


Abstract
Radioactive imaging agents, like diagnostic drugs generally, undergo a drug development process that parallels that of therapeutic agents, with similar development times but substantially lower development costs and substantially smaller postapproval markets. Although rapid advances in genetic and expression profiling are furthering the development of expensive pharmacotherapies targeted to small patient populations, the commercial development of imaging agents for small patient populations is blocked by the limited revenues available with current per-dose pricing and the relatively small numbers of imaging procedures that would be performed. A wide-ranging discussion on the best approaches to allow new diagnostic imaging agents to become part of the health-care system, and benefit the patient, is needed.
Medical imaging is an important tool in biomedical research and clinical practice, providing time-dependent information on structural and anatomic features, physiologic function, and the presence of molecules such as receptors and enzymes. Imaging can play a role in initial diagnosis, in stratifying patients for therapy, and in determining response to therapy, with the promise of limiting the use of expensive therapies that fail to benefit specific individuals. Imaging agents are regulated as diagnostic drugs, a class that includes radiotracers, MR contrast agents, ultrasound agents, CT iodinated contrast agents, and fluorescent probes for optical imaging.
CLINICAL TRANSLATION
There are 2 general views of what is meant by “clinical translation.”
Academic View
Academic biomedical researchers (and some of their grant-providing sponsors) consider clinical translation to be either the first use of a new chemical entity in humans or a study demonstrating a new use of a compound previously used in humans. Publications, the basis of grant funding, follow such studies and increase our body of scientific knowledge.
Costs associated with the first use of a new chemical entity in humans vary greatly with the imaging modality chosen. Radioactive imaging probes are administered at trace amounts and are considered unlikely to elicit a pharmacologic or physiologic response. Nonradioactive imaging agents (MR imaging, CT, ultrasound, fluorescent probes) are administered at higher doses often capable of producing toxicologic or pharmacologic effects. With high-dose imaging agents, studies of absorption, distribution, metabolism, and elimination (ADME) are required and parallel those of therapeutics. Radioactive agents, used in trace amounts, are considered to disappear by radioactive decay and ADME studies are not required. In addition, the high doses of nonradioactive imaging agents increases the number, duration, and cost of toxicity studies, which typically require scale-up and manufacture of the drug substance even before the first patient has been dosed. Stability studies required of nonradioactive diagnostic drugs also add to costs. However, investigational new drug (IND)–related costs are a small fraction of total development costs, since the major fraction of new drug application (NDA) costs results from clinical studies, particularly phase III studies. Although preclinical ADME studies are not required for radioactive drugs, and toxicity studies are easier because of low efficacious doses, total development costs for radioactive and nonradioactive diagnostic agents are similar.
New chemical entities of SPECT or PET imaging agents can use an exploratory IND (eIND) for Food and Drug Administration (FDA) approval or institutional Radioactive Drug Research Committee approval. The eIND permits the use of up to 5 nonpharmacologically active, chemically related agents at microdoses, enabling a better selection of compounds for further study. At least one animal toxicology study following good laboratory practices is required for a new chemical entity filed under an eIND. If drug development continues after an eIND, a full IND must replace the eIND. Thus, whereas an eIND can lessen the cost of the first use in humans, it has little bearing on the total development costs of imaging agents. Total development costs consist of the IND filing, phase I–III clinical studies, and the NDA filing. Radioactive Drug Research Committees are most commonly applied for new studies using compounds that have a history of prior human use.
Investor/Economic View
From the investor’s point of view (and from the health-care system’s point of view), clinical translation means sustained sales and an impact of a diagnostic drug on patient care. To examine this view of clinical translation, an understanding of the economic issues surrounding diagnostic drugs is required. A discussion of these issues, and a comparison between diagnostic and therapeutic drug development costs, can be found in a 2006 paper by Nunn (1) that is still accurate today.
COSTS OF DEVELOPING DIAGNOSTIC DRUGS
Both development costs and sales revenues are lower for diagnostic than therapeutic drugs because diagnostic drugs are used for a small number of imaging procedures per patient and per disease and because per-dose prices for diagnostic drugs are limited. For diagnostic drugs, development times are 8–10 y (vs. an average of 12.9 y for therapeutics) and total development costs are between $100 million and $200 million (vs. $850 million for therapeutics). For diagnostic drugs, worldwide sales vary between $100 million and $400 million per year, with the top radiodiagnostic drugs (such as 99mTc-Myoview [tetrofosmin] by GE Healthcare, 99mTc-Cardiolite [sestamibi] by Lantheus Medical, and 18F-FDG by many manufacturers) having sales that fall within this revenue range. Hoffman et al. (2) use the same dollar figures as Nunn (1) and have arrived at similar conclusions regarding diagnostic drug development times. Revenues for therapeutic drugs vary widely, but many successful therapeutic drugs generate revenues of $1 billion per year or greater, far larger than the maximal revenues generated by diagnostic drugs.
Imaging researchers see the costs associated with first-in-human studies as the major barrier to clinical translation. The total costs of IND-generated phase I and II studies have been estimated to be between $1 million and $10 million (3). Although formidable when compared with a typical R01 National Institutes of Health grant at $200,000–$400,000 direct per year, investors with access to major capital markets focus not on the $10 million needed to generate phase I/II data but on total development costs and the prospects for profitability after an NDA approval. Prospects for post-NDA profitability are dependent on the price per dose and the number of doses that might be sold.
PRICES OF DIAGNOSTIC DRUGS
Diagnostic drugs provide information and, although they can help guide therapy, are of no direct therapeutic benefit. Diagnostic drugs are priced to be a relatively small fraction of the total cost of an imaging procedure, which includes access to and operation of imaging equipment, patient care, and professional image interpretation as well as the diagnostic drug. In addition, no marketing approval for a diagnostic drug has yet been obtained under orphan drug regulations. (The FDA reviews more drugs as possible orphans than are approved as orphan drugs.) Historically, the pricing of diagnostic drugs, even those with patent protection, has been modest relative to the total costs of an imaging procedure. This combination of factors (providing information only, historical pricing patterns, lack of an orphan drug status) limits the per-dose prices of diagnostic drugs such that commonly used major diagnostic drugs sell for less than $500 per dose. Diagnostic drugs without competition, such as the newly approved Amyvid (florbetapir; Eli Lilly and Co.) for imaging β-amyloid, can be priced above this level. However, pricing for florbetapir may not be sustainable as other 18F-labeled β-amyloid imaging agents under development become available (4). Although patent protection plays a role in increasing the price of diagnostic drugs, it has not enabled per-dose pricing so high that the cost of the drug becomes the major component of the imaging procedure. In some cases, compositional patents on diagnostic drugs have a limited impact because a variety of structurally different compounds can be used with similar results (e.g., iodinated CT contrast medium or gadolinium chelates).
SUSTAINABILITY OF DIAGNOSTIC DRUGS IN THE MARKETPLACE
Sales needed for sustainability arise from the frequency of use multiplied by the cost per dose. To attain the necessary frequency of use, the approved indication must be for a condition of high prevalence, such as suspected dementia, or be used for biomarkers that are common to a variety of conditions. Diagnostic drugs must also meet high safety standards.
Since diagnostic drugs can be given to young or healthy patients, a mantra of “first, do no harm” generates a special burden for this class of pharmaceuticals. The acute safety of diagnostic drugs can be evaluated as a severity or incidence profile termed an adverse reaction profile. Adverse reaction profiles vary with the rules set for them, such as the duration of the observation period after injection, the criteria for the categories of severity, and the stringency of the criteria for determining when an effect is drug-related. Diagnostic drugs typically produce serious-adverse-reaction profiles at a frequency of less than 1 per 10,000 uses. Carcinogenicity and mutagenicity, common among cancer therapeutics, are unacceptable with cancer diagnostic drugs, since a diagnostic drug can be used with non–cancer-bearing individuals. Factors determining the sustainability of diagnostic drugs are summarized in Figure 1.
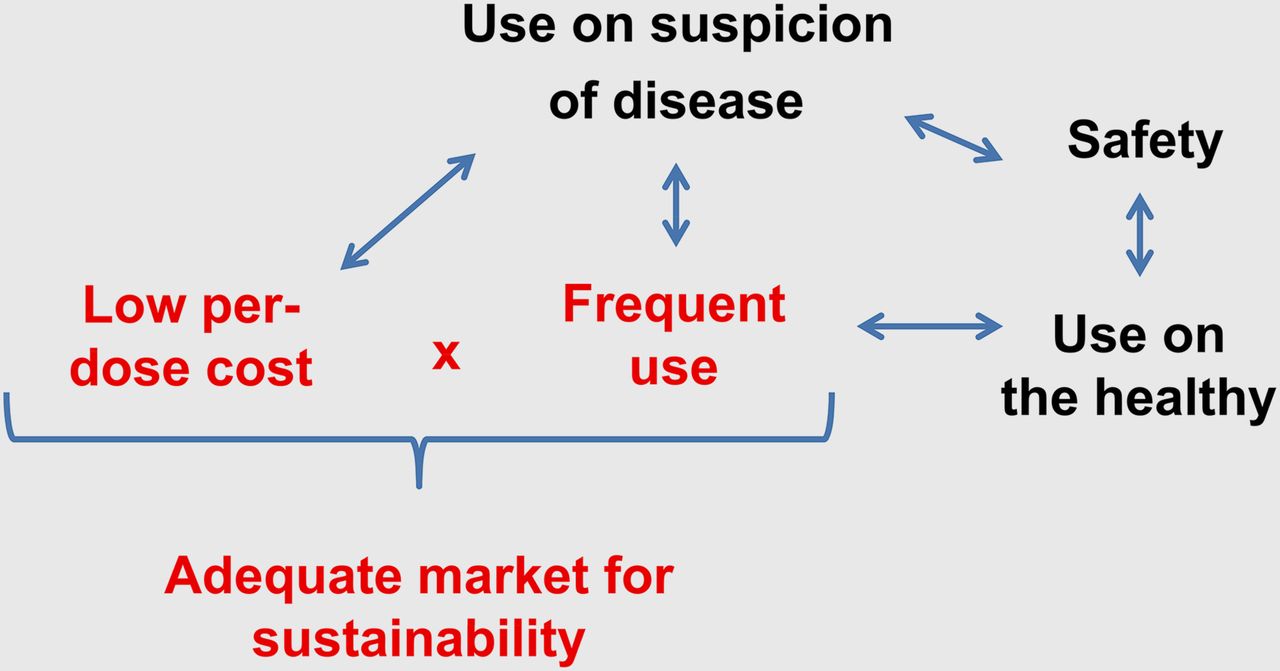
Sustainability of diagnostic drugs in marketplace. Use on suspicion of disease leads to use in healthy individuals and need for low frequency of adverse reactions. Adequate market size is obtained through single indication of high prevalence or multiple indications, coupled with relatively low cost per dose. There has been no orphan drug option for diagnostic drugs, which would allow an infrequent use and a high price per dose.
With this background, a rule of thumb to evaluate the potential profitability of developing a compound into a diagnostic drug has been postulated by Nunn (1): do peak-year sales equal or exceed total expected development costs? This implies that the yearly worldwide sales of a diagnostic drug would need to be on the order of $100 million to $200 million per year. To appreciate the issues of long-term profitability and sustainability, consider that in order to achieve a hypothetical revenue of $100 million per year, and assuming a price of $1,000 per dose (which is high for a diagnostic drug), 100,000 examinations per year would be required worldwide. Hence, diagnostic drugs need to be useful for a wide range of conditions (and eligible for reimbursement for those conditions) to generate the examination frequency needed for profitability.
In recent years, both SPECT agents and MR contrast agents have undergone postapproval withdrawals from the U.S. marketplace. Four such drugs are Feridex IV (ferumoxides; AMAG Pharmaceuticals), Teslascan (mangafodipir; GE Healthcare), Myoscint (imciromab; Centacour), and AcuTect (technetium; CIS Bio International). To avoid the immense waste of resources associated with drug approval and subsequent withdrawal, a better understanding is needed—both of barriers to the initial clinical use of diagnostic drugs and of the requirements a diagnostic drug faces—for profitability and sustainability in the marketplace. The markets for diagnostic drugs must be carefully considered and understood to avoid postapproval product discontinuation.
CLINICAL BENEFIT AND BREADTH OF INDICATION FOR DIAGNOSTICS
Two key questions are essential for assessing diagnostic drugs: does the information provided change patient management, and what is the patient population (healthy or unhealthy) that should receive the agent? Regulatory agencies may argue that for the best proof of efficacy, clinical studies should use a well-defined, narrow patient population. Although a narrow approved indication is perhaps the most scientifically rigorous approach, investors fearing that such an indication implies a small market may hence discontinue investing in the drug.
This scenario, albeit coupled with safety concerns, was played out with ferumoxides (Combidex in the United States and Sinerem in the European Union) in 2005 (5). Combidex (Sinerem) was an MR contrast agent used for determining the metastatic status of lymph nodes. The FDA wanted additional clinical studies for visualizing anatomically distinct groups of lymph nodes (e.g., head and neck, pelvic) corresponding to a narrow indication range. In contrast, the company sought a broad pan–lymph node indication based on studies that combined results from lymph nodes in a variety of anatomic locations. (6). In the wake of this disagreement, further drug development was halted.
AVENUES FOR DEVELOPMENT OF CLINICAL DIAGNOSTIC PROBES
Three nonexclusive avenues for the future development of diagnostic drugs are focusing on the tandem development of diagnostic drugs and therapeutic agents and on applications with large markets imaging widely useful disease biomarkers.
Applications with Large Markets
Because the prices charged for a single use of a diagnostic drug are limited, the frequency of use becomes the central component in defining economic sustainability. The needed frequency can be achieved by focusing on highly prevalent conditions, which include neurodegeneration or metabolic disorders such as diabetes (for which markers of β-cell mass and function are desperately needed).
The newly FDA-approved PET tracer Amyvid (florbetapir), which binds in the brain the aggregated β-amyloid peptides that are a hallmark of Alzheimer disease (AD), is an interesting case. Florbetapir (or other amyloid-binding agents) could be used to evaluate the efficacy of plaque-modulating therapies or to allow stratification of the patient population entering a clinical trial for a new AD drug, avoiding the inclusion of patients with other dementias that might dilute the overall response. Thus, florbetapir was approved before effective treatment options are available for AD but might play a key role in the development of effective therapies. However, whether decreasing the plaque load, as derived from florbetapir imaging, will translate into clinical and functional benefit remains to be demonstrated.
Given the number of new AD diagnoses each year, the estimated market for imaging agents varies between less than $100 million per year and $600 million per year, depending on whether health-care insurance will cover the costs (7). In view of the fact that other companies are developing analogous products, the market share for any AD agent is likely to be on the order of 30%–50% of the total market size.
Imaging Biomarkers Common to Many Diseases
The pharmaceutical industry is facing major issues of future profitability, in part because the approval rate for new molecular entities has stagnated for almost 60 y whereas average costs per new molecular entity (NME) have increased exponentially at a rate of 13% per annum (8). This has led regulatory authorities to launch efforts such as the Critical Path Initiative of the FDA, an effort to ensure that scientific discoveries translate more rapidly and efficiently into approved drugs (9). Within the Critical Path Initiative, biomarkers (including imaging biomarkers) can play a central role. The FDA defines a biomarker as “a characteristic that is subjectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” (9).
A wide array of imaging methodologies and imaging agents has been proposed as biomarkers for indications such as assessing the efficacy of cancer therapies (Table 1 of the paper by Rudin (10)). These include measurement of tumor glucose utilization using 18F-FDG in combination with PET, cell proliferation by assessing 18F-fluorothymidine uptake with PET, vascular permeability as a readout of angiogenesis using dynamic contrast-enhanced MRI with gadolinium chelates as contrast agents, inflammatory processes by monitoring the infiltration of immune cells labeled with magnetic nanoparticles, or cell apoptosis by monitoring the accumulation of labeled annexin-V using either SPECT or PET. Such imaging assays for generic hallmarks of tumors (11) have been efficiently used as early readouts of therapy response in clinical proof-of-concept studies (10). They might also be of value for guiding intervention, particularly when expensive therapies are being considered.
Tandem Development of a Diagnostic Drug and a Therapeutic Drug
Two trends in drug development may supply the motivation for the tandem development of imaging agents and therapeutic agents sponsored by “Big Pharma.” The first is the use of genetic and protein expression profiles to subdivide broad disease states and tailor pharmacotherapies to relatively small patient populations. The second is the decreasing number (or stagnating number) of new chemical entities being approved. There can be a considerable incentive for the pharmaceutical industry to use diagnostic imaging in the preapproval development of a therapeutic drug, either for patient selection or for managing the responses of individual patients.
Tandem development of diagnostic and therapeutic agents can be considered in light of the considerable need for improved tools in drug development and because of the relative development costs of diagnostics and therapeutics. For example, the cost of a phase I/II study with a new imaging agent (<$10 million) might validate the development of a therapeutic with total development costs of more than $850 million. However, if the imaging method is crucial to patient selection in the preapproval phase of therapeutic agent development, how can it be made available after approval of the therapeutic? The complete tandem development of a new diagnostic and new therapeutic agent might be undertaken since total development costs for diagnostics ($100 million–$200 million) are well below those of therapeutics ($850 million). If a diagnostic drug enabled the approval of a large-market therapeutic agent, one that otherwise would have failed to gain approval, complete tandem development might be economically feasible.
CONCLUSION
Imaging agents can play an important role in patient selection and in determining an individual’s response to new, expensive pharmacotherapies. Before drug approval, imaging agents might enhance the chances of drug approval by assisting in patient selection. After drug approval, imaging agents might limit the use of expensive therapies to those patients who will respond. However, the approval procedure for diagnostic drugs, including nuclear imaging agents, currently parallels that for therapeutic agents with comparable development times. The limited revenues available with current per-dose pricing and numbers of imaging procedures performed needs to foster a wide-ranging discussion on approaches for the codevelopment of diagnostic imaging agents and therapeutic agents. In addition, postapproval revenues need to be carefully considered to enable diagnostic drugs to become sustainable after approval and therefore benefit society as part of routine medical practice.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jan. 28, 2013
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication September 18, 2012.
- Accepted for publication December 17, 2012.





{kind=link}