


Abstract
Triple-negative breast cancer (TNBC) is associated with poor survival. Chemotherapy is the only standard treatment for TNBC. The prevalence of BRCA1 inactivation in TNBC has rationalized clinical trials of poly(adenosine diphosphate ribose) polymerase (PARP) inhibitors. Similarly, the overexpression of epidermal growth factor receptor (EGFR) rationalized anti-EGFR therapies in this disease. However, clinical trials using these 2 strategies have not reached their promise. In this study, we used EGFR as a target for radioimmunotherapy and hypothesized that EGFR-directed radioimmunotherapy can deliver a continuous lethal radiation dose to residual tumors that are radiosensitized by PARP inhibitors and chemotherapy. Methods: We analyzed EGFR messenger RNA in published gene expression array studies and investigated EGFR protein expression by immunohistochemistry in a cohort of breast cancer patients to confirm EGFR as a target in TNBC. Preclinically, using orthotopic and metastatic xenograft models of EGFR-positive TNBC, we investigated the effect of the novel combination of 177Lu-labeled anti-EGFR monoclonal antibody, chemotherapy, and PARP inhibitors on cell death and the survival of breast cancer stem cells. Results: In this first preclinical study of anti-EGFR radioimmunotherapy in breast cancer, we found that anti-EGFR radioimmunotherapy is safe and that TNBC orthotopic tumors and established metastases were eradicated in mice treated with anti-EGFR radioimmunotherapy combined with chemotherapy and PARP inhibitors. We showed that the superior response to this triple-agent combination therapy was associated with apoptosis and eradication of putative breast cancer stem cells. Conclusion: Our data support further preclinical investigations toward the development of combination therapies using systemic anti-EGFR radioimmunotherapy for the treatment of recurrent and metastatic TNBC.
Breast cancer is the leading cause of cancer-related mortality among women worldwide, and 30%–40% of breast cancer patients will develop metastatic disease. Currently, no targeted therapies are used as the standard of care for triple-negative breast cancer (TNBC), which is defined by the lack of expression of human epidermal growth factor receptor 2 (HER2), estrogen receptor, and progesterone receptor. This aggressive disease, relative to other breast cancer subtypes, is associated with a higher incidence of recurrence and distant metastasis and a poorer survival (1). Conventional cytotoxic chemotherapy remains the only treatment option for TNBC.
In 2 large studies of breast cancer patients (2,3), 17%–24% of patients were identified as having TNBC by immunohistochemistry. Triple-negative status still identifies a heterogeneous group with significant differences in molecular markers and survival. The basal-like subtype of TNBC, which stains positively for high-molecular-weight cytokeratins or epidermal growth factor receptor (EGFR) by immunohistochemistry, represents 53%–67% of TNBC (2–4). Interestingly, although basal-like TNBC has the poorest outcome (3,4), patients with these tumors have a disease-free survival benefit with adjuvant chemotherapy (2,4).
Several studies have reported high expression of EGFR by immunohistochemistry in 40%–70% of TNBC patients (2,4–6), with high expression correlating with poor outcome and decreased disease-free survival (5). Elevation of EGFR messenger RNA (mRNA), as well as activation of the EGFR pathway in gene expression microarray studies, is a characteristic of basal-like breast cancer (2,7) and is associated with poor prognostic signatures in almost all basal-like tumors (8). Thus, EGFR represents a clinically relevant target in basal-like TNBC. Another target for TNBC is poly(adenosine diphosphate ribose) polymerase-1 (PARP1). The high prevalence of inactivation of BRCA1, which is involved in the homologous recombination DNA repair pathway, in TNBC supports combining chemotherapy or radiotherapy with PARP inhibitors. A synthetic lethality approach using PARP inhibition has been the subject of recent reviews (9).
Although both EGFR and PARP may be attractive targets for breast cancer drugs, EGFR inhibition (antibodies or small inhibitors) or PARP inhibition has shown limited success in treating TNBC (1). In this study, we used EGFR as a target for radioimmunotherapy in EGFR-positive TNBC. We hypothesized that anti-EGFR monoclonal antibody (mAb) labeled with the therapeutic radionuclide 177Lu would deliver a lethal and continuous radiation dose to radiosensitized tumor cells remaining after chemical tumor debulking and PARP inhibition.
MATERIALS AND METHODS
EGFR Gene and Protein Expression in Breast Cancer Patients
Gene expression array data from 5 studies (GSE12276, GSE21653, GSE2990, GSE3494, and GSE2034) were obtained from the Gene Expression Omnibus. The datasets were analyzed for EGFR mRNA expression across the intrinsic subtypes as described in Supplemental Figure 4 (supplemental materials are available online only at http://jnm.snmjournals.org).
For immunohistochemical analysis of EGFR, approval was obtained from the Human Research Ethics Committee of the Royal Brisbane and Women’s Hospital and the University of Queensland to use human breast tumor samples for research. Tissue microarrays were enriched for high-grade invasive ductal carcinomas for which estrogen receptor, progesterone receptor, and HER2 immunohistochemical data were also available. Standard immunohistochemistry was performed on tissue microarray sections to evaluate EGFR protein expression (Supplemental Methods).
Orthotopic and Metastatic Breast Xenograft Models
Cell culture conditions and the production and radiolabeling of the anti-EGFR mouse mAb (clone 225), precursor to the derived chimeric human:murine antibody C225 commercially known as cetuximab, are detailed in the Supplemental Methods. The Animal Ethics Committee of the Queensland Institute of Medical Research gave approval for use of the mice. We used 2 orthotopic mouse models using the TNBC MDA-MB-231 cell line and the patient-derived TNBC xenograft HCI-002. We also used an experimental metastasis model established from intravenous injection of the MDA-MB-231 cell line (Supplemental Methods).
For the orthotopic mammary fat pad model, treatments were initiated when tumors were 50 ± 1 mm3 as calculated by caliper measurement of the longest (a) and shortest (b) diameters and the following equation: tumor volume (mm3) = a/2 × b2. For the metastasis model, treatments were initiated 14 d after inoculation, when metastases were established. Treatment groups consisted of 5 mice each for the MDA-MB-231 orthotopic model (3 independent experiments were performed), 10 mice each for the patient-derived HCI-002 orthotopic model, and 5–7 mice each in the MDA-MB-231 established metastasis model (2 independent experiments were performed). Docetaxel, doxorubicin, and the PARP inhibitor rucaparib (Pfizer’s PF-01367338) were purchased from Selleck Chemicals LLC and diluted in a 5% solution of d-glucose in phosphate-buffered saline for intraperitoneal injection. Docetaxel and doxorubicin were injected as single doses, whereas PARP inhibitor was injected as 5 daily doses. For radioimmunotherapy, 177Lu-anti-EGFR mAb in phosphate-buffered saline was injected in the tail vein 24 h after chemotherapy. Control mice received vehicle solutions, and EGFR control mice were injected with unlabeled anti-EGFR mAb. Mouse weights and tumor volumes were measured twice per week in the first 3 wk of treatment and then once per week.
Cell Death Assays in In Vivo and Ex Vivo Analyses
Activation of caspase-3 in vivo was detected by live animal imaging on day 7 after treatments using the VivoGlo Caspase-3/7 substrate (Promega Corp.). Images were acquired 10 min after intraperitoneal injection of the substrate (25 mg/kg).
Standard immunoblotting was performed using lysates prepared from freshly isolated tumors from mice 2 d after administration of 177Lu-anti-EGFR mAb. Membranes were probed with antibodies against phosphorylated DNA-dependent protein kinase (DNA-PK) at serine 2056 (Abcam), DNA-PK, RAD51 (Santa Cruz Biotech), and β-actin (Sigma-Aldrich) and developed using Chemilucent Plus (Millipore).
Additional apoptosis and cell death assays were performed ex vivo on tumors 3 and 7 d after treatments. Single-cell suspensions were prepared by collagenase B digestion as described previously (10) for annexin V/7-AAD apoptosis assays and DNA-content cell cycle assays (Invitrogen, Life Technologies) as per manufacturer instructions and analyzed using a FACSCalibur flow cytometer (BD Biosciences). Formalin-fixed, paraffin-embedded tumors were used to prepare 7-μm tissue sections for hematoxylin and eosin staining or immunohistochemistry with rabbit IgG against cleaved PARP1 (1:100 of stock; Promega) as described previously (10). Slides were scanned using ScanScope TX (Aperio Technologies).
The percentage of breast cancer stem cells (BCSCs) in vitro was investigated using a previously published method (11). Briefly, the cells were treated with single agents or combinations as described in the Supplemental Methods, and after 2 rounds of 3-d treatment, the cells were collected and stained (30 min at 4°C) with anti-CD44 PE-Cy5-mAb, anti-CD24 PE-mAb, and anti-EpCAM FITC-mAb (all from BD Biosciences) as per manufacturer instructions. In vivo, the tumors were excised after treatments to prepare single-cell suspensions as described earlier. Single-cell suspensions were stained with CD44, CD24, and EpCAM antibodies before analysis using the FACSCalibur flow cytometer.
Statistical analysis was performed with Prism, version 5.0 (GraphPad Software). The types of tests used are stated in the figure legends.
RESULTS
EGFR mRNA and Protein in Basal-Like TNBC
We found that EGFR mRNA levels are significantly higher in basal-like breast cancer than in luminal A and B and HER2 molecular subtypes but not in normal-like breast cancer (Supplemental Fig. 4). We also used immunohistochemistry on tissue microarrays of a sporadic breast cancer cohort and found that a higher percentage of TNBC (39%) than of non-TNBC (14%) was positive for EGFR (P = 0.005, Table 1).
Immunohistochemistry Scores for EGFR Staining in Cohort of Breast Cancer
Combination Therapy with 177Lu-Anti EGFR mAb
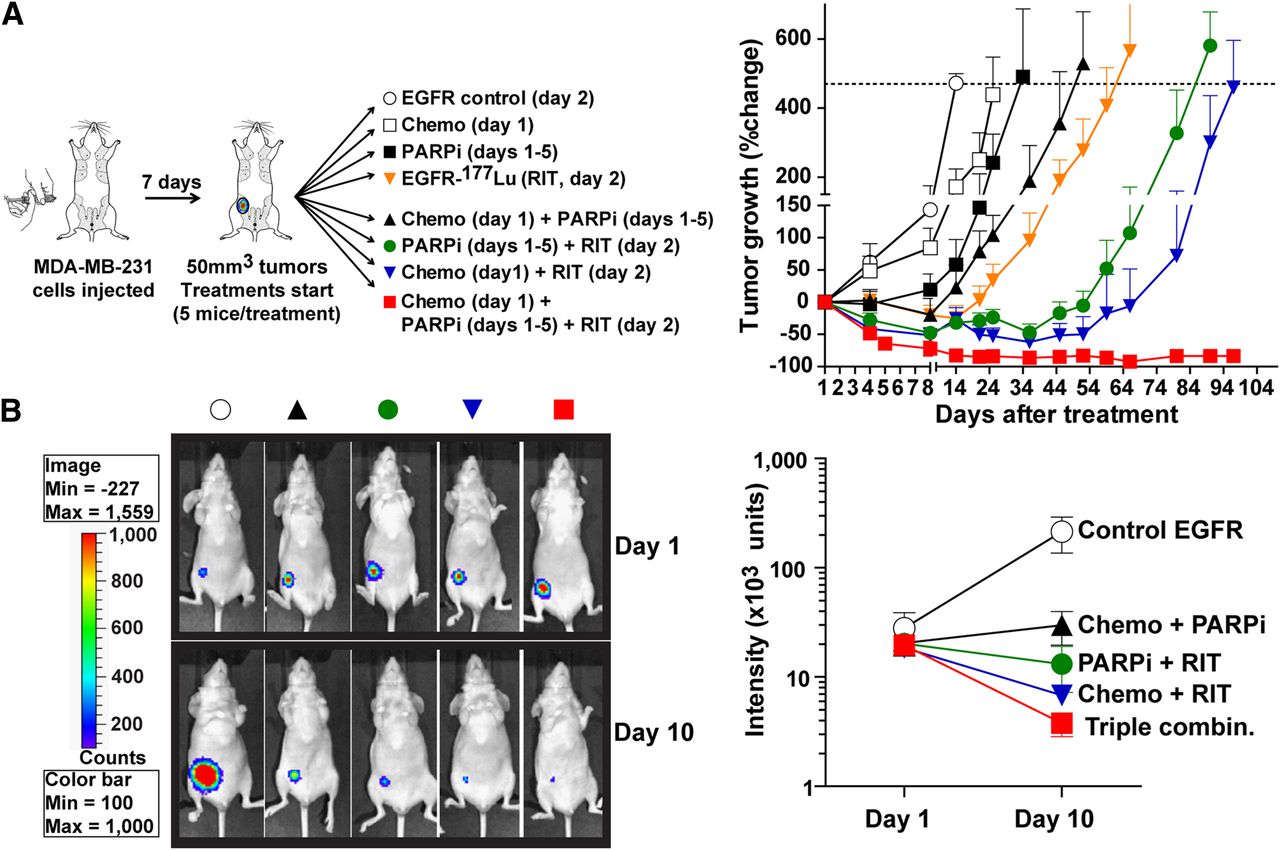
On the basis of toxicity and efficacy studies (Supplemental Results), we determined that chemotherapy with 2 mg of docetaxel and 10 mg of doxorubicin per kilogram and radioimmunotherapy with 300 MBq of 177Lu-anti-EGFR per kilogram were suitable to investigate with PARP inhibitor. Indeed, as shown in Fig. 1, 5 daily doses of PARP inhibitor at 2 mg/kg potentiated chemotherapy (tumor growth delay, 38 d for chemotherapy and PARP inhibitor vs. 11 d for chemotherapy alone) and 177Lu-anti-EGFR radioimmunotherapy (tumor growth delay, 76 d for PARP inhibitor and radioimmunotherapy vs. 52 d for radioimmunotherapy alone). Chemotherapy potentiated 177Lu-anti-EGFR radioimmunotherapy (tumor growth delay, 83 d for chemotherapy and radioimmunotherapy vs. 52 d for radioimmunotherapy alone). Treatment with naked anti-EGFR mAb alone, which acted as a control for radioimmunotherapy-containing treatments, had no effect on tumor growth, which was the same as in mice with untreated tumors. Strikingly, the combination of 177Lu-anti-EGFR radioimmunotherapy with chemotherapy and PARP inhibitor (triple-agent combination) eradicated the established xenografts, with no recurrences observed up to 120 d after treatment (Fig. 1). The efficacy of this triple-agent combination was confirmed by bioluminescence imaging, as minimal signal was observed on day 10 (Fig. 1B). From a total of 15 mice treated with the triple-agent combination, only 1 mouse showed residual disease, and it did not progress (6.7%); 14 mice (93.3%) were completely tumor-free up to 120 d.

Eradication of MDA-MB-231 mammary fat pad xenografts using combination of EGFR-directed radioimmunotherapy with chemotherapy and PARP inhibitor. Female nude BALB/c mice bearing MDA-MB-231 mammary fat pad tumors (5 mice per group) either were left untreated as vehicle controls or were treated with unlabeled anti-EGFR mAb (control EGFR), doxorubicin (10 mg/kg) and docetaxel (2 mg/kg) chemotherapy (chemo), PARP inhibitor at 2 mg/kg daily for 5 d (PARPi), or 177Lu-anti-EGFR mAb at 300 MBq/kg (RIT). Any treatment without radioimmunotherapy included unlabeled anti-EGFR mAb as control. Mouse weights and tumor volumes were monitored. No significant weight loss was observed. (A) Treatment schedules and tumor growth curves. Data are average percentage change (±SEM) in tumor volume. (B) Representative bioluminescence images of mice and mean luminescence intensities (±SEM) on days 1 and 10 after treatment (Supplemental Fig. 1 shows images of all mice).
Biologic Effects of the Triple-Agent Combination In Vivo
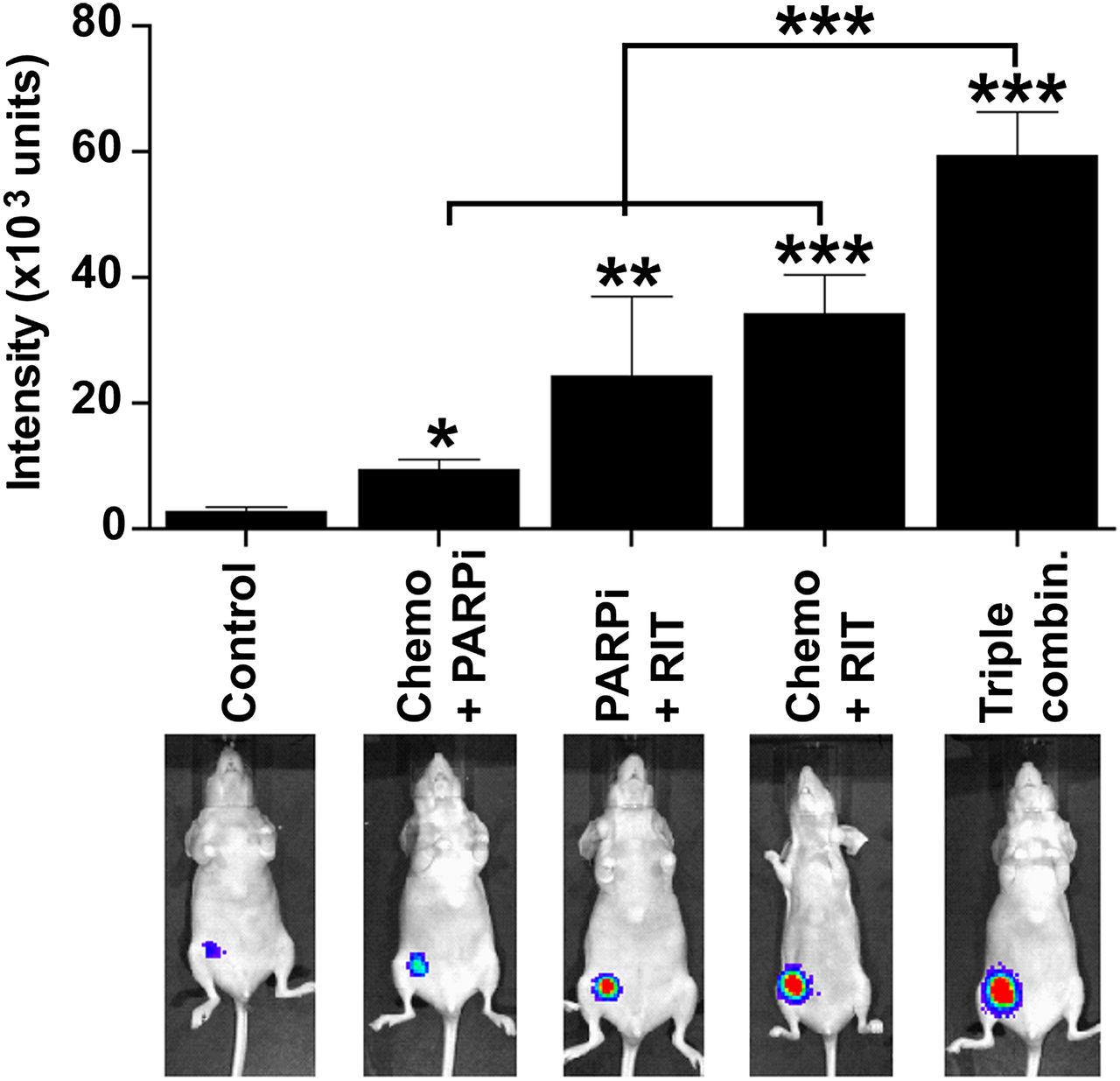
Next, we used bioluminescence imaging with caspase-3/7 substrate and found that the triple-agent combination induced significantly higher caspase activation in vivo than did other combinations (Fig. 2).
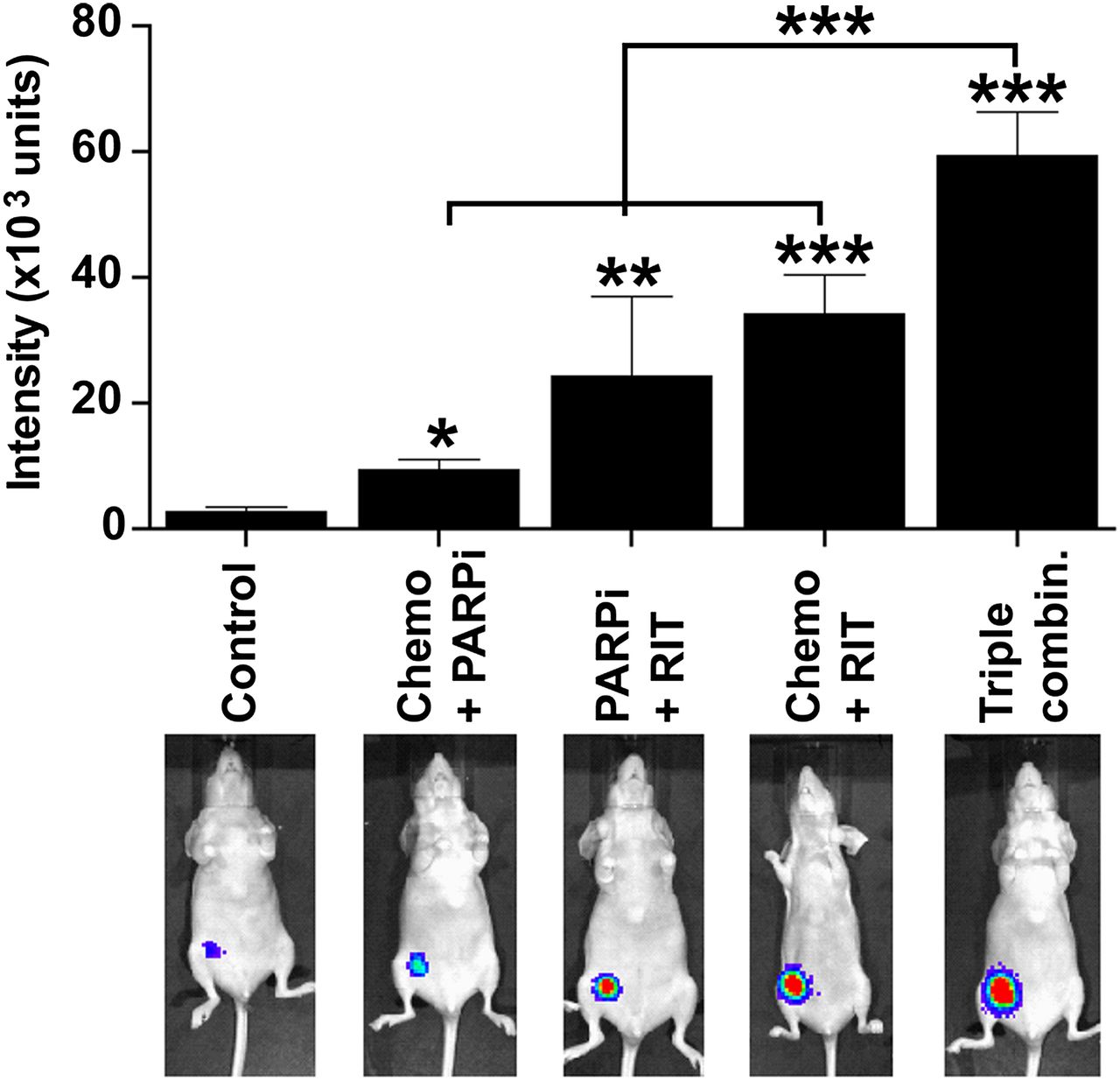
In vivo bioluminescent imaging of caspase-3/7 activation after treatment. Female nude BALB/c mice bearing luciferase-expressing mammary fat pad xenografts of MDA-MB-231 were left untreated or treated as indicated. Treatment doses, schedule, and routes of administration were as described in legend to Figure 1. Seven days after treatment (5 mice per group), mice were injected with VivoGlo caspase 3/7 substrate (25 mg/kg) and imaged 10 min later. Mean luminescence intensities (±SEM) for caspase-3 activation are summarized in bar graph. Representative images are shown for each treatment (Supplemental Fig. 2 shows images of all mice). *P < 0.05, **P < 0.01, ***P < 0.001 (1-way ANOVA with Tukey multiple-comparisons posttest).
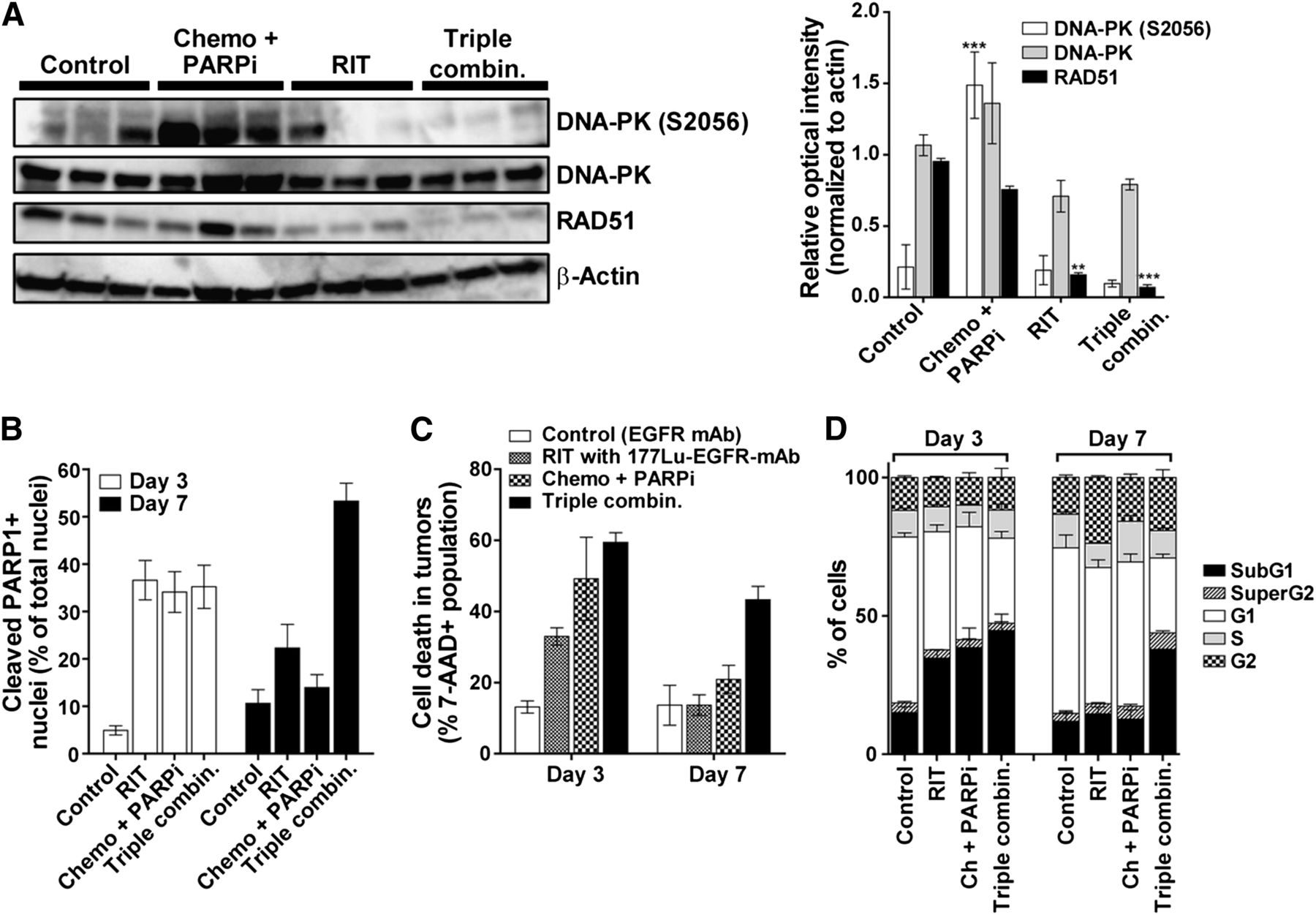
We excised tumors 2 d after radioimmunotherapy to quantify the expression of activated DNA-PK (phosphorylated at serine 2056 [DNA-PK S2056]) and RAD51 protein, central players in the DNA double-strand break repair pathways. As shown in Fig. 3A, DNA-PK was activated (S2056 phosphorylation) in tumors treated with chemotherapy and PARP inhibitor; however, this phosphorylation was inhibited when EGFR-directed radioimmunotherapy was added. In the case of RAD51, chemotherapy and PARP inhibitor did not affect the protein level, but RAD51 protein was significantly reduced in tumors from mice treated with the triple-agent combination. These data suggest that the inhibition of central DNA repair proteins contributed to the efficacy of the triple-agent combination. Additional in vitro studies revealed that this combination caused a delay in the repair of double-strand breaks (Supplemental Results).
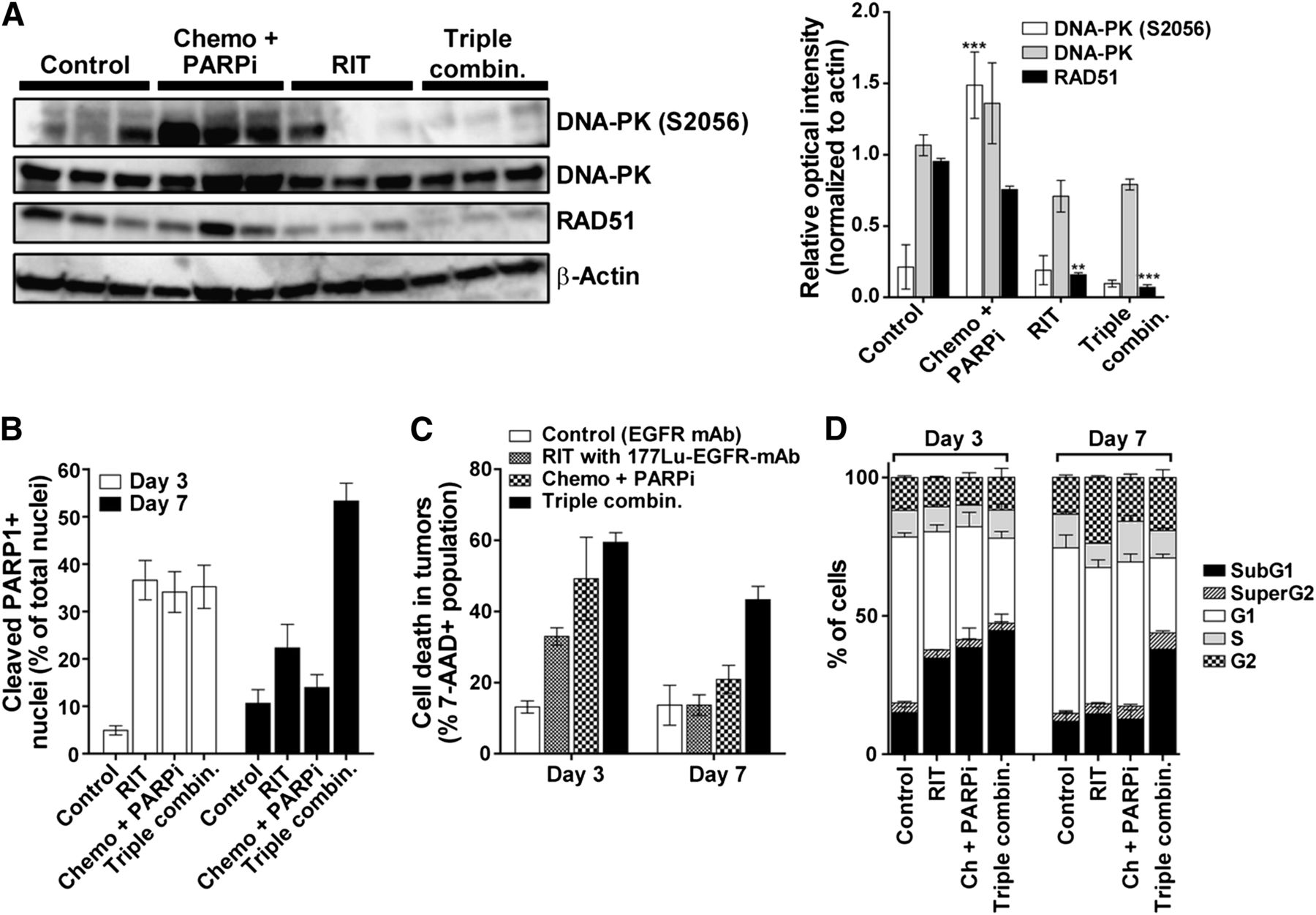
Ex vivo investigation of DNA repair proteins and cell death after combination therapy. (A) Immunoblot analysis of tumors excised from mice 2 d after administration of 177Lu-anti-EGFR radioimmunotherapy. Bar graph depicts mean relative intensity of specific bands normalized to loading control β-actin (±SEM, 3 mice per group). Asterisks denote statistical differences in comparison to tumors from control mice. (B) Quantification of cleaved PARP1 detected by immunohistochemistry of tumors on days 3 and 7 after treatments (Supplemental Fig. 6 presents representative hematoxylin and eosin and cleaved PARP1 immunohistochemistry images). Data are mean (±SEM) percentage of nuclei staining positively for cleaved PARP1 to total number of nuclei (hematoxylin counterstain). (C and D) Quantification of percentage cell death using 7-AAD vital dye (C) and proportion of cells in different cell cycle phases by FACSCalibur analysis (D) in single-cell suspensions prepared from tumors at 3 and 7 d after treatment (3 tumors/d/treatment). Ch and chemo = chemotherapy; combin. = combination; PARPi = PARP inhibitor; RIT = radioimmunotherapy. ***P < 0.001, **P < 0.01 (2-way ANOVA).
To determine the consequence of DNA repair inhibition, we further examined tumor responses ex vivo. In agreement with studies of caspase activation in vivo, ex vivo studies showed significantly higher cleavage of PARP1 (an event mediated by active caspase-3) that persisted up to day 7 when mice were treated with the triple-agent combination (Fig. 3B and Supplemental Fig. 6). Flow cytometry analysis showed high levels of persistent cell death after treatment with the triple combination as shown by lack of cell viability staining (Fig. 3C) and sub-G1 accumulation (Fig. 3D), corroborating the findings of the apoptosis studies.
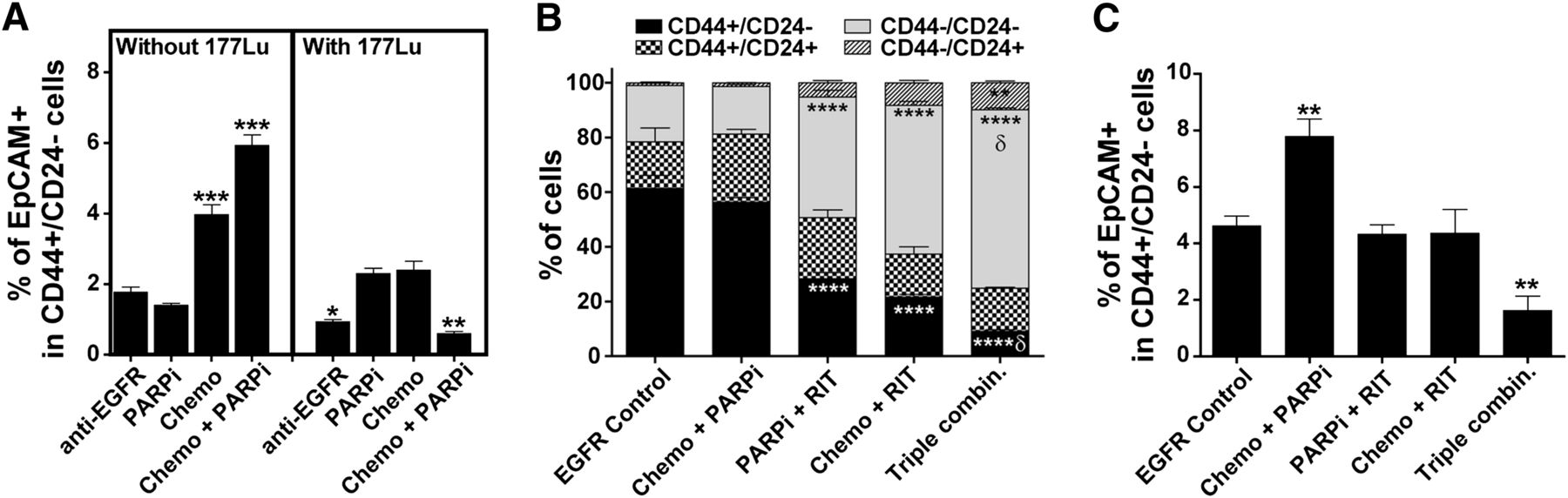
BCSCs with the CD44+/CD24−/EpCAM+ profile are enriched by chemotherapeutics in vitro (12). We found that treatment of MDA-MB-231 cells (a TNBC cell line) with unlabeled anti-EGFR mAb did not enrich these cells. The treatment of MDA-MB-231 cells with chemotherapy alone and in combination with PARP inhibitor significantly increased the percentage of BCSCs in vitro (Fig. 4A, left panel). In contrast, treatment with anti-EGFR mAb labeled with 177Lu alone significantly reduced the percentage of CD44+/CD24−/EpCAM+ cells, and more so when combined with chemotherapy and PARP inhibitor (Fig. 4A, right panel). In vivo, we found that MDA-MB-231 xenograft tumors remaining on day 10 after treatment with the triple-agent combination contained significantly fewer CD44+/CD24− cells and more CD44−/CD24− cells than did tumors in control and other treatment arms (Fig. 4B). The loss of CD44 results in defective luminal–myoepithelial cell–cell adhesion and correlates to loss of epithelial–stromal interactions (13)—functions that may affect tumor formation. Importantly and consistently with the in vitro results, the combination of chemotherapy and PARP inhibitor significantly enriched for CD44+/CD24−/EpCAM+ cells in the mammary fat pad xenografts (Fig. 4C). Although the combination of EGFR-directed radioimmunotherapy with chemotherapy or PARP inhibitor did not significantly affect the percentage of these cells, the triple-agent combination significantly reduced the percentage of CD44+/CD24−/EpCAM+ in residual tumors.
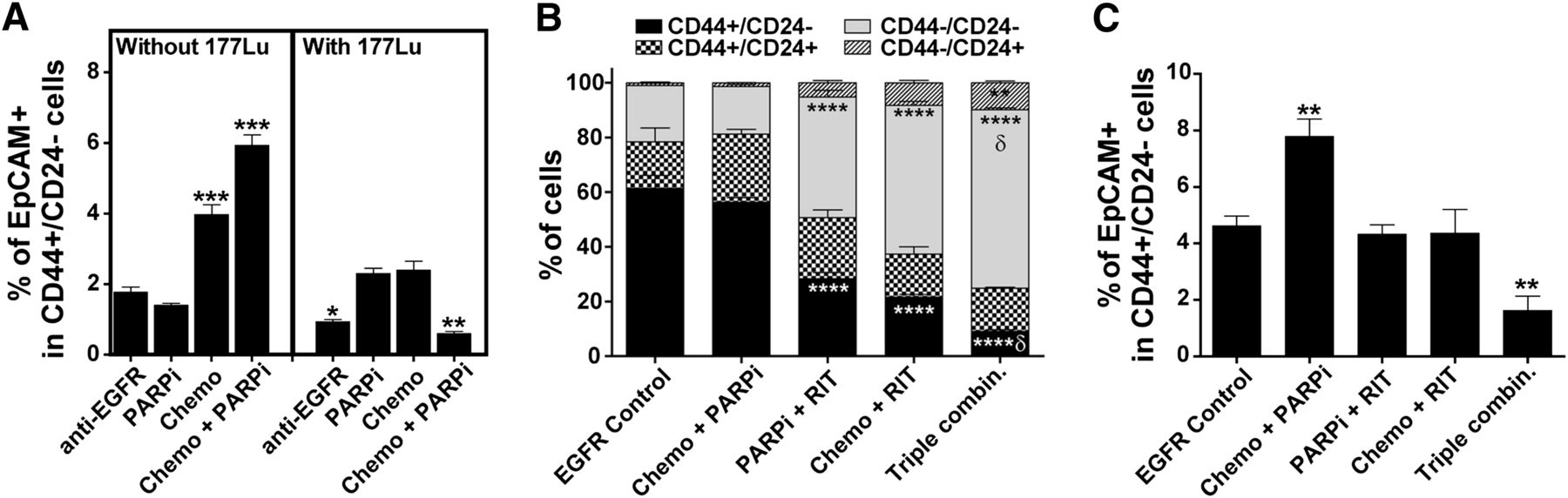
In vitro and in vivo demonstration that radioimmunotherapy eliminated BCSCs. (A) Quantification of BCSCs (CD44+/CD24−/EpCAM+) in vitro after various treatments as detected by flow cytometry analysis (3 mice per group). Flow cytometric quantification of CD44/CD24 (B) and CD44+/CD24−/EpCAM+ (C) BCSC populations ex vivo using MDA-MB-231 mammary fat pad xenografts isolated on day 10 after treatment of mice (5 mice per group). Supplemental Figure 8 shows representative staining results. Chemo = chemotherapy; combin. = combination; PARPi = PARP inhibitor; RIT = radioimmunotherapy. ***P < 0.001, **P < 0.01 (compared with tumors treated with unlabeled anti-EGFR mAb; 2-way ANOVA). δP < 0.01 (for 3-agent combination compared with 2-agent combinations; 2-way ANOVA).
Treatment of Patient-Derived Grafts and Established Metastases
As shown in Fig. 5A, the triple-agent combination successfully stopped recurrence of the primary patient-derived TNBC graft (HCI-002) established in the mammary fat pads in a similar manner to the MDA-MB-231 xenograft model. Moreover, in a separate model, the triple-agent combination therapy also eliminated experimental metastases. As shown in Fig. 5B, pulmonary metastases of MDA-MB-231 cells were evident by bioluminescence imaging at 14 d after injection, which became day 0 before combination treatments were administered. Although initial responses were observed in all mice treated with EGFR-directed radioimmunotherapy in combination with PARP inhibitor or chemotherapy, the triple-combination treatment eliminated metastases in 100% of the mice (Fig. 5B). We did not observe any recurrences in mice (total of 15 mice, 2 independent experiments) that were followed up to 42 d after treatment with the triple-agent combination.
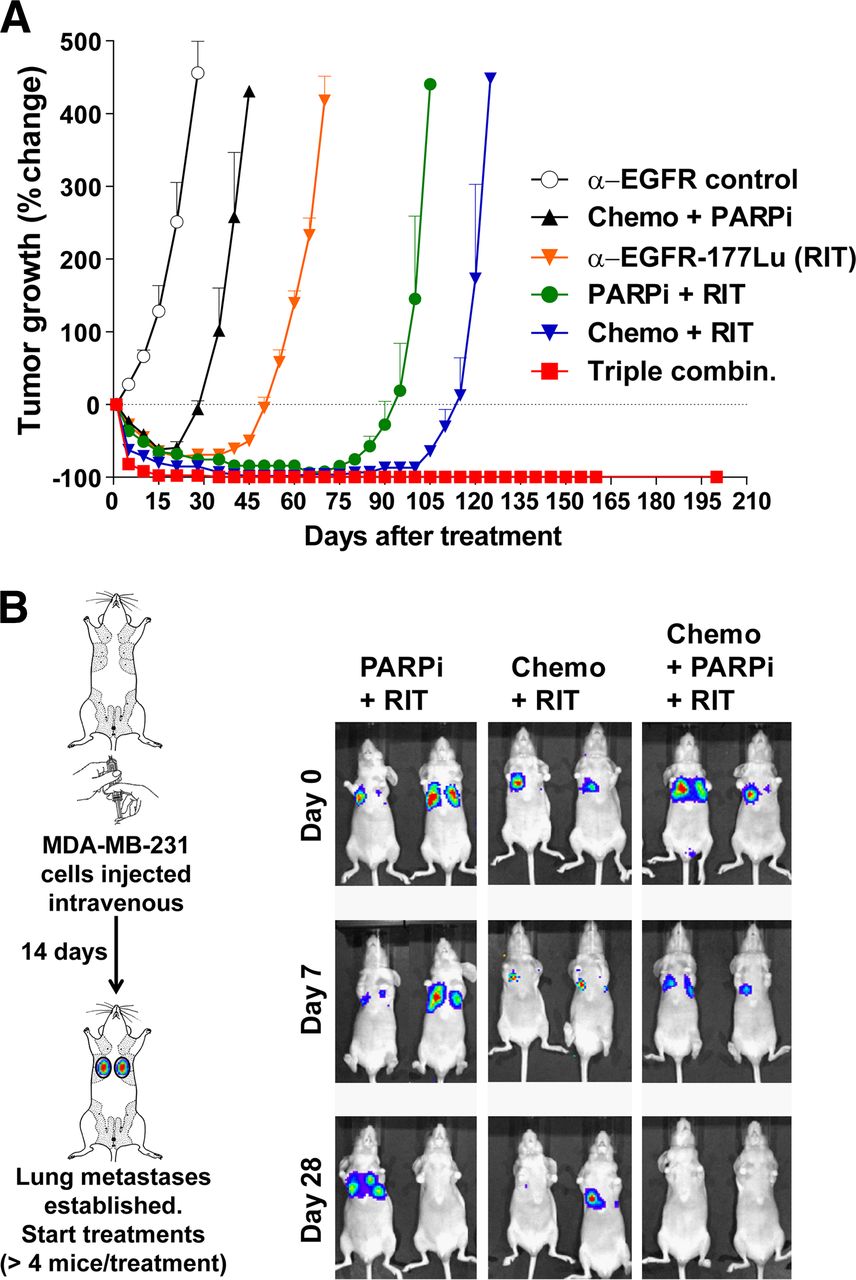
Treatment of patient-derived orthotopic grafts and established MDA-MB-231 metastases using triple-combination therapy. (A) Mice bearing patient-derived HCI-002 grafts in mammary fat pads were treated as described in Figure 1 (10 mice per group), and tumors were monitored for growth using caliper measurements. Triple-agent combination stopped recurrences observed in all other treatment arms. (B) In separate model, established metastases (14 d after intravenous injection of MDA-MB-231 cells) were visualized by bioluminescent imaging. Treatments were initiated immediately after imaging (day 0), and mice were imaged weekly thereafter. Combination of EGFR-directed radioimmunotherapy with PARP inhibitor or with chemotherapy showed transient antitumor effect on lung metastases, whereas triple-agent combination eliminated these metastases, with no recurrences observed up to 42 d after treatment. Only representative images are shown; Supplemental Fig. 8 shows images of all mice.
DISCUSSION
This preclinical study supported the contention that EGFR-directed radioimmunotherapy in combination with anthracycline/taxane-based chemotherapy and PARP inhibition is effective against EGFR-positive orthotopic and metastatic TNBC models. The combinations of chemotherapy and PARP inhibitors or chemotherapy and anti-EGFR mAbs/inhibitors are currently at the forefront of clinical trials in metastatic breast cancer and TNBC (1). To our knowledge, this was the first study in which EGFR-directed radioimmunotherapy was used alone or in combination in breast cancer.
The biodistribution and excellent selective tumor targeting of radiolabeled anti-EGFR mAbs in mice are well established (14–16). The uptake of radioimmunoconjugates in the bone (femur), kidneys, and other normal organs was invariably low (<5% injected dose/g), and the highest nontumor uptake was seen in the liver (∼10% injected dose/g), but this reduced with time. The tumor uptake consistently ranged between 20% and 40% injected dose/g and increased with time, showing more than 4-fold tumor-to-liver ratios. 90Y-labeled panitumumab showed a significant antitumor effect against head and neck cancer xenografts, with limited toxicity to normal organs (17). On the basis of the tumor uptake in these studies, we can estimate that the dose of 177Lu-anti-EGFR mAb delivered to the tumors ranges between 12 and 25 Gy per gram of tumor within the first 7 d of treatment. This excellent biodistribution profile explains the lack of toxicity in our experimental groups, the extensive level of cell death in tumors we observed up to 7 d, and the eventual elimination of these tumors, which was associated with the elimination of BCSCs.
The anti-EGFR 225 mAb we used does not bind mouse EGFR, nor does cetuximab or panitumumab. This lack of specificity is described in the literature and was confirmed by us. Unfortunately, a caveat of preclinical models using human tumor xenografts and human-specific antibodies is that toxicity to normal organs of the murine host may not be accurately determined. Nevertheless, several lines of evidence support the notion that EGFR-targeted radionuclide therapy may be clinically feasible. First, the biodistribution of mAb against mouse EGFR (7A7) has been investigated in non–tumor-bearing and immunocompetent mice, in which uptake in liver and other normal organs was well below 10% injected dose/g at 48 h after injection (18). In a phase I clinical trial performed using the original anti-EGFR 225 mAb for safety and imaging (19), the most important observation was the lack of toxicity on the liver, kidneys, or bone marrow in patients who received the 225 mAb (up to 300 mg, including 40 mg of 111In-labeled 225 mAb). This lack of toxicity was observed in the clinical trials performed in the early 1990s using other anti-EGFR mAbs.
Stronger evidence for the clinical utility of anti-EGFR–directed radioimmunotherapy comes from clinical trials in glioblastoma. In the earliest trial, patients were treated with systemic doses (1.48–5.19 GBq [40–140 mCi]) of 131I-anti-EGFR mAb (clone EGFR1). With the 3.7-GBq (100-mCi) injection, the estimated dose to tumors was 12.5 Gy and the estimated dose to bone marrow was 1.4 Gy. No acute toxicities were encountered in any patients, and more important, the authors did not observe any impairment in liver or renal function tests (20). The dose rate from the β-emission of 131I (0.10449 Gy⋅g/MBq⋅h) is higher than the dose rate of 177Lu (0.07641 Gy⋅g/MBq⋅h). In the second phase II clinical trial, a total of 192 patients with glioblastoma were treated with 125I-anti-EGFR mAb (clone 425) over a course of 3 weekly intravenous injections of 1.8 GBq (total of 5.4 GBq). No patient required hospitalization or had grade 3 or 4 toxicities with the administration of 125I-EGFR mAb (21). EGFR-targeted radioimmunotherapy seems not yet to have been investigated clinically in other cancers, including breast cancer. Encouragingly, a phase I clinical trial (NCT01384253, registered July 2011) of 212Pb-labeled trastuzumab as a targeted α-particle anti-HER2 radioimmunotherapy for metastatic disease is recruiting patients with HER2-positive cancers.
There is a strong rationale for targeting EGFR for radioimmunotherapy in TNBC. EGFR is expressed in at least half of TNBC cases, and EGFR has an emerging role in the maintenance of BCSCs, which are associated with disease progression, recurrence, and metastasis (22). The high content of the putative BCSC phenotype, CD44+/CD24−, in tumors from basal-like breast cancer patients correlates positively with elevated EGFR expression (23,24). Functionally, EGFR, HER2, and CD44 form complexes in a high percentage of metastatic breast carcinomas (25). This functional interaction regulates shedding, internalization, and motogenic (inducing cellular movement) effects of CD44 in vitro and in vivo and can be inhibited by antibodies against EGFR (26). Inhibition of EGFR signaling, unlike chemotherapy (27), reduces the percentage of CD44+/CD24− cells and disrupts mammosphere formation in cultures (28). Altogether, the literature supports the involvement of EGFR signaling in maintaining BCSCs. However, TNBC cell lines lack sensitivity to EGFR inhibitors (inhibitory concentration of 50% in the micromolar range) and anti-EGFR antibodies (inhibitory concentration of 50% > 100 μg/mL) (29), which has been attributed to pathway activation downstream of EGFR through PI3K/AKT, loss of PTEN, MEK/ERK/RAS, or localization of EGFR in lipid rafts (30,31). Accordingly, we hypothesized that delivering a therapeutic radionuclide via an anti-EGFR antibody would deplete CD44+/CD24− BCSCs despite downstream compensatory signaling. Indeed, our in vitro data showed that 177Lu-anti-EGFR radioimmunotherapy (10 μg/mL) significantly reduced the number of BCSCs when used alone and more so when combined with chemotherapy and PARP inhibitor. Corroborating these findings, the percentage of BCSCs was significantly reduced in tumors when treated with a single dose of 177Lu-anti-EGFR radioimmunotherapy. Our data suggest that the high expression of EGFR can be used to target lethal doses of radiation to BCSCs without the need to modulate the EGFR-related canonical signaling networks.
EGFR nuclear translocation, a novel pathway independent of canonical signaling pathways, increases DNA repair after cisplatin and ionizing radiation treatment via increased nuclear retention and activation of DNA-PK (32), which is a critical protein in the nonhomologous end-joining DNA repair pathway. However, EGFR nuclear translocation is inhibited by cetuximab and gefitinib (an EGFR tyrosine kinase inhibitor), which sequester DNA-PK in the cytoplasm thus leading to suppression of DNA-PK activity (33,34). Moreover, EGFR inhibitors decrease RAD51 protein stability and mRNA levels and sequester BRCA1 in the cytoplasm (35,36), which are major players in the homologous recombination DNA repair pathway, thus potentiating the cytotoxicity of chemotherapeutics and radiation (36,37). To this end, we hypothesized that reduced activity of crucial DNA repair proteins would sensitize tumor cells to 177Lu-anti-EGFR radioimmunotherapy. In agreement, we found that expression levels of activated DNA-PK (DNA-PK S2056) and RAD51 protein were lower in tumors from mice treated with 177Lu-anti-EGFR than in tumors from mice treated with chemotherapy and PARP inhibitor. Therefore, incorporating a low dose of radiolabeled anti-EGFR mAb in the triple-agent combination treatment modulated the direct nuclear EGFR signaling pathway sufficiently to impair DNA repair.
The acknowledged efficacy of combined-modality therapy using external-beam radiotherapy and chemotherapy for treating inoperable, locally advanced cancers (38) depends on the radiosensitizing effects of the coadministered cytotoxic drugs (39). Several reports indicate that chemotherapy can sensitize tumors to targeted radionuclide therapy such as radioimmunotherapy (40). In our study, chemotherapy alone significantly potentiated the antitumor effect of 177Lu-anti-EGFR radioimmunotherapy. Radiosensitization by PARP inhibition has also been reported previously (39). PARP inhibition in our study potentiated not only chemotherapy but also the antitumor effect of 177Lu-anti-EGFR radioimmunotherapy in TNBC models. As far as we know, this is the first report to show that PARP inhibitor augments the effect of low-dose radionuclide therapy.
CONCLUSION
Our preclinical data support further development of anti-EGFR–directed radioimmunotherapy against EGFR-positive TNBC. We observed antitumor activity from a single dose of anti-EGFR–directed radioimmunotherapy alone that was greater than that of the chemotherapy and PARP inhibitor combined. More important, adding anti-EGFR radioimmunotherapy to chemotherapy and PARP inhibitor cured tumor-bearing mice whereas in mice treated with any 2-agent combination, tumors reemerged after initial regression. Mechanistically, tumor relapse in these mice may originate from persistent BCSCs, which in contrast had been eliminated after triple-agent combination treatment. Our results support further investigation of the efficacy of anti-EGFR–directed radioimmunotherapy in rational combinations toward the clinical development of this approach.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by the Cure Cancer Foundation Australia/Priority-Driven Collaborative Cancer Research Fund (grant 631924), the 2011 Rio Tinto Ride to Conquer Cancer Fund (grant 1017028), and National Health and Medical Research Council (grant 442903). No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Apr. 5, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication July 19, 2012.
- Accepted for publication December 27, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- How shall we treat early triple-negative breast cancer (TNBC): from the current standard to upcoming immuno-molecular strategies
- Combining Chk1/2 Inhibition with Cetuximab and Radiation Enhances In Vitro and In Vivo Cytotoxicity in Head and Neck Squamous Cell Carcinoma
- Gemcitabine and CHK1 Inhibition Potentiate EGFR-Directed Radioimmunotherapy against Pancreatic Ductal Adenocarcinoma
- Targeted Chemoradiation in Metastatic Colorectal Cancer: A Phase I Trial of 131I-huA33 with Concurrent Capecitabine