover, in isolated perfused rodent livers no evidence of hydroxylation has been shown, and in nearly all animal species studied most of the GSK14822160 parent was recovered from the urine (32). This is supported by the pharmacokinetic determination of the GSK14822160 clearance and circulating half-life, which when modeled are in excess of 2,177 mL/h and 30.5 h, respectively, in humans (derived from (32)). Taken together, these data suggest that metabolite corrections will have minimal impact on kinetics if the imaging time is substantially shorter than the apparent half-life of the parent.
Estimates of VT for 5 of the 8 regions analyzed were significantly elevated by 1.6- to 2.1-fold over saline controls (depending on brain region), again supporting the rationale that the microglia were activated and P2X7R upregulated by lipopolysaccharide treatment. Regional analysis of lipopolysaccharide-treated mice preblocked with authentic standard GSK1482160 showed trends similar to that of whole-brain analysis, with all but one region showing significant decreases in VT with treatment. These data clearly illustrate the receptor dependence of regional binding in microglia activated via peripheral lipopolysaccharide treatment and argue that these changes in binding are independent of lipopolysaccharide-mediated changes in blood–brain barrier integrity (33).
The current study provides compelling data that support 11C-GSK1482160 as a radiopharmaceutical for targeting P2X7R, which is expressed on the cell surface membranes of myeloid-derived cells (i.e., macrophages, monocytes, and microglia) during activation via adenosine-triphosphate (4,5). In particular, P2X7R has been implicated in microglial proliferation and phagocytosis of the injured site (6,7) and is a common mechanism among diseases such as Alzheimer disease, Parkinson disease, Huntington disease, frontotemporal dementia, atrophic lateral sclerosis, multiple sclerosis, and traumatic brain injury (11–13). Several single-nucleotide polymorphisms of the P2X7R have been reported, which include gain-of-function Ala348Thr, Thr357Ser, and Gln460Arg modifications, and have been suggested to alter both protein structure and function (34,35). Depending on allelic copy and penetrance, these gain-of-function single nucleotide polymorphisms may result in greater susceptibility to neurodegenerative inflammatory diseases. Moreover, the Glu496Ala single-nucleotide polymorphism has been reported to result in loss of function (36,37) and may result in greater resistance to inflammatory diseases. Depending on the number of single-nucleotide polymorphisms, penetrance, and existing risk factors, being able to monitor P2X7R density noninvasively via 11C-GSK1482160 may prove beneficial to a wide array of inflammatory conditions and resulting disease.
CONCLUSION
The initial characterization of 11C-GSK1482160 shows high affinity and favorable association–disassociation kinetics for P2X7R. In vivo PET/CT tracer kinetics revealed that VT followed expected trends for lipopolysaccharide-treated mice and was completely displaceable with an excess of cold blocking. Taken collectively, these results suggest that 11C-GSK1482160 is suitable for use as a novel radioligand targeting P2X7R and as a biomarker of neuroinflammation.
DISCLOSURE
This work was supported by the Advanced Imaging Research and Technology Development (AIRTD) grant, to Paul Territo, from the Indiana Institute for Biomedical Imaging Sciences (IIBIS) of the Indiana University Department of Radiology and Imaging Sciences. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Oct. 20, 2016.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication July 19, 2016.
- Accepted for publication September 15, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Increased regional P2X7R uptake detected by [18F]GSK1482160 PET in a tauopathy mouse model
- Plcg2M28L interacts with high fat-high sugar diet to accelerate Alzheimers disease-relevant phenotypes in mice
- The 677C>T variant in methylenetetrahydrofolate reductase causes morphological and functional cerebrovascular deficits in mice
- A modest increase in 11C-PK11195-PET TSPO binding in depression is not associated with serum C-reactive protein or body mass index
- Preclinical Evaluation and Nonhuman Primate Receptor Occupancy Study of 18F-JNJ-64413739, a PET Radioligand for P2X7 Receptors
- P2X7 PET Radioligand 18F-PTTP for Differentiation of Lung Tumor from Inflammation
- 18F-JNJ-64413739, a Novel PET Ligand for the P2X7 Ion Channel: Radiation Dosimetry, Kinetic Modeling, Test-Retest Variability, and Occupancy of the P2X7 Antagonist JNJ-54175446