


Abstract
The balance between proliferation and cell death is pivotal to breast tumor growth. Because of a combination of environmental and genetic factors leading to activation of oncogenes or inactivation of tumor suppressor genes, these processes become deregulated in cancer. PET imaging of proliferation, angiogenesis, and DNA damage and repair offers the opportunity to monitor therapeutic efficacy to detect changes in tumor biology that may precede physical size reduction and simultaneously allows the study of intratumoral and intertumoral heterogeneity.
This review examines recent developments in breast cancer imaging using novel probes. The probes discussed here are not licensed for routine use and are at various stages of development ranging from preclinical development (e.g., the DNA repair marker γH2AX) to clinical validation in larger studies (such as the proliferation probe 3′-deoxy-3′-18F-fluorothymidine [18F-FLT]). In breast cancer, most studies have focused on proliferation imaging mainly based on 18F-labeled thymidine analogs. Initial studies have been promising; however, the results of larger validation studies are necessary before being incorporated into routine clinical use. Although there are distinct advantages in using process-specific probes, properties such as metabolism need careful consideration, because high background uptake in the liver due to glucuronidation in the case of 18F-FLT may limit utility for imaging of liver metastases.
Targeting angiogenesis has had some success in tumors such as renal cell carcinoma; however, angiogenesis inhibitors have not been particularly successful in the clinical treatment of breast cancer. This could be potentially attributed to patient selection due to the lack of validated predictive and responsive biomarkers; the quest for a successful noninvasive biomarker for angiogenesis could solve this challenge. Finally, we look at cell death including apoptosis and DNA damage and repair probes, the most well-studied example being 18F-annexin V; more recently, probes that target caspase endoproteases have been developed and are undergoing early clinical validation studies.
Further clinical studies including analysis of test–retest variability are essential to determine sensitivity and future utility of these probes in breast cancer.
The balance between cell proliferation and cell death is crucial for tumor survival. Although breast cancer treatments are becoming more sophisticated, to allow for precise targeting of specific alterations in tumor cells, the ultimate goal of all anticancer therapies remains the same: to diminish tumor proliferation and increase tumor cell death. Because of the lack of specificity of 18F-FDG for detection of selective targets and effect of benign processes such as inflammation, investigators have developed novel PET biomarkers that may in the future help select patients for therapy and be accurate early predictive indicators of disease response (1). The probes in this section remain at relatively early stages of clinical development and are not in routine clinical use, because they require further validation in larger clinical studies.
Over recent years, there has been a rapid expansion in our knowledge of the molecular biology of breast cancer; this has resulted in the identification of new therapeutic targets and improvements in the long-term survival of cancer patients (2). The probes here are proposed to capture events downstream of well-known signaling cascades such as via estrogen receptor (ER) and human epidermal growth factor receptor 2. Of the various topics included here, proliferation imaging is the most advanced, and most studies have concentrated on thymidine analogs as surrogates for DNA synthesis. Changes in uptake may reflect antitumor activity, which would otherwise not be detected by standard anatomic techniques as some newer therapies may be cytostatic and therefore not cause a rapid change in tumor volume (3), rendering anatomic response assessment criteria such as RECIST inappropriate (3,4). The high costs of novel tracer development, together with the ever-changing priorities of pharma, often result in probes being abandoned before even reaching the clinical validation stage.
BREAST CANCER PROLIFERATION PROBES
Thymidine is the only nucleotide incorporated into DNA but not RNA, therefore most proliferation markers have concentrated mainly on thymidine analogs, because theoretically these should provide a reliable surrogate measure for DNA but not RNA synthesis. Thymidine is a substrate for thymidine kinase 1 (TK1), E.C. 2.7.1.21, and therefore a marker of activity of the thymidine salvage pathway. Proliferation measurements in breast cancer are normally performed using immunohistochemistry for the Ki-67 labeling index; criteria for assessment exist, but there is considerable variability between different laboratories and measurements could be affected by tumor heterogeneity (5,6).
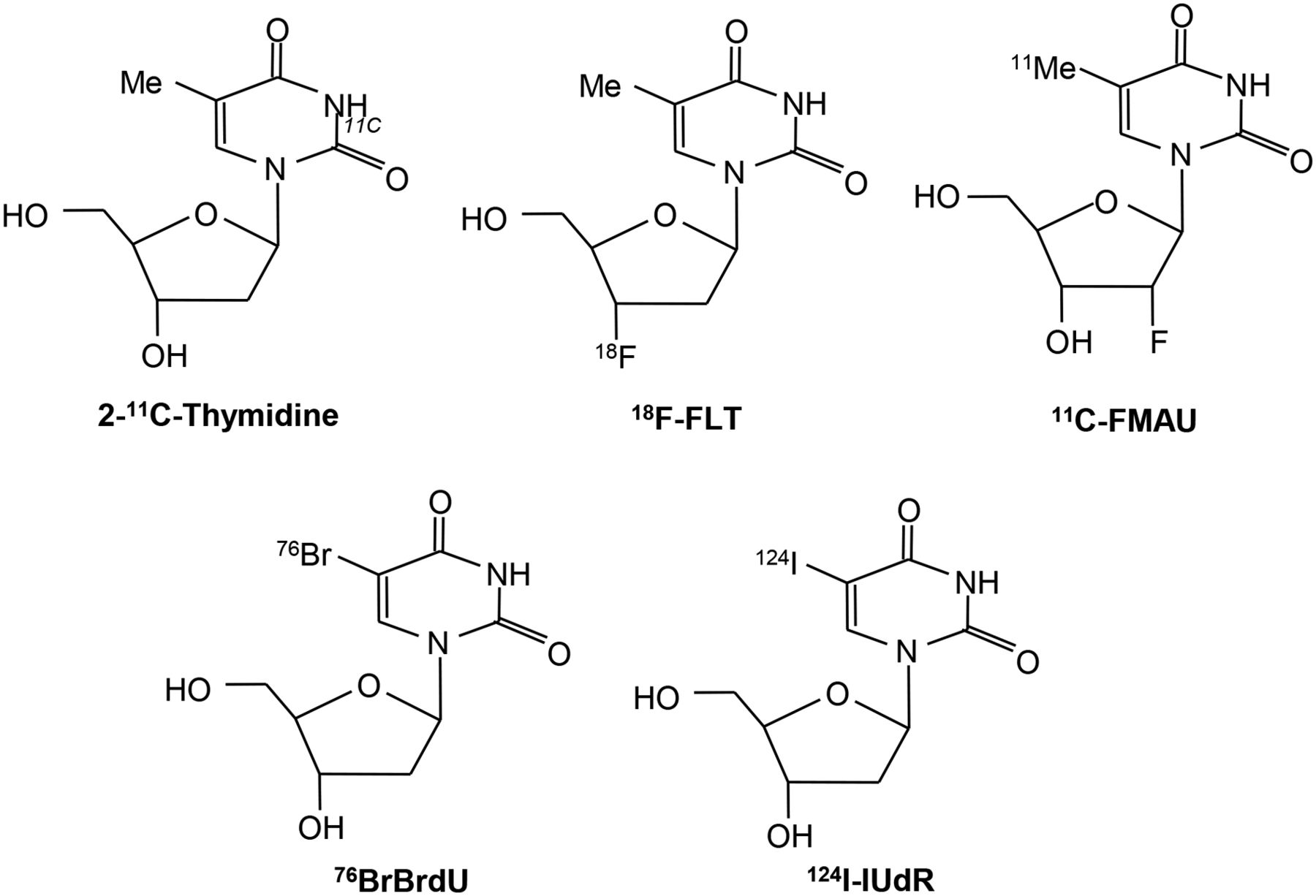
2-11C-thymidine was one of the first proliferation probes. Promising results were seen in early studies, however, this probe has generally been abandoned because of the short half-life of 11C, its rapid metabolism, and complex modeling analysis that is required (7–11). Second-generation probes using 18F have been developed, and these are shown diagrammatically in Figure 1 (12,13). Most experience has been with 3′-deoxy-3′-18F-fluorothymidine (18F-FLT). Transport of 18F-FLT into the cell occurs via passive diffusion and by the equilibrative nucleoside transporter (ENT1), which is expressed in higher numbers in response to drugs such as 5-fluorouracil (which act to inhibit thymidylate synthase, thereby increasing the activity of the salvage pathway) (Fig. 2) (14). Reduced expression of the human ENT1 transporter in breast cancer was found to be associated with poor clinical outcome and prognosis (15). Knockout of the tumor suppressor gene p53 in cells reduces sensitivity to radiation and is associated with increased 3H-FLT uptake, increased cells in S-phase, and increased TK1 activity (16). FLT is not a substrate for thymidine phosphorylase, but competing thymidine is, and this may go some way to explain why thymidine phosphorylase expression is related to FLT uptake.
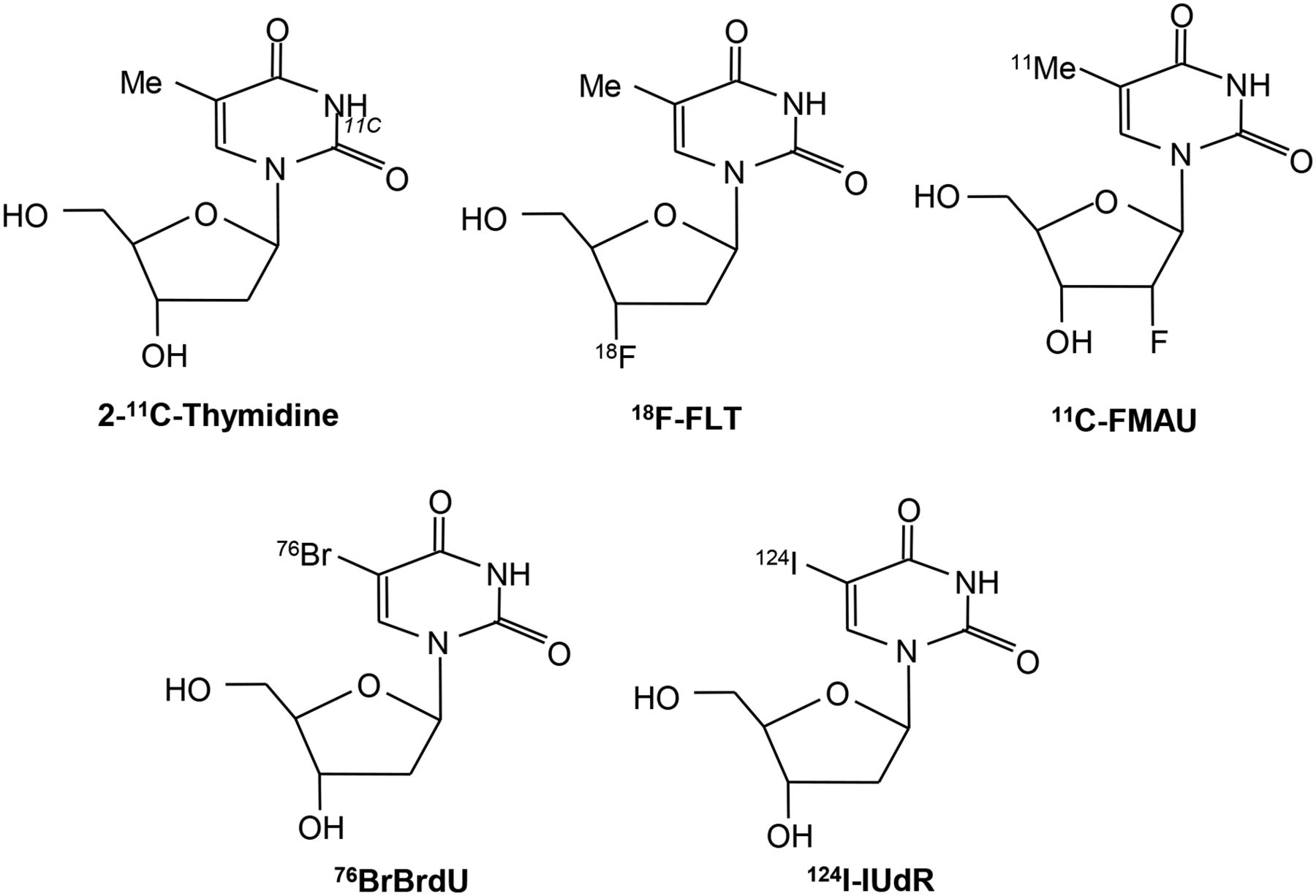
Structure of radiolabeled thymidine and related analogs used for imaging proliferation.

Thymidine and 18F-FLT transport via ENT1 transporter and thymidine salvage pathway.
Shields et al. performed the first 18F-FLT PET study in a patient with non–small cell lung cancer (17). Correlation with immunohistochemistry measures of proliferation such as Ki-67 has been shown for several tumor types including breast cancer (18–20). The limitations of 18F-FLT PET are in imaging the liver and the bone marrow because 18F-FLT undergoes glucuronidation in the liver, leading to a high background uptake level, and in the bone marrow there is high uptake of 18F-FLT, which is attributed to physiologic proliferation. 18F-FLT does benefit from a favorable metabolism profile, because it is metabolized slowly, with 71% still present as the parent compound at 90 min after injection (19).
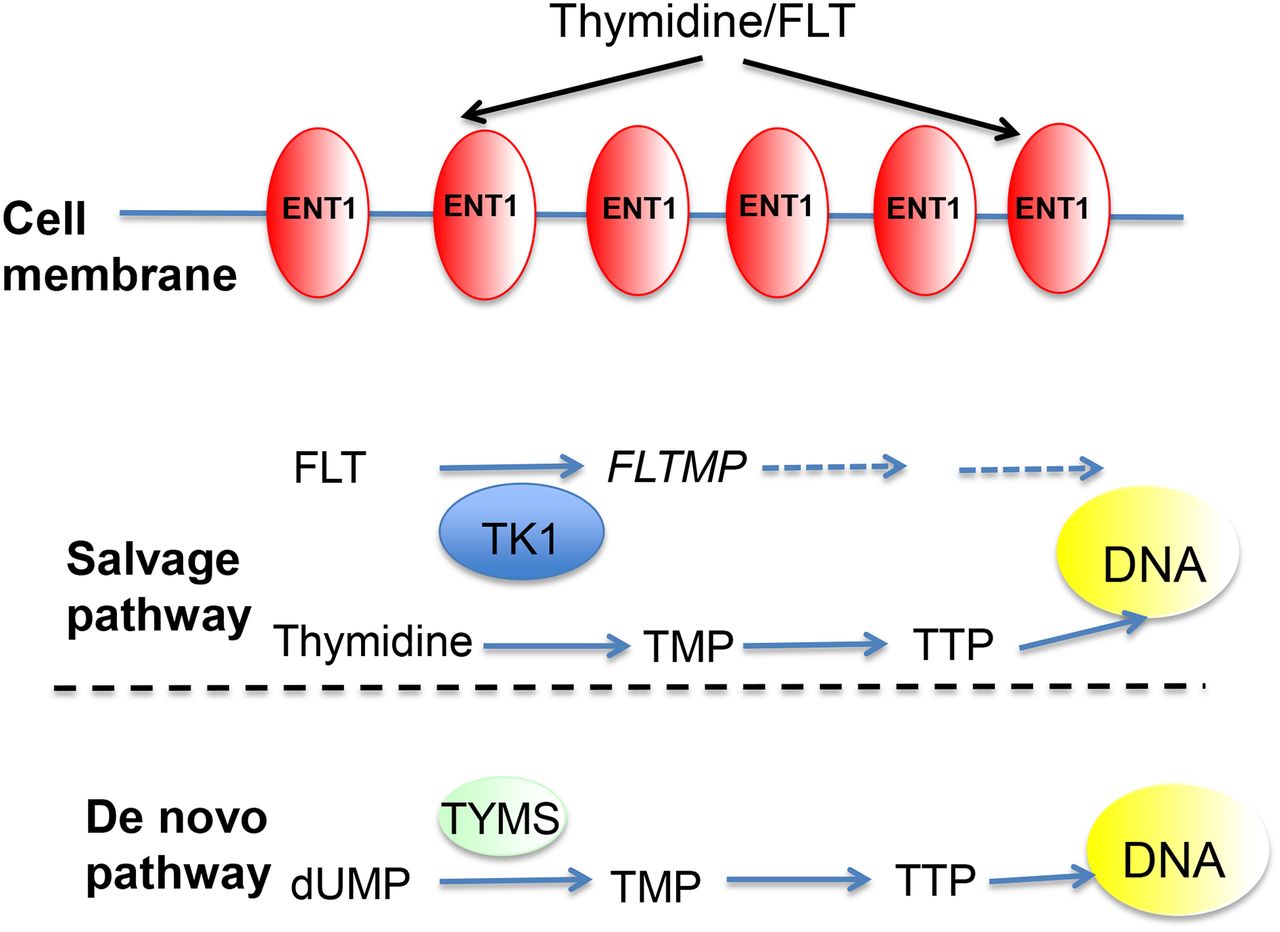
In addition to being a marker of proliferation, very early measurements of 18F-FLT uptake in response to thymidylate synthase (TYMS) inhibitors such as capecitabine may represent a pharmacodynamic readout of this pathway, and an initial study suggests that failure to increase 18F-FLT at an early time point may be predict resistance (21). Later measurements in studies with longer post-treatment intervals (wk) show decreases in 18F-FLT after various cytotoxic chemotherapy regimens and are thought to reflect a decrease in tumor proliferation. These data are summarized in Table 1. A representative image of 18F-FLT before and 1 wk after the first cycle of breast cancer chemotherapy is shown in Figure 3. To date, the number of patients in these studies remains small; more definitive answers should come from the several studies that are currently under way to assess the utility of 18F-FLT response assessment to both chemotherapy and endocrine therapy (www.clinicaltrials.gov). Studies have shown that 18F-FLT uptake is repeatable in both breast and lung cancers (22,23). Sohn et al. found that a reduction of greater than 10.9% for FLT SUVmax could be used to predict therapeutic tumor response (23).
Clinical Trials Involving 18F-FLT PET in Breast Cancer

18F-FLT PET images of grade 3 invasive ductal breast carcinoma (arrow) with scans obtained at baseline and 10 d after FEC chemotherapy. High uptake is also seen in vertebral bone marrow at baseline, reflecting physiologic proliferation, and decreased at 10 d, reflecting hematologic effects of chemotherapy.
1-(2′-deoxy-2′-fluoro-b-d-arabinofuranosyl) thymine (FMAU) is another thymidine analog that has been developed for use in PET studies. One study of various tumors showed good tumor–to–normal-tissue ratios, with an average SUV in breast cancer of 2.17; low uptake was noted in the bone marrow, but there was physiologic uptake noted in the liver (24). In preclinical studies, FMAU uptake was 5–10 times lower in highly proliferating tumors such as triple-negative breast cancer than 18F-FLT, and this was attributed to FMAU being a preferential substrate for thymidine kinase 2 compared with TK1 (25). Unlike TK1, thymidine kinase 2 is constitutively expressed and independent of the cell cycle, whereas TK1 is increases in G1/S phase (26).
Novel 18F-benzamide analogs, which bind to sigma 2 (σ2) receptors, are also being developed as proliferation probes and have just completed first-in-patient studies. The precise physiologic function of these receptors is being evaluated but is thought to relate to potassium and calcium ion channel transport; density of σ2 receptors is higher in proliferating than nonproliferating cells, in some cases up to 10-fold. In a preclinical breast cancer model, expression of σ2 receptors was related solely to proliferation (27). The σ2 receptor binding site is thought to be located in the progesterone receptor membrane component 1 (28). Shoghi et al. have assessed 18F-ISO-1 in 2 rodent models of breast cancer, with uptake correlating with proliferation (measured by bromodeoxyuridine) and growth rate (measured by MR) (28).
Original probes started with 11C labels, and recently Dehdashti et al. explored 18F-ISO-1 (N-(4-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-2-(2-18F-fluoroethoxy)-5-methylbenzamide) in a first-in-patient study including 13 patients with breast cancer (29). Tumor SUVmax and tumor-to-muscle ratio correlated with Ki-67; however, there was no correlation between the tumor–to–normal-tissue uptake ratio and Ki-67. The study revealed high uptake in the liver and pancreas, possibly due to metabolism and receptor density, respectively. A breast cancer–specific study using 18F-ISO-1 is currently recruiting patients in Pennsylvania and is due to complete in 2018.
CHOLINE
MR spectroscopy studies in the 1980s revealed altered choline metabolite profiles in tumors (30). Choline is a natural product in the body, an essential component of the phospholipid bilayer in cell membranes, and necessary for fatty acid synthesis (31). Oncogenic transformation of mammary epithelial cells with the erbb2 (human epidermal growth factor receptor 2) oncogene results in altered choline metabolism (32). CTL1 and OCT3 transport choline intracellularly, where it becomes phosphorylated by the enzyme choline kinase α (CHK, E.C. 2.7.1.32) using adenosine triphosphate as a phosphate donor to form phosphocholine and is then metabolized to phosphatidylcholine via cytidine diphosphate-choline in the Kennedy pathway. In patients, CHK activity is increased by 38.5% in tumor compared with normal tissue and correlates with tumor grade (33). CHK is essential for heregulin-induced growth of breast cancer cells, and inhibition of CHK in tumor cells by the specific inhibitor MN58b leads to apoptosis (34). Most clinical studies have been in prostate cancer; however, promising results using 11C-choline have been reported in breast cancer patients, in which tumor uptake was reproducible and inhibition of mitogen-activated protein kinase by trastuzumab decreased uptake (Fig. 4) (35). 11C-choline and 18F-FLT PET uptake were compared in 21 patients with ER-positive breast cancer, and a correlation between both tracers and with Ki-67 was found. Physiologic liver uptake may limit the ability to study liver metastases.

11C-choline PET image of cervical lymph node metastasis from breast cancer.
Recent developments include 18F-fluoroethylcholine (36) and dueterated choline analogs 18F-fluoro-[1,2- (2)H(4)]choline (18F-D4-choline and 11C-D4-choline); preclinical studies suggest that the compounds have similar sensitivities for tumor detection. 18F-D4-choline is more resistant to metabolism to betaine than the other 2 compounds, which may enable future clinical imaging studies without metabolite correction (37–39). Initial radiodosimetry and biodistribution studies in healthy volunteers showed that 18F-D4-choline undergoes renal and hepatobiliary excretion (40); these fluorinated compounds have yet to be assessed in breast cancer.
ACETATE
Under metabolic stress, cancer cells switch from glucose to acetate as the main nutrient to provide lipids and fatty acids, and breast cancers are sensitive to fatty acid synthesis inhibition (41,42). Acetate is transported into cancer cells via monocarboxylate transporters and is ligated with Co-A by acetyl-CoA synthetase (ACSS, EC 6.2.1.1), forming acetyl-CoA, which can subsequently be used to synthesize cholesterol and fatty acids; alternatively acetate can undergo oxidation in mitochondria via the tricarboxylic acid cycle to CO2 and H20. ACSS2 is upregulated in breast cancers in hypoxia, and 40% of invasive ductal carcinomas express ACSS2 (41). 11C-acetate has been explored mainly in prostate cancer, although it may be useful for detecting breast cancer liver metastases (43). ACSS inhibitors are being developed as well as 18F-fluoroacetate; however, there is a lack of evidence for these probes in breast cancer. In addition, acetate uptake may not be specific, because acetate is used for protein acetylation (41). Further research needs to determine the relationship between acetate uptake and physiologic functions within the cell.
AMINO ACIDS
Protein synthesis is essential for cell survival and is upregulated in cancer. The main amino acid that has been used for PET studies is methionine labeled with 11C, as l-methyl-11C-methionine (11C-MET), because of its ease of synthesis and high radiochemical yields. Analysis of 11C-MET uptake is complicated by the number of nonprotein metabolites. Only limited data relate to clinical studies in breast cancer, including a study of 13 metastatic breast cancer patients in which patients were scanned after 1 mo of endocrine therapy or combination chemotherapy, where reductions in uptake were seen in responding patients, but the numbers of patients were too small to be definitive (44–46). Most studies that have been performed in brain tumors, also for those in which the physiologic uptake in the bone marrow was studied, may limit the ability of 11C-MET PET for imaging metastatic disease. In addition, radiolabeled tyrosine (l-11C-tyrosine and 18F-fluoroethyltyrosine) and the SPECT tracer 3-123I-iodo-α-methyltyrosine have been developed. Whether uptake truly reflects protein synthesis or amino acid transport via the L system needs to be considered. More recently, (4S)-4-(3-18F-fluoropropyl)-l-glutamate (18F-FSPG), a novel 18F-labeled glutamate derivative for PET imaging, was used to study the xc- transporter, which exchanges cysteine in exchange for glutamate, in breast cancer patients and non–small cell lung cancer patients. Although 18F-FSPG uptake was demonstrated in 3 of 5 breast cancer lesions, the 2 that were missed were over 7 cm in size; thus, the importance of molecular subtype of breast cancer in relation to 18F-FSPG uptake needs to be determined (47).
So far the probes discussed image cell growth and synthetic functions; the nature of the cell microenvironment is also crucial for cell survival.
ANGIOGENESIS
Folkman characterized angiogenesis as being fundamental for tumor growth beyond 2 mm in 1971 (48). Since then, our understanding of the molecular biology of angiogenesis has advanced significantly, leading to the development of licensed therapeutic agents. Surprisingly, in the clinic there is no validated predictive biomarker for the selection of antiangiogenic therapy (49). Agents that target angiogenesis can be divided into 3 categories: those that prevent binding of growth factors to their receptors, such as the vascular endothelial growth factor–neutralizing antibody bevacizumab; the multitargeted tyrosine kinase inhibitors (such as pazopanib, sunitinib, and sorafenib); and the matrix metalloproteinase inhibitors such as marimistat (50). Bevacizumab was previously approved for breast cancer because of improved progression-free survival, but the Food and Drug Administration later withdrew this indication because of no clear benefit in overall survival (51).
Imaging probes have been directed to targets expressed on or binding to vascular endothelial cells or to the extracellular matrix. 89Zr-bevacizumab was used to study 23 patients with breast tumor lesions, patients were imaged 4 d after administration because of the probe’s half-life of 78.4 h, and significant differences were observed between breast tumor and normal breast uptake and also between luminal A and luminal B tumors (luminal B had higher uptake) but the radiotracer was less sensitive for the detection of nodal metastases (only 4 of 10 lesions were detected) (52).
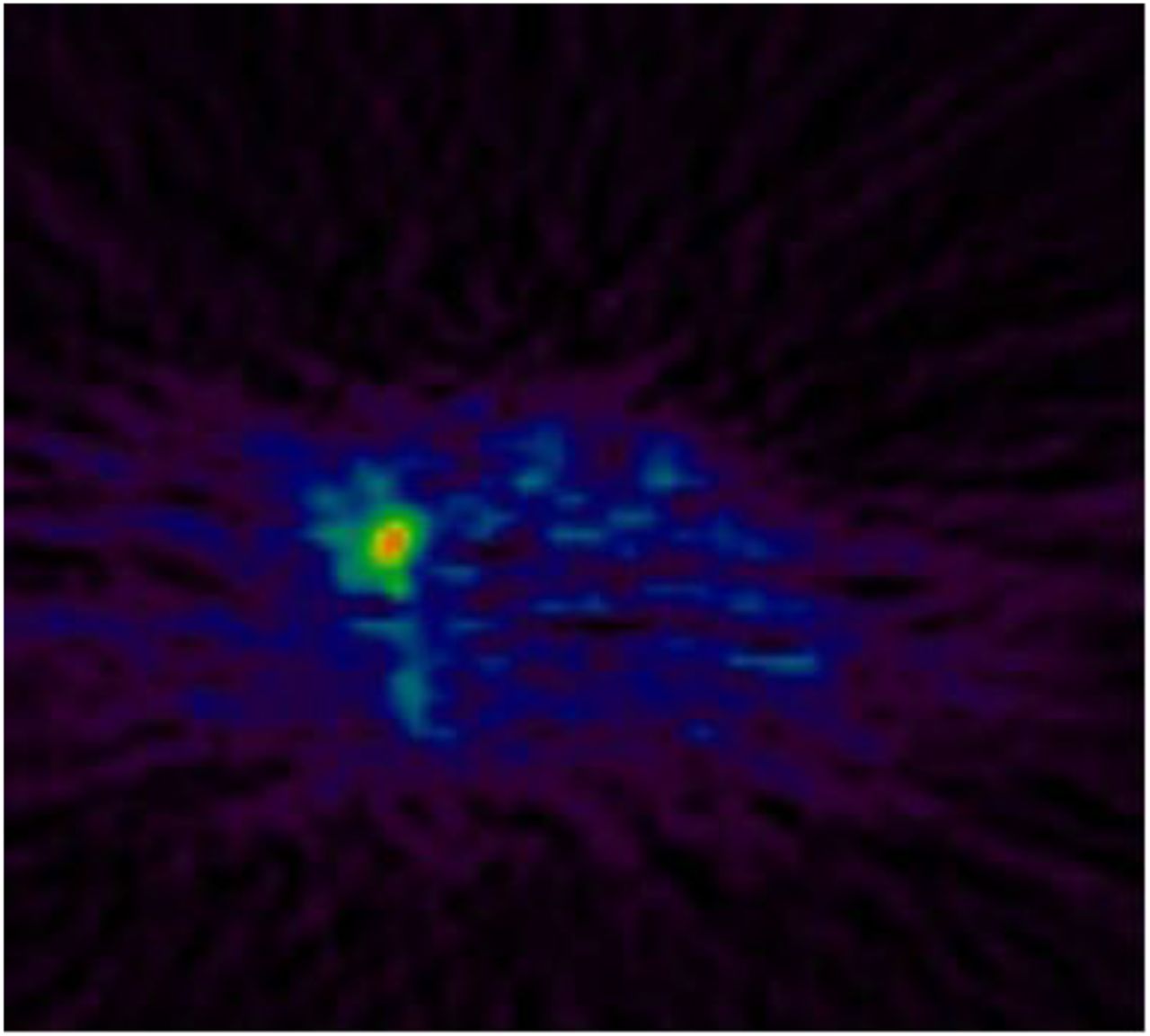
The integrins are heterodimeric proteins that regulate the cellular microenvironment and stroma (53,54). The αvβ3 integrin is involved in angiogenesis and is expressed on newly developing blood vessels and some tumor cells (55). RGD (arginine-glycine-aspartame) peptides are known to bind to the αvβ3 with high affinity (11 nM), and preclinical models using vascular endothelial growth factor inhibitors have shown reduced radiolabeled RGD-peptide after treatment (56). 18F-fluciclatide (previously named 18F-AH111585) is safe and well tolerated, with acceptable dosimetry (57). A study in 7 patients with metastatic breast cancer showed good tumor–to–normal-tissue ratios. Liver lesions were viewed as regions of hypointense uptake due to high background signal, presumably due to physiologic metabolism; areas of heterogeneity of uptake were seen in larger lesions with increased peripheral uptake and decreased uptake centrally, in keeping with active tumor growth and necrosis, respectively (Fig. 5) (22).

18F-fluciclatide PET images of metastatic breast cancer.
Beer et al. have studied 18F-galacto-RGD for imaging of the αvβ3 integrin, designed with a sugar moiety to enhance excretion; promising results were seen in breast cancer and other cancers (58). Uptake of 18F-galacto-RGD correlated with immunohistochemistry for the αvβ3 integrin and microvessel density in snap-frozen tissue (58). 18F-galacto-RGD synthesis is complex, requiring multiple steps; a second-generation compound, 68Ga-NODAGATHERANOST, has been assessed in 2 patients, one of whom had breast cancer, and shows some promise (59). For 18F-FPPRGD2, which was recently developed by the Gambhir group to study αvβ3 integrin expression and compared with 18F-FDG uptake in 8 patients with breast cancer, high specificity was seen, although high liver and renal uptake were seen due to metabolism and excretion (60).
HYPOXIA IMAGING
Hypoxia in breast cancer cells may support the acquisition of the stem cell phenotype (61) and for many years has been attributed as a major factor to resistance to both chemotherapy and radiotherapy (62). Tumor hypoxia develops as result of poor arteriolar supply, high O2 consumption rate, disorganized tumor vasculature, slow blood flow, and vascular shunting. In many tumors, these are associated with upregulation of hypoxia-inducible factor-1 α, which stimulates several downstream targets, resulting in increased glycolysis, angiogenesis, and resistance to apoptosis.
Hypoxia imaging has focused mainly on radiolabeled nitroimidazoles; relatively few studies have been performed in breast cancer, as most have involved brain tumors using 18F-misonidazole (18F-FMISO). A recent study compared 18F-FDG and 18F-FMISO PET in postmenopausal women with stage II–IV ERα-positive breast cancer before and after 3 mo of treatment with the aromatase inhibitor letrozole. 18F-FMISO was superior to 18F-FDG in predicting resistance to endocrine therapy; this study did not show a correlation between tumor hypoxia-inducible factor-1 α and uptake of 18F-FMISO (63). The disadvantages of 18F-FMISO PET include a long biologic half-life, imaging 2–3 h after radiotracer injection, metabolism, and slow clearance from the blood (63).
Second-generation compounds including 18F-fluoroazomycin arabinoside, which is more hydrophilic than 18F-FMISO, and 18F-2-nitroimidazol-pentafluoropropyl acetamide, which is more lipophilic, have been developed (64), as has the nonnitroimidazole compound copper(II)diacetyl-bisN (4)-methylthiosemicarbazone (Cu-ATSM), which can be labeled with either 60Cu or 64Cu, but so far none has been explored in breast cancer (64). Lysl oxidase expression correlates with tissue hypoxia, and a new 18F-labeled probe has been developed and studied in ER-positive and triple-negative breast tumor xenografts with good results (65).
DNA DAMAGE
DNA double-strand breaks (DSBs) are lethal, and detection of DSBs is useful for detection of cell death. After a DSB induced by exogenous or endogenous DNA damage, the protein H2AX becomes phosphorylated by kinases, such as ataxia-telangiectasia mutated in the PI3K pathway, in the first step of the DNA repair process, forming γH2AX complexes in a 1:1 ratio to DSBs. Antibodies to γH2AX labeled with fluorescent probes are commonly used for laboratory assays of cell death. 111In-anti-γH2AX-TAT immunoconjugate was used as a SPECT probe for detecting DNA damage in mice during mammary oncogenesis and showed changes before MR imaging (66). 89Zr-anti-γH2AX-TAT immunoconjugates have also shown promising results in a preclinical breast cancer model with 8-fold-higher uptake in irradiated γH2AX cells than nonirradiated controls (67).
APOPTOSIS
Imaging apoptosis may be a useful biomarker of drug sensitivity or resistance. Most experience is with radiolabeled annexin V, which detects phosphatidylserine expression on the cell surface where it has transferred from the cytoplasmic surface of the cell membrane. However, this process is not unique to apoptosis and can occur in other forms of cell death; even in apoptosis, some cell lines do not express phosphatidylserine (68,69). Other disadvantages include slow clearance, leading to high background due to the large size (36 kDa). 18F-annexin V showed a 3- to 10-fold rise in liver cells, which correlated with histologic measures of apoptosis using a terminal deoxynucleotidyl transferase assay (70). Lower-molecular-weight probes, including 18F-labeled 2-(5-fluoro-pentyl)-2-methyl-malonic acid (ML-10), that undergo selective membrane binding and intracellular transport, have been developed, and these are thought to be selective for apoptosis; further efforts are necessary to determine their specific applications to breast cancer (71).
Caspases are cysteine aspartate–specific proteases activated by the intrinsic and extrinsic apoptotic pathways. The executioner caspases 3 and 7 have been sought as imaging probes. Isatin sulfonamides bind to caspase 3 and 7 with nanomolar affinity, and a new 18F-labeled probe, 18F-ICMT-11, correlates with cellular caspase 3 activation in vitro. A healthy volunteer study has shown acceptable radiation dosimetry, with predominantly hepatobiliary and renal excretion; a current study is now assessing the effects of chemotherapy on 18F-ICMT-11 uptake in breast cancer (72).
Other tracers in the pipeline include 18F-fluorobenzyl triphenylphosphonium, a voltage sensor detector for mitochondrial dysfunction during apoptosis.
CONCLUSION
PET is useful for studying proliferation in breast cancer using thymidine analogs such as FLT and shows promise for imaging of cell death. Probes of angiogenesis, DNA repair, and cell death remain at earlier stages of development and require validation in patients. Factors such as radiotracer metabolism need careful consideration when imaging liver metastases. Further clinical studies are now required to compare the utility of these tracers in patients with 18F-FDG, and to determine their attributes in the various molecular subtypes of breast cancers.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. Laura Kenny is funded by an NIHR Clinician scientist fellowship (CS09/009). The PET images were provided from studies funded by Professor Eric Aboagye’s MRC programme grant and GE Healthcare (fluciclatide). No other potential conflict of interest relevant to this article was reported.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 24, 2015.
- Accepted for publication November 25, 2015.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}