


Abstract
The tumor-homing property of mesenchymal stem cells (MSCs) allows targeted delivery of therapeutic genes into the tumor microenvironment. The application of sodium iodide symporter (NIS) as a theranostic gene allows noninvasive imaging of MSC biodistribution and transgene expression before therapeutic radioiodine application. We have previously shown that linking therapeutic transgene expression to induction of the chemokine CCL5/RANTES allows a more focused expression within primary tumors, as the adoptively transferred MSC develop carcinoma-associated fibroblast-like characteristics. Although RANTES/CCL5-NIS targeting has shown efficacy in the treatment of primary tumors, it was not clear if it would also be effective in controlling the growth of metastatic disease. Methods: To expand the potential range of tumor targets, we investigated the biodistribution and tumor recruitment of MSCs transfected with NIS under control of the RANTES/CCL5 promoter (RANTES-NIS-MSC) in a colon cancer liver metastasis mouse model established by intrasplenic injection of the human colon cancer cell line LS174t. RANTES-NIS-MSCs were injected intravenously, followed by 123I scintigraphy, 124I PET imaging, and 131I therapy. Results: Results show robust MSC recruitment with RANTES/CCL5-promoter activation within the stroma of liver metastases as evidenced by tumor-selective iodide accumulation, immunohistochemistry, and real-time polymerase chain reaction. Therapeutic application of 131I in RANTES-NIS-MSC–treated mice resulted in a significant delay in tumor growth and improved overall survival. Conclusion: This novel gene therapy approach opens the prospect of NIS-mediated radionuclide therapy of metastatic cancer after MSC-mediated gene delivery.
Effective control of metastatic disease represents a central challenge in the treatment of solid tumors. Colorectal cancer is the third most common cancer worldwide, with the liver being the most common metastatic site. Although the prognosis of resectable colorectal liver metastases was improved in recent years, 5-y survival rates after resection are still not higher than 25%–40%, and survival rates have remained poor for unresectable disease (1).
An emerging tumor therapy approach is based on the use of adoptively transferred mesenchymal stem cells (MSCs) as vehicles engineered to express specific reporter or therapy genes (eMSC). This approach uses the innate ability of MSCs to migrate to damaged tissue in the course of tissue repair (2). Tumors are seen by the body as chronic, never healing wounds that drive continuous tissue remodeling, thus providing the basis for the marked tumor tropism of MSCs (3). We and others have harnessed this biology using eMSC to deliver anticancer gene products into tumor stromal environments (4–10). Adoptively transferred MSCs are actively recruited to growing tumors where they contribute to the formation of the benign stromal compartment containing a variety of cell types, including endothelial cells, smooth muscle cells, and pericytes/carcinoma-associated fibroblasts (CAFs) that provide important support for the growth of solid tumors. However, MSCs also contribute to normal tissue homeostasis and therefore may also emigrate to nontumor environments under these conditions. To reduce potential side effects of transgene expression in nontumor-associated tissues, the therapeutic utility of eMSCs has been enhanced by the use of tumor stroma–specific gene promoters linked to unique differentiation pathways activated as MSCs respond to tumor microenvironments. For example, the Tie2 promoter/enhancer is activated as MSCs respond to angiogenic signals within tumor environments (7). MSCs engineered to express the suicide gene herpes simplex virus thymidine kinase (HSV-TK) under control of the Tie2 promoter/enhancer showed selective expression of transgenes after systemic injection of the eMSC into mice with growing breast, pancreatic, or liver tumors (7,9). Although this approach was effective in targeting large primary tumors, the Tie2-based strategy showed low efficacy in the treatment of tumor metastases. An alternative targeting approach was therefore developed using the RANTES/CCL5 promoter to drive transgene expression in MSCs. In the course of differentiation into CAFs, MSCs induce expression of the chemokine RANTES (11,12). eMSC engineered to express HSV-TK under control of the RANTES promoter in concert with ganciclovir treatment led not only to a significant reduction in the growth of primary pancreatic carcinoma but also dramatically reduced incidence of metastases in a pancreatic cancer model (6). Importantly, it was not possible to distinguish if this therapy effect was due to an overall reduction in metastases or direct targeting of individual metastases.
More recently, to enhance the therapeutic potential of MSC-mediated gene therapy, a more advanced therapy gene was applied that makes use of the selective delivery of radionuclides for imaging and therapy. The sodium iodide symporter (NIS) protein is responsible for the active uptake of iodide from the blood into the thyroid gland and, as such, forms the molecular basis for the diagnostic and therapeutic use of radioiodine in the management of differentiated thyroid cancer (13). One of the major advantages of NIS as a therapy gene is its dual function as therapy and reporter gene (13–16). MSCs engineered to express NIS can be monitored by whole-body imaging using 123I scintigraphy/SPECT or 124I PET allowing direct, noninvasive in vivo imaging of MSC biodistribution and functional transgene expression (16,17). Importantly, a robust therapeutic effect can be delivered through 131I or 188Re application (4,5). In the present study, the efficacy of RANTES-based eMSC targeting of NIS expression was evaluated in an experimental mouse model of colon cancer liver metastases. The model was designed to exclude potential effects on the primary tumor to provide information about the efficacy of this therapeutic approach on smaller, metastatic tumor targets.
MATERIALS AND METHODS
Cell Culture
The establishment and characterization of MSCs has been described previously (4,6,18). These cells have been previously shown to maintain their capacity to differentiate and show migration that parallels that seen with primary cells (4,6,18). The ease of expansion of clonal populations allows well-controlled experimental studies.
Plasmids and their synthesis as well as the establishment of stably transfected cell lines have been described previously (4,5).
The human colon carcinoma cell line LS174t (CCL188; American Type Culture Collection) was cultured in RPMI (Invitrogen/Life Technologies) supplemented with 10% fetal bovine serum (v/v; PAA) and 1% penicillin/streptomycin. The cell lines were maintained at 37°C and 5% CO2 in an incubator with 95% humidity.
Establishment of Hepatic Colon Cancer Metastases Mouse Model
The experimental protocol was approved by the regional governmental commission for animals (Regierung von Oberbayern). Fully anesthetized female CD-1 nu/nu mice (Charles River Laboratories) were placed in the right lateral position; after skin disinfection, a 0.5-cm cut was made at the left subcostal region. Tumor cell suspension (50 μL; 1 × 106 cells in 1 × phosphate-buffered saline) was then injected at the upper splenic pole using a 27-gauge needle. Abdominal wall and skin were then sutured separately. Forty-eight hours later, the abdominal wall was reopened at the same site and a splenectomy was performed, followed by suturing of the abdominal wall and skin. Mice were pre- and posttreated with carprofen (5 mg/kg) to minimize wound pain. The induction of liver metastases with this technique was highly reproducible; histologic liver studies showed that 90% of injected mice developed disseminated liver metastases. Animals were maintained under specific pathogen-free conditions with access to mouse chow and water ad libitum.
MSC Application and Radionuclide Biodistribution Studies In Vivo
Application of eMSC was initiated 5 d after intrasplenic tumor cell injection, before metastatic disease was macroscopically visible. In addition, mice were pretreated with thyroid hormone L-T4 (levothyroxine) as described previously (4). Wild-type (WT)-MSCs or RANTES-NIS-MSCs were applied via the tail vein (5 × 105 cells/500 μL of phosphate-buffered saline). Two groups of mice were treated as follows: 3 intravenous applications of RANTES-NIS-MSC in 3-d intervals (n = 22) or 3 intravenous applications of WT-MSC in 3-d intervals (n = 7). As an additional control, in a subset of mice injected with RANTES-NIS-MSC (n = 7) the specific NIS-inhibitor sodium-perchlorate (NaClO4, 2 mg/mouse) was injected intraperitoneally 30 min before radionuclide administration. Forty-eight hours after the last MSC application, 18.5 MBq of 123I was injected intraperitoneally and radionuclide biodistribution was assessed using a γ camera equipped with a UXHR collimator (Ecam) as described previously (19,20). Small-animal PET imaging was performed in a subgroup of mice treated with RANTES-NIS-MSC (n = 8) receiving a dose of 12 MBq of 124I 48 h after the last MSC application, as described previously (4).
Ex Vivo Analyses of Tumor and Nontarget Tissues
For ex vivo biodistribution studies, mice were injected with RANTES-NIS-MSCs (n = 15) or WT-MSCs (n = 5) as described above, followed by intraperitoneal injection of 18.5 MBq of 123I. A subset of RANTES-NIS-MSC–injected mice (n = 5) was treated with NaClO4 before radionuclide administration as an additional control. Three hours after radioiodine injection, the levels of 123I in the liver and nontarget organs were analyzed ex vivo as described previously (4,5).
At the end of the imaging procedure, immunohistochemical and immunofluorescence staining procedures for the expression of NIS and SV40 large T Ag as well as quantification of cellular proliferation (Ki-67) and blood vessel density (CD31) were performed as described previously (5).
The analysis of NIS messenger RNA (mRNA) expression using quantitative real-time polymerase chain reaction (PCR) was performed at the end of the imaging procedure as outlined above. Total RNA was isolated from the liver and other tissues using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s recommendations, and quantitative real-time PCR was performed as described previously (21).
Radioiodine (131I) Therapy Studies In Vivo
After a 10-d L-T4 pretreatment as described previously (4,5), 2 groups of mice were established for which each mouse received 55.5 MBq of 131I (sodium iodide; GE Healthcare Buchler GmbH) 48 h after 3 RANTES-NIS-MSC (RANTES-NIS-MSC + 131I, n = 15) or WT-MSC (WT-MSC + 131I, n = 15) applications given in 2-d intervals (each 5 × 105 cells/500 μL of phosphate-buffered saline). Twenty-four hours later, we applied 2 more rounds of 2 MSC (5 × 105 cells) injections (in 2-d intervals), followed by 55.5 MBq of 131I 48 h later (Fig. 1A). As control, 1 further group of mice was treated with saline instead of 131I after injection of RANTES-NIS-MSC (n = 15). By protocol, mice were sacrificed when healthy liver tissue reached less than 20%, or in the case of weight loss of more than 10% of initial weight, or when impairment of breathing, drinking, or eating behavior was observed.
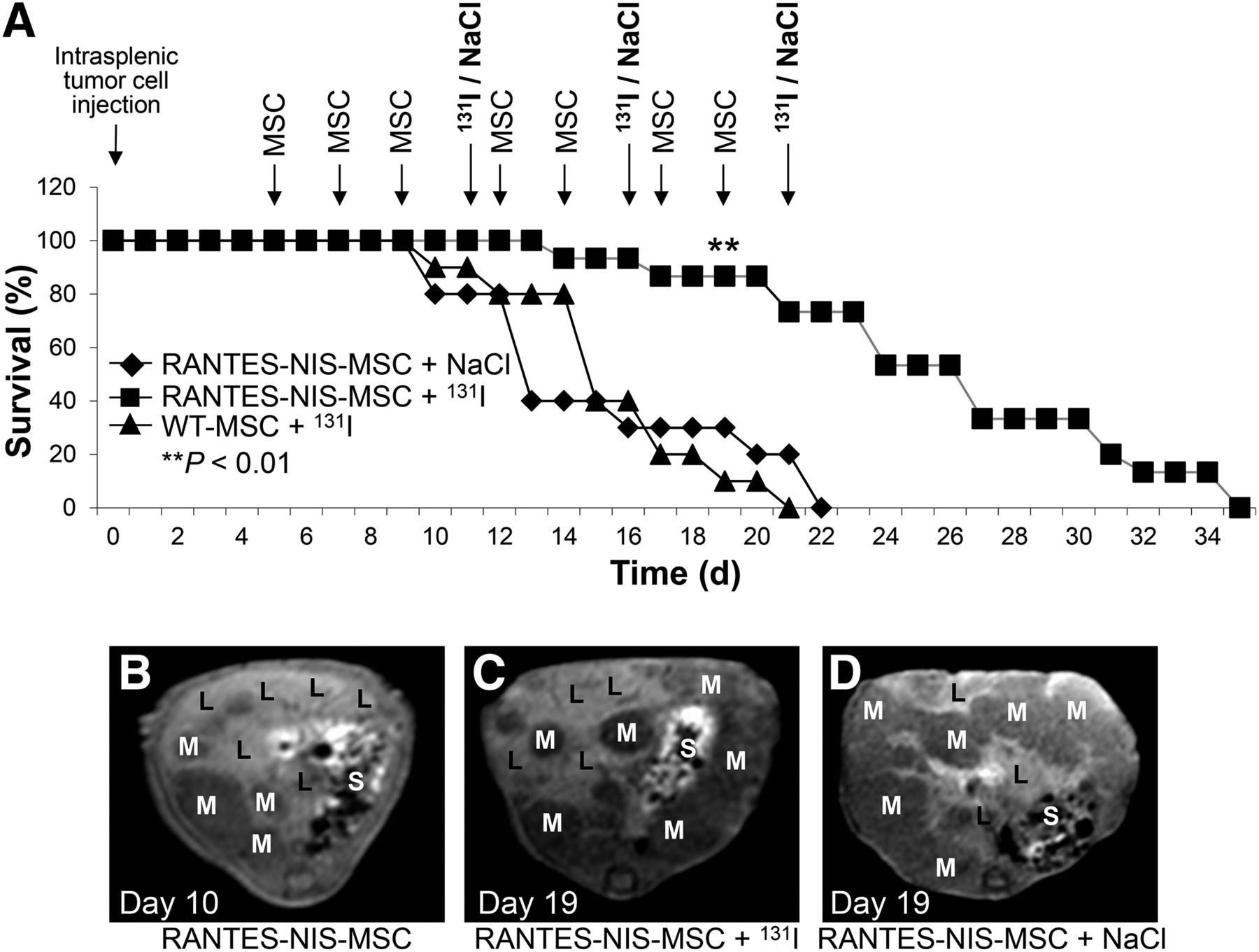
Mice received 3 rounds of RANTES-NIS-MSC or WT-MSC applications followed by 131I (55.5 MBq) administration. Further control group received saline instead of 131I. Therapeutic application of 131I after injection of RANTES-NIS-MSCs resulted in significantly improved survival as compared with control groups (A). MR imaging monitoring of metastases growth showed small metastases in liver with hepatic tumor load of approximately 40% at day 10 after intrasplenic LS174t cell injection (after 3 MSC applications and before first 131I injection) (B). Exponential tumor growth was observed in control group (RANTES-NIS-MSC + NaCl), with tumor load of at least 90% at day 19 (D) in contrast to therapy group (RANTES-NIS-MSC + 131I) (C), which revealed delayed metastases growth, with tumor load of approximately 60% at day 19 after intrasplenic tumor cell injection. L = liver; M = metastases; S = stomach.
MR Imaging
For MR imaging examinations, animals were anesthetized by intraperitoneal injections of ketamine (Inresa Arzneimittel) (100 mg/kg of body weight) and xylazine (Bayer) (10 mg/kg of body weight), and a 27-gauge tail vein catheter was placed for subsequent contrast medium administration. MR imaging was performed on a clinical 3-T system (Magnetom Skyra; Siemens Healthcare) with animals prone, using a clinical wrist coil (Siemens Healthcare). MR images were acquired using a brief imaging protocol with T1 sequences in axial and coronal views for optimal delineation of intrahepatic tumor manifestations. T1-weighted fast low-angle shot (FLASH) 3-dimensional sequences were acquired before contrast and after a standardized intravenous manual bolus injection of 100 μL of gadolinium-based contrast medium (Primovist; Bayer Healthcare AG). Sequence details were a repetition time of 5.74 ms, echo time of 2.26 ms, slice thickness of 1 mm, flip angle α of 10°, matrix size of 416 × 416, reconstructed matrix of 832 × 832, field of view of 150 × 150 mm, spatial resolution of 0.18 × 0.18 × 1 mm, 80 slices, and acquisition time of 142 s.
Statistical Methods
The statistical significance of in vitro experiments was tested using the Student t test. The statistical significance of the in vivo experiments has been calculated using the Mann–Whitney U test.
RESULTS
Radioiodine In Vivo Imaging Studies
To evaluate the ability of eMSC to target early stages of colon cancer liver metastases, eMSC injections via the tail vein were started 5 d after intrasplenic injection of the human colon cancer cells LS174t, before visible metastases developed; biodistribution of RANTES-NIS-MSC was then determined by 123I scintigraphy. Enhanced iodide uptake activity was detected in the liver area 2 h after injection of 18.5 MBq of 123I (Fig. 2A), resulting in a maximum uptake of 12.1 ± 2.6 percentage injected dose per gram (%ID/g) with a biologic half-life of 2.9 h and a tumor-absorbed dose of 63.2 mGy/mBq (Supplemental Fig. 1 [supplemental materials are available at http://jnm.snmjournals.org]). In contrast, after injection of WT-MSC no liver-specific iodide accumulation was detected (Fig. 2C). To further demonstrate NIS specificity of iodide uptake in the liver region, a subset of RANTES-NIS-MSC–treated mice received NaClO4 30 min before radioiodine injection, resulting in a complete blockade of iodide accumulation in liver metastases, in addition to the thyroid gland and stomach (Fig. 2B), which represent physiologically NIS-expressing organs. The high iodide accumulation seen in the bladder is a result of renal iodide excretion. To improve the resolution of radioiodine uptake in the liver area, and to more clearly differentiate between hepatic and gastric iodide accumulation, additional PET imaging studies were performed using 124I. Three-dimensional data were subsequently generated using iterative reconstructions of list-mode data (0–40 min), which provided better anatomic definition. Forty-eight hours after the last RANTES-NIS-MSC administration, 12 MBq of 124I were applied, and selective iodide accumulation was detected in single metastases 3 h after 124I application (Figs. 2D and 2E). In 70% of RANTES-NIS-MSC–treated mice, a maximum uptake of 16.2 ± 3.5 %ID/g was measured in single nodules.
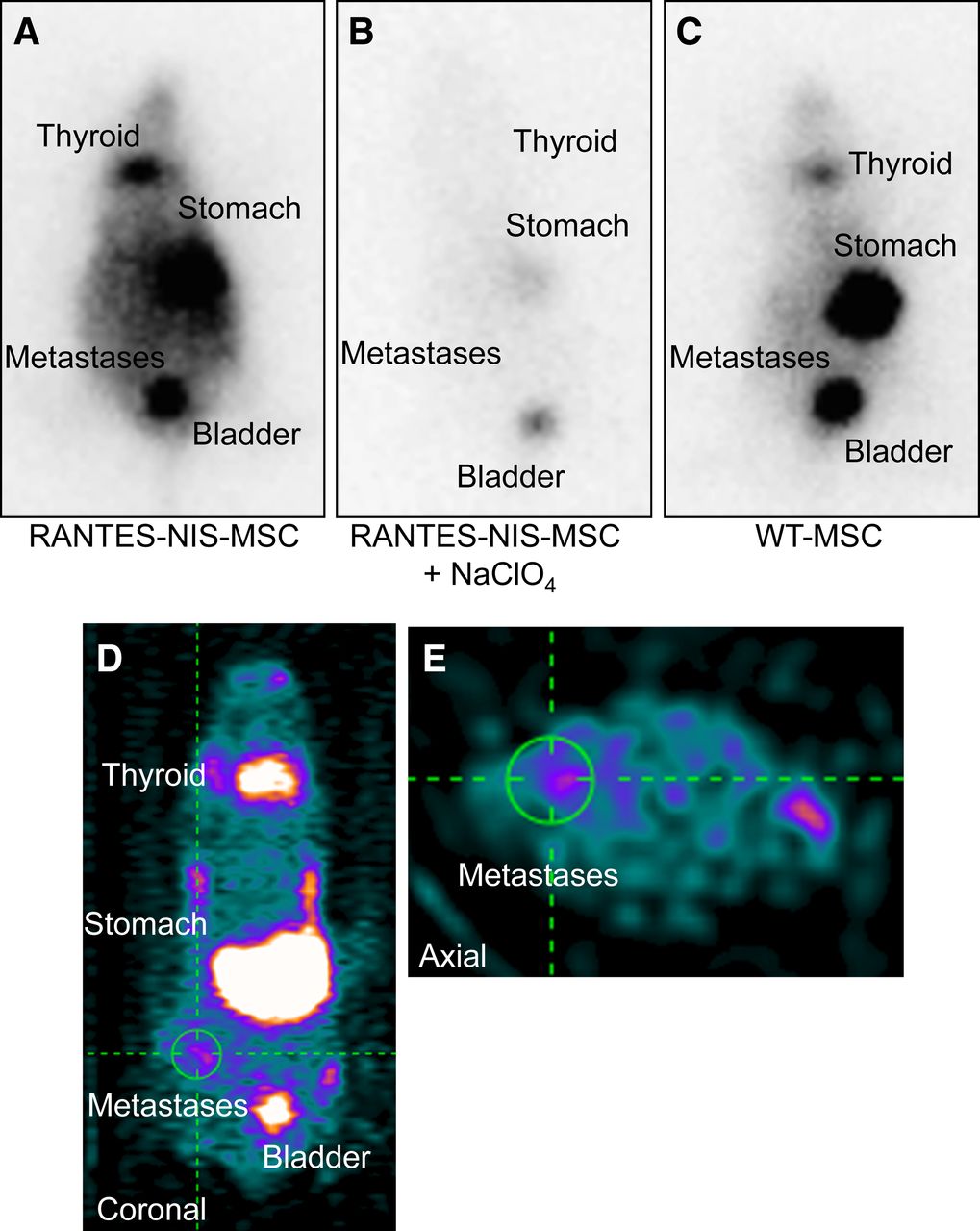
Systemic injection of RANTES-NIS-MSCs resulted in enhanced iodide accumulation in livers of mice harboring colon cancer metastases as shown by 123I γ-camera imaging (A) and small-animal 124I PET imaging (D and E) 2 h after radionuclide injection, which was completely blocked upon treatment with NIS-specific inhibitor NaClO4 (B). Intravenous injection of WT-MSCs did not result in enhanced radioiodine accumulation in liver region (C).
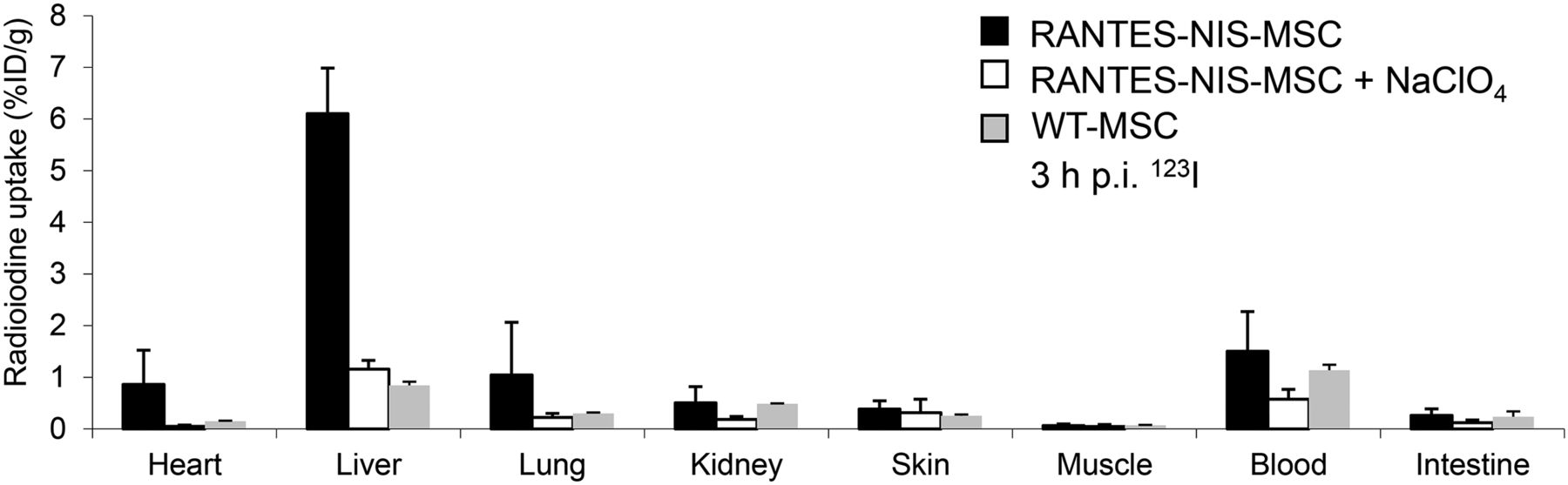
Ex Vivo Radioiodine Biodistribution Study
Ex vivo γ counter analysis of radioiodine biodistribution was performed, confirming increased iodide uptake (∼6.1 ± 1.1 %ID/g) in the liver of RANTES-NIS-MSC–treated mice 3 h after 123I injection (Fig. 3). In contrast, mice injected with WT-MSCs showed no significant hepatic iodide uptake, and no significant iodide uptake levels were observed in nontarget organs (Fig. 3). In both groups, the thyroid gland and the stomach accumulated approximately 40% and 39% 123I injected dose per organ, respectively, resulting from endogenous expression of NIS in these organs (data not shown). In additional control mice injected with RANTES-NIS-MSC, administration of the competitive inhibitor perchlorate resulted in a blockade of iodide uptake in the liver (Fig. 3) as well as in the thyroid gland and stomach (data not shown).
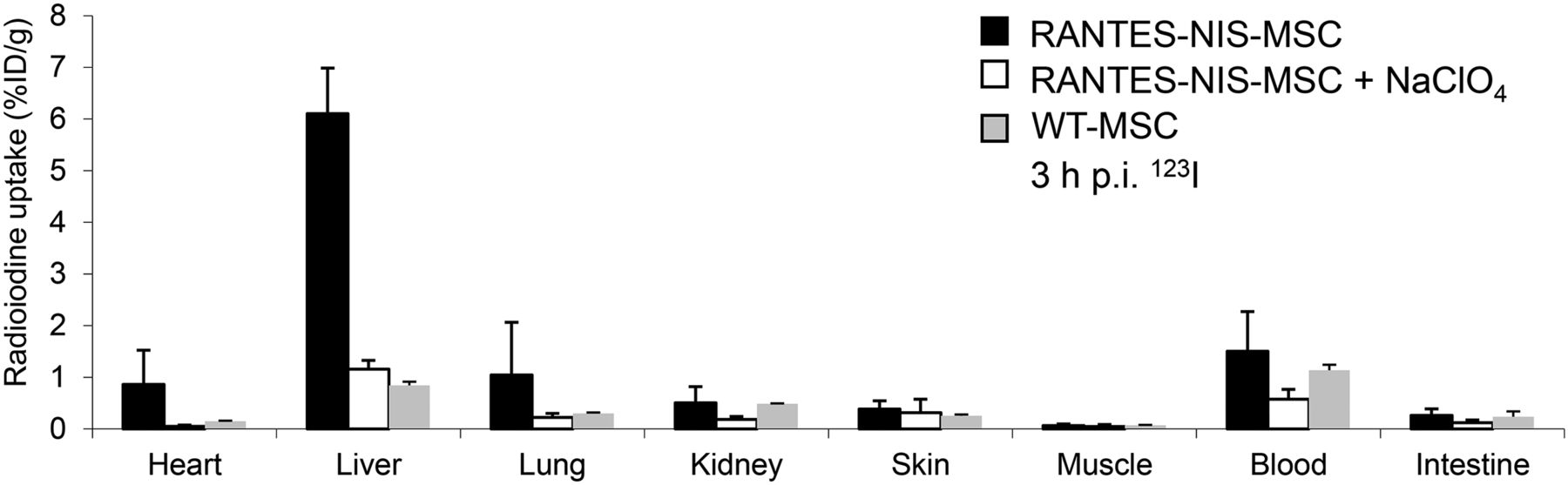
Ex vivo 123I biodistribution studies revealed hepatic iodide uptake of approximately 6.1 ± 1.1 %ID/g, whereas no significant radioiodine uptake was measured in nontarget organs, after pretreatment with perchlorate or after systemic injection of WT-MSCs. Results are reported as %ID per organ ± SD. p.i. = after injection.
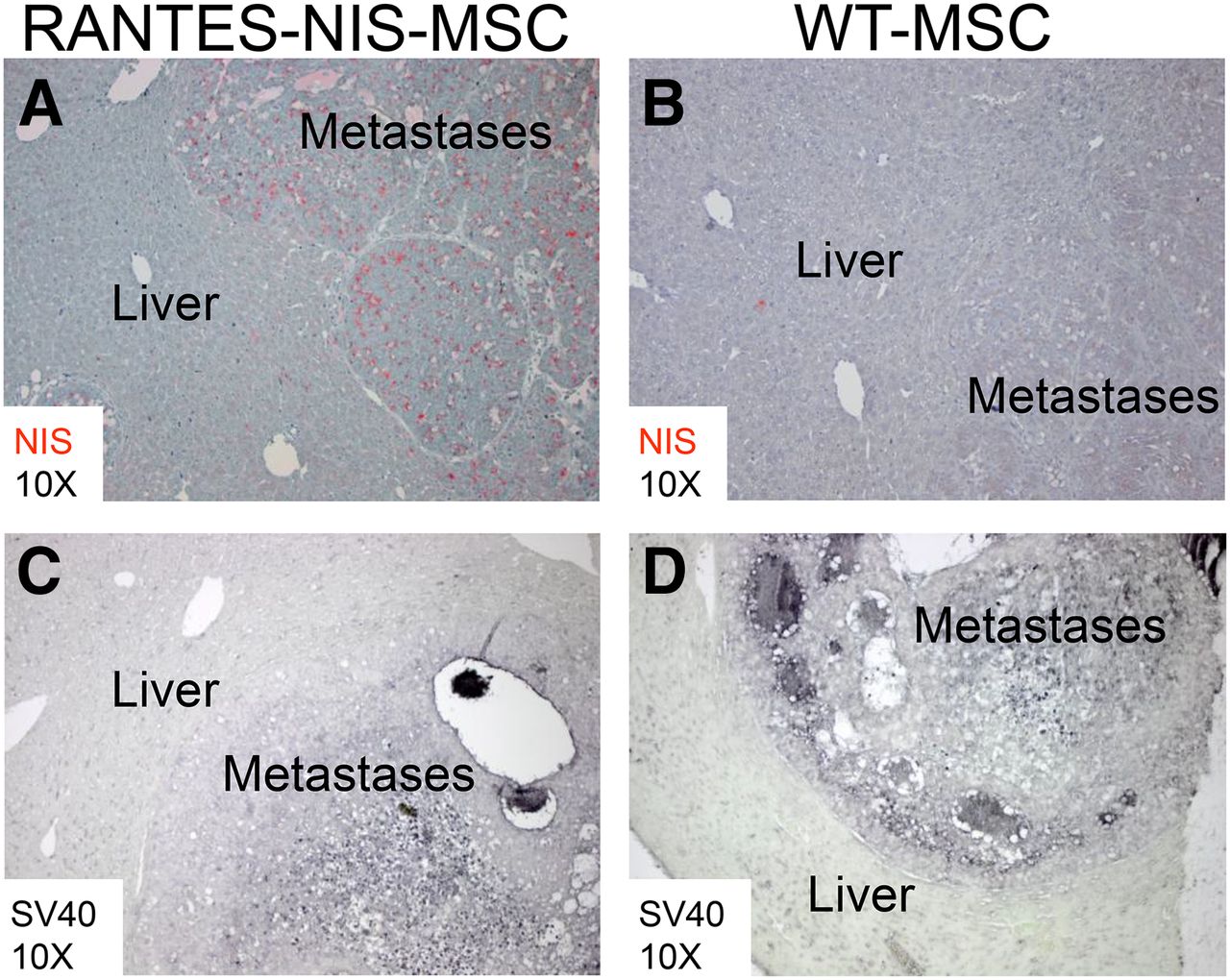
Immunohistochemical Analysis of eMSC/Transgene Biodistribution
SV40 large T Ag was used to immortalize the MSCs and could thus be used as a target to assess the biodistribution of eMSCs, whereas human NIS-specific antibodies allowed the determination of RANTES promoter–induced NIS transgene expression. Paraffin-embedded tissues from the liver and additional nontarget organs (lung, kidneys) were processed for immunohistochemical staining using both sets of antibodies. In liver metastases of mice treated with RANTES-NIS-MSCs, NIS-specific immunoreactivity was confined to metastatic nodules, and no NIS-specific staining was seen in the surrounding normal liver tissue (Fig. 4A). This biodistribution pattern was confirmed by SV40 large T Ag–specific staining (Fig. 4C). Systemic injection of WT-MSCs showed no NIS-specific immunoreactivity in metastases or in normal liver tissue (Fig. 4B). However, SV40 large T Ag staining still demonstrated active WT-MSC recruitment into the metastatic nodules (Fig. 4D), thus supporting tumor-selective eMSC recruitment. Immunofluorescence analysis confirmed the colocalization of NIS and SV40 large T Ag staining in the stroma of liver metastases of tumor-bearing mice after the injection of RANTES-NIS-MSC (Supplemental Figs. 2A–2C).
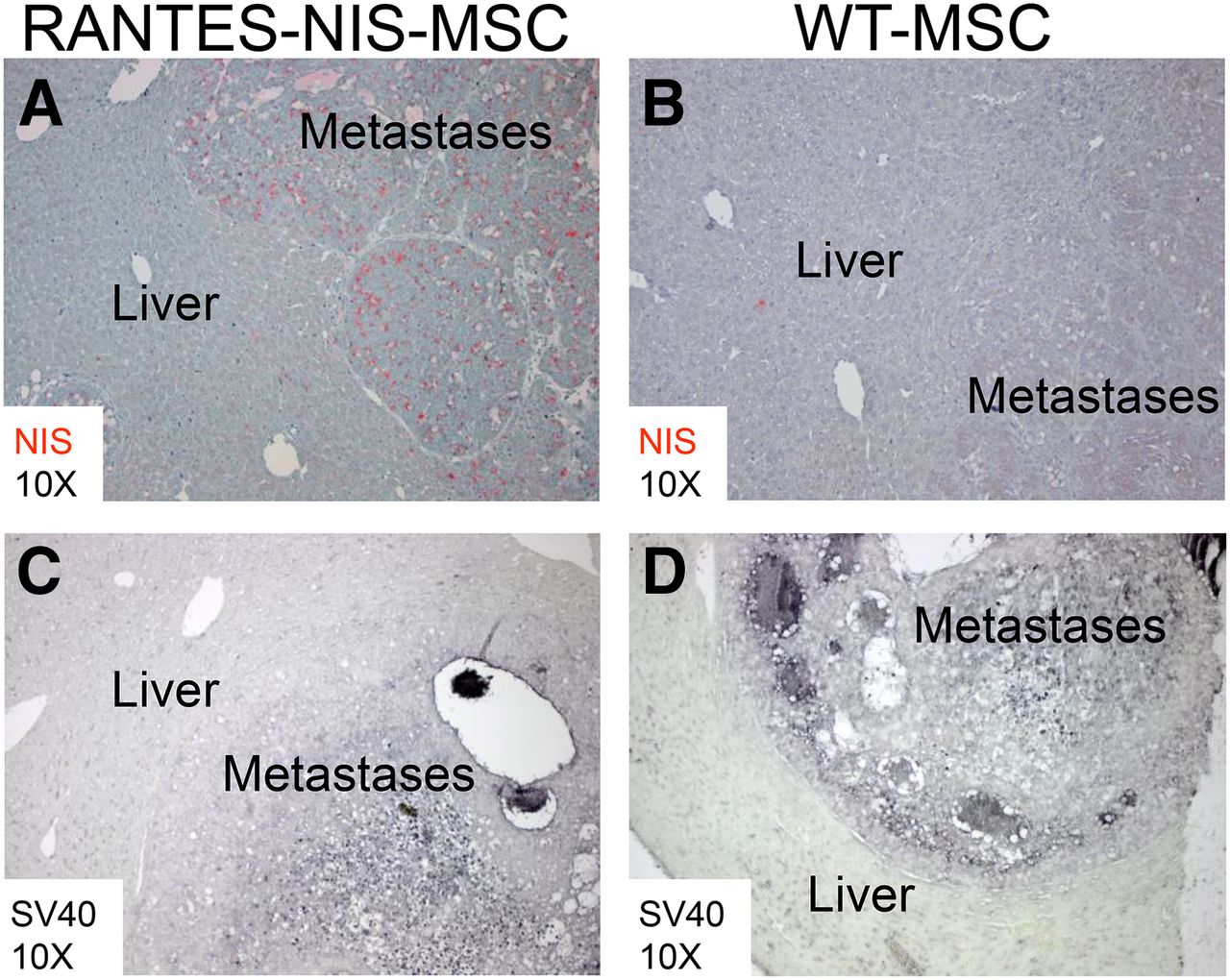
After systemic RANTES-NIS-MSC injection, strong NIS-specific immunoreactivity was detected confined to metastatic tissue (A), whereas normal liver tissue did not show NIS-specific immunostaining. No NIS-specific immunoreactivity was detected after systemic injection of WT-MSCs (B). SV40 large T Ag staining demonstrated presence of RANTES-NIS-MSC (C) and WT-MSC (D) in hepatic colon cancer metastases.
In nontarget organs such as the lung or kidney, no NIS-specific or SV40 large T Ag–specific staining was detected in mice treated with RANTES-NIS-MSCs (Supplemental Figs. 2D–2G).
Analysis of NIS mRNA Expression
Metastatic livers from mice treated with RANTES-NIS-MSCs revealed a 7.6-fold increased level of NIS mRNA expression as compared with metastatic livers from mice treated with WT-MSCs. As expected, additional treatment with the competitive NIS inhibitor perchlorate (NaClO4) had no influence on NIS mRNA expression in metastases-bearing mice injected with RANTES-NIS-MSC (7.4-fold increase). In contrast, nontarget organs such as the lung or kidney showed no NIS mRNA expression in RANTES-NIS-MSC– or WT-MSC–treated mice (Supplemental Fig. 3).
131I Therapy Study
The therapeutic effect of 131I was assessed using the therapy regimen optimized in previous studies (4,5). The protocol is based on 3 cycles of eMSC injections followed by 131I (Fig. 1A). After systemic injection of RANTES-NIS-MSC followed by 131I injection, a significantly improved survival of up to 14 d was observed as compared with control groups (Fig. 1A).
The growth of metastases was monitored by MR imaging starting before the first 131I application after 3 injections of eMSCs (10 d after intrasplenic tumor cell injection) when the tumor load in the liver was moderate (∼40%) (Fig. 1B). Over time, an exponential tumor growth was seen in the control groups (RANTES-NIS-MSC + NaCl or WT-MSC + 131I) as compared with the therapy group (RANTES-NIS-MSC + 131I), which showed a significantly reduced tumor growth. MR images at day 19 showed a reduced hepatic tumor load of approximately 60% in the therapy group (RANTES-NIS-MSC + 131I) (Fig. 1C) as compared with a tumor load of at least 90% in the control group (RANTES-NIS-MSC + NaCl) (Fig. 1D). Mice in the control groups did not survive long enough to receive the final 131I or saline application.
At the end of the experiments, animals were sacrificed and their organs removed and characterized by immunofluorescence analysis for cellular proliferation (Ki-67, green) and blood vessel density (CD31, red) markers. The results showed striking differences between therapy and control groups (Supplemental Figs. 4A–4C). Mice treated with RANTES-NIS-MSCs followed by 131I showed decreased proliferation (Ki-67, 23.3% ± 2.5%) and blood vessel density (CD31, 3.1% ± 0.2%), whereas control mice treated with RANTES-NIS-MSC followed by NaCl or treated with WT-MSCs followed by 131I revealed high blood vessel density (RANTES-NIS-MSC + NaCl, CD31: 6.6% ± 0.7%; WT-MSC + 131I, CD31: 7.8% ± 0.8%) and high levels of proliferation (RANTES-NIS-MSC + NaCl, Ki-67: 73.8% ± 6.9%; WT-MSC + 131I, Ki-67: 76.5% ± 6,7%) (Supplemental Figs. 4D and 4E).
DISCUSSION
In the present study, an emerging gene therapy approach was evaluated in which the therapy gene was expressed in the context of eMSC-based protocols—specifically for the treatment of metastatic disease. eMSCs are excellent gene delivery vehicles based in part on their relative ease of engineering and expansion in vitro. Importantly, these cells also show a remarkable natural tropism for solid tumors (22,23). An array of therapy proteins has already been demonstrated to be successfully delivered to tumor environments using eMSC, including interferon-γ, TNF-related apoptosis-inducing ligands, IL-12, the chemokine CX3CL1, and various suicide genes (4,5,24,25). Recent studies have shown that adoptively applied MSCs can also efficiently home to tumor metastases, opening the door to the potential use of eMSC for treatment of metastatic disease (26–29).
In a recent study, using experimental models of melanoma, breast, and hepatoma tumors, the authors applied an IL-12–based eMSC approach that showed reduced progression of metastases at mid stage of development and even regression at later stages after an extended course of intravenous immunotherapy using IL-12 gene–engineered MSCs (29). In a model of metastatic breast cancer, Zhao et al. showed that human neural stem cells engineered to constitutively secrete the suicide gene carboxylesterase were also able to target tumor metastases in multiple organs including the liver, lymph nodes, and lung (27).
The downside of gene therapy using traditional suicide genes is that even though they act through bystander killing, their effects are limited to the cells most proximal to the transgene-expressing cells. In contrast, NIS-targeted radioiodine therapy is associated with a higher degree of bystander killing effect that results from the crossfire effect of the β-emitter 131I and from the radiation-induced biologic bystander effect. This therapy concept has been effectively used for the treatment of differentiated thyroid cancer for almost 70 y and still represents one of the most effective systemic anticancer radiotherapies available to the clinician today.
The application of the NIS gene as a combined imaging/therapy gene has been an area of expanded research in various tumor settings over the past few years (13–17). The ability to noninvasively monitor the biodistribution of NIS expression after systemic gene delivery provides an essential extension to the clinical setting. A large number of NIS-compatible radioactive tracers are available including 123I for scintigraphy/SPECT and 124I- and 18F-tetrafluoroborate (30) for PET imaging. Finally, NIS transgene expression allows the delivery of a robust therapeutic effect through 131I or 188Re application (5,20,31–36). We have demonstrated the flexibility of this approach in studies using oncolytic viruses and nonviral nanoparticles equipped with the NIS gene to facilitate noninvasive in vivo vector biodistribution imaging as well as 131I-based radiotherapy after systemic application (37,38).
In previous studies, we demonstrated a proof of concept of systemic NIS gene delivery using MSCs as delivery vehicles with subcutaneous xenograft mouse models (4,5). Systemic application of eMSCs expressing NIS under control of the tumor stroma–associated RANTES promoter (RANTES-NIS-MSC) led to a significant radioiodide accumulation in a subcutaneous liver cancer xenograft model in nude mice, resulting in a dramatic delay of tumor growth and improved survival by therapeutic 131I application (5). The potential therapeutic efficacy of MSCs engineered to express NIS using the RANTES promoter in the treatment of tumor metastases represents a clinically important open issue. Here, a liver metastasis model of colon cancer was used to analyze the biodistribution of MSC recruitment and NIS-mediated radioiodine accumulation.
By 123I γ camera and 124I PET imaging, NIS-eMSCs were shown to specifically home to liver metastases and induce RANTES promoter–driven NIS transgene expression, resulting in a tumor-selective radioiodine accumulation. While 123I scintigraphic imaging showed a diffusely elevated radioiodine uptake in the hepatic area, 124I PET imaging allowed a 3-dimensional analysis of NIS-mediated radioiodine accumulation with higher resolution showing iodide uptake confined to metastatic nodules. Ex vivo analysis of radioiodine biodistribution confirmed NIS-specific iodine uptake confined to the liver, which was further validated by quantitative PCR analysis and immunohistology, demonstrating strong NIS and SV40 large T Ag immunoreactivity strictly confined to metastatic tissue without expression in normal liver tissue or nontarget organs. In our previous study in the subcutaneous liver cancer xenograft mouse model with normal liver, we have also not seen any hepatic NIS and SV40 large T Ag immunoreactivity after systemic application of RANTES-NIS-MSC, demonstrating that MSC recruitment into liver metastases is not the result of significant liver pooling of MSCs (5).
In therapy studies, a significant reduction of tumor load in the liver was observed from at least 90% in the control groups to approximately 60% in the therapy group starting with 40% tumor load before the first 131I application. This reduction was associated with a significantly extended life span, which is highly significant considering the aggressive growth of tumor metastases in this model. These findings correlate with markedly reduced proliferation and blood vessel density in the tumors of the therapy group.
In contrast to our study, previous studies of gene therapy of metastatic disease analyzed therapeutic efficacy ex vivo. Zischek et al. showed a reduced level of metastases in a pancreatic carcinoma mouse model by ex vivo analysis of tumor growth after treatment with MSCs expressing the HSV-TK suicide gene (6). With the translation from animal to human studies in mind, in the present study we used contrast-enhanced MR imaging to monitor the growth of hepatic colon cancer metastases over time in vivo. MR imaging is emerging as a powerful method to investigate the potential effects of treatment approaches in mice. The contrast between tumor and normal tissue was enhanced by a liver-specific contrast agent (Primovist), which allowed an optimal evaluation of treatment effects in hepatic metastases (39).
We are aware that with the mouse model used in this study based on metastases of human colon cancer cells to the liver in athymic nude mice, the influence of the immune system on MSC biodistribution as well as on tumor growth and therapeutic efficacy of MSC-mediated NIS radionuclide therapy cannot be investigated and will be addressed in future studies. MSCs are known to have immunosuppressive functions that may influence therapeutic efficacy. However, previous studies from our group using syngeneic mouse models with well-defined tumor antigens have convincingly demonstrated tumor-selective homing of MSCs without changes of growth dynamics of the tumor after systemic application of control MSCs (6,9).
CONCLUSION
Noninvasive 123I γ camera and 124I PET imaging demonstrated remarkable metastatic selectivity of MSC recruitment and NIS expression driven by the tumor stroma–specific RANTES/CCL5 promoter after systemic MSC application. The tumor stroma–targeted iodide uptake confined to liver metastases was strong enough for a significant reduction of metastases growth monitored in vivo by MR imaging, resulting in significantly improved survival. These data therefore convincingly demonstrate the enormous potential of MSC-mediated NIS gene radionuclide therapy in metastatic cancer.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This study was supported by grants from the Deutsche Forschungsgemeinschaft (Bonn, Germany)—SFB 824 (project C8) and SP 581/6-1 (SPP 1629)—and by a grant from the Wilhelm-Sander-Stiftung (2008.037.1). No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We are grateful to Sissy M. Jhiang, Ohio State University, Columbus, Ohio, for supplying the full-length human NIS complementary DNA and to Roswitha Beck, Rosel Oos, and Andreas Delker (Department of Nuclear Medicine, Ludwig-Maximilians-University, Munich, Germany) and Matthias Moser (Department of Clinical Radiology, Laboratory for Experimental Radiology, University Hospital Munich, Munich, Germany) for their assistance with imaging and therapy studies.
Footnotes
Published online Mar. 5, 2015.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 28, 2014.
- Accepted for publication January 12, 2015.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Targeting Novel Sodium Iodide Symporter Interactors ADP-Ribosylation Factor 4 and Valosin-Containing Protein Enhances Radioiodine Uptake
- Radiation-Induced Amplification of TGFB1-Induced Mesenchymal Stem Cell-Mediated Sodium Iodide Symporter (NIS) Gene 131I Therapy
- A Novel Approach for Image-Guided 131I Therapy of Pancreatic Ductal Adenocarcinoma Using Mesenchymal Stem Cell-Mediated NIS Gene Delivery
- Role of iodide metabolism in physiology and cancer
- Thyroid hormones and tetrac: new regulators of tumour stroma formation via integrin {alpha}v{beta}3