


Abstract
One of the pathways leading to androgen independence in prostate cancer involves upregulation of insulinlike growth factor type 1 receptor (IGF-1R). Radionuclide imaging of IGF-1R in tumors might be used for selection of patients who would most likely benefit from IGF-1R–targeted therapy. The goal of this study was to evaluate the feasibility of in vivo radionuclide imaging of IGF-1R expression in prostate cancer xenografts using a small nonimmunoglobulin-derived binding protein called an Affibody molecule. Methods: The IGF-1R-binding ZIGF1R:4551 Affibody molecule was site-specifically conjugated with a maleimido derivative of DOTA and labeled with 111In. The binding of radiolabeled ZIGF1R:4551 to IGF-1R–expressing cells was evaluated in vitro and in vivo. Results: DOTA-ZIGF1R:4551 can be stably labeled with 111In with preserved specific binding to IGF-1R–expressing cells in vitro. In mice, 111In-DOTA-ZIGF1R:4551 accumulated in IGF-1R–expressing organs (pancreas, stomach, lung, and salivary gland). Receptor saturation experiments demonstrated that targeting of DU-145 prostate cancer xenografts in NMRI nu/nu mice was IGF-1R–specific. The tumor uptake was 1.1 ± 0.3 percentage injected dose per gram, and the tumor-to-blood ratio was 3.2 ± 0.2 at 8 h after injection. Conclusion: This study demonstrates the feasibility of in vivo targeting of IGF-1R–expressing prostate cancer xenografts using an Affibody molecule. Further development of radiolabeled Affibody molecules might provide a useful clinical tool for stratification of patients with prostate cancer for IGF-1R–targeting therapy.
The insulinlike growth factor (IGF) axis includes 2 insulinlike growth factors (IGF-1 and IGF-2), 2 transmembrane tyrosine kinase receptors (IGF-1R and IGF-2R), and 6 IGF binding proteins (IGFBP-1 to IGFBP-6) (1,2). An increasing amount of evidence implicates that this axis, in particular IGF-1R signaling, plays a role in malignant cell transformation, cancer progression, and metastatic spread of different types of tumors. Increased IGF-1R signaling has been shown to result in activation of growth-promoting intracellular signaling pathways, including the ras-raf-MAPK and PI3K cascades (1–3). Elevated IGF-1R expression has also been shown to confer resistance to different anticancer therapies in preclinical studies (4–6)—an effect that can be reversed by inhibition of IGF-1R signaling. For these reasons, IGF-1R signaling inhibitors have been developed to be used for cancer therapy (7,8).
In prostate cancer, upregulation and activation of IGF-1R is considered as a part of outlaw signaling pathway, leading to activation of androgen receptors and transcription of target genes in the absence of or at insufficient concentrations of androgens—that is, androgen independence (9–11). Accordingly, IGF-1R inhibition using monoclonal antibodies, small-molecule tyrosine kinase inhibitors, or antisense oligonucleotides is regarded as a promising strategy for treatment of androgen-independent prostate cancer (12,13). Both preclinical and clinical data suggest that an elevated expression level of IGF-1R in tumors is a prerequisite for response to IGF-1R–targeting therapy (14–17). Monitoring of the IGF-1R expression level in androgen-independent prostate cancer lesions is therefore important for the selection of patients who would most likely benefit from anti–IGF-1R treatment.
Histologic evaluation of tumor samples would be the most accurate approach to stratify patients for anti–IGF-1R therapy. However, this method is associated with invasive acquisition of multiple biopsies from a patient. In addition, the accuracy of biopsy-based methods is limited: samplings may be nonrepresentative because of intratumoral expression heterogeneity, and molecular target level may be discordant in primary tumors and metastases. Radionuclide molecular imaging offers an alternative and allows repeated noninvasive evaluation of tyrosine kinase expression both for patient stratification and for monitoring expression-level changes in response to therapy (18). This approach would apparently be attractive for in vivo determination of IGF-1R status of disseminated malignant tumors.
Recently a new generation of molecular imaging probes, made by the coupling of a radiolabel to a scaffold-based affinity protein, has been shown to give promising results (19). One such class of scaffold proteins is the Affibody molecules, which are structurally based on one of the independently folding IgG-binding domains of staphylococcal protein A (20,21). Thirteen surface-exposed amino acids on one face of the molecule have been subjected to randomization, resulting in a library from which variants with desired specificity can be selected by biopanning. This process has led to the isolation of several high-affinity binders to different target antigens (21,22). One example is Affibody molecules binding to HER2 with high affinity; these molecules have demonstrated high-contrast imaging of HER2-expressing xenografts in preclinical models (23) and imaging of HER2-expressing metastases in the clinic (24). Direct preclinical comparison of biodistribution data for radiolabeled Affibody molecules and the anti-HER2 monoclonal antibody trastuzumab showed that Affibody molecules provide much better contrast and specificity of radionuclide molecular imaging of HER2 expression (25).
We have recently reported on the development of a new Affibody ligand, Z4:40, which specifically binds to IGF-1R with a high affinity of 1.9 ± 0.2 nM (26). This high affinity would be sufficient for imaging of molecular targets that are abundantly expressed. However, the level of IGF-1R, which is associated with tumor response to anti-IGF-1R therapy, is fairly low (10,000–30,000 receptors per cell) (14,15). Therefore, affinity maturation was undertaken (Graslund, unpublished data, 2010), resulting in the Affibody molecule ZIGF1R:4551 having enhanced affinity for IGF-1R.
The goal of this study was to investigate the feasibility of radionuclide-based IGF-1R imaging in vivo using ZIGF1R:4551.
MATERIALS AND METHODS
Instrumentation
Radioactivity was measured using an automated γ-counter with a 7.6 cm (3-in) NaI(Tl) detector (1480 Wizard; Wallac Oy). Distribution of radioactivity along the thin-layer chromatography strips was measured on a Cyclone Storage Phosphor System and analyzed using the OptiQuant image analysis software (PerkinElmer). Cells were counted using an electronic cell counter (Beckman Coulter).
Characterization of Affibody Molecule ZIGF1R:4551 and Conjugation with DOTA
ZIGF1R:4551 was obtained by affinity maturation of the Affibody molecule Z4:40. The affinity maturation will be described elsewhere. A single cysteine was introduced at the C-terminus of ZIGF1R:4551 to enable site-specific labeling using thiol-directed conjugation chemistry. The identity of ZIGF1R:4551 was confirmed by high-performance liquid chromatography with mass spectrometry, CD spectra were recorded, and melting point was determined, as described earlier (27).
For affinity determination, Oregon green (OG) was site-specifically conjugated to ZIGF1R:4551 as described by Lundberg et al. (28). The affinity of ZIGF1R:4551 for IGF-1R–expressing MCF-7 cells was determined by fluorescence-activated cell sorting by making a 3-fold dilution series comprising 12 different concentrations of ZIGF1R:4551-OG, ranging between 50 μM and 2.8 pM. The fluorescence signal was plotted as a function of ZIGF1R:4551-OG concentration, and the equilibrium association constant was retrieved from the inflection point of the curve.
For site-specific labeling, maleimide-monoamide-DOTA (MMA-DOTA; 1,4,7,10-tetraazacyclododecane-1,4,7-tris-acetic acid-10 maleimidoethylacetamide) (Macrocyclics) was conjugated to the C-terminal cysteine, as described by Ahlgren et al. (27). The conjugated Affibody molecule, designated DOTA-ZIGF1R:4551, was lyophilized.
Radiolabeling and In Vitro Evaluation of 111In-DOTA-ZIGF1R:4551
DOTA-ZIGF1R:4551 was reconstituted in 0.2 M ammonium acetate buffer, pH 5.5 (purified of metal impurities using Chelex 100 resin [Bio-Rad Laboratories]) to a concentration of 0.75 μg/μL. An aliquot, containing 60 μg of DOTA-ZIGF1R:4551, was mixed with 60 MBq of 111In-chloride (Covidien). The mixture was incubated for 30 min either at 50°C or at 90°C and then analyzed using 50-771 Dark Green Tec-Control Chromatography instant thin-layer chromatography strips (Biodex Medical Systems) eluted with 0.2 M citric acid. To assess labeling stability, 111In-DOTA-ZIGF1R:4551 (labeled at 90°C) was incubated with a 1,000-fold excess of ethylenediaminetetraacetic acid (EDTA) for 4 h and then analyzed using instant thin-layer chromatography.
The specificity of 111In-DOTA-ZIGF1R:4551 for binding to IGF-1R–expressing cells after labeling at 60°C and 90°C was evaluated using 2 prostate cancer cell lines, DU-145 and PC-3, and the cervical carcinoma A431 cell line. All cell lines were purchased from American Type Tissue Culture Collection via LGC Promochem. An in vitro specificity test was performed according to the methods described earlier (27). Briefly, a solution of 111In-DOTA-ZIGF1R:4551 (at 1 nM) was added to 6 Petri dishes (∼106 cells in each). For blocking, a 100-fold excess of nonlabeled DOTA-ZIGF1R:4551 was added 15 min before radiolabeled conjugates to saturate the receptors. The cells were incubated during 2 h in a humidified incubator at 37°C. Thereafter, the medium was collected, the cells were detached by trypsin-EDTA solution, and the radioactivity in cells and medium was measured, to enable calculation of the fraction of cell-bound radioactivity.
An internalization of 111In-DOTA-ZIGF1R:4551 by DU-145 cells was performed according to the method described earlier (27). Briefly, the cells (∼106 per dish) were incubated with the labeled compound (1.5 nM) at 37°C, 5% CO2. At predetermined time points (1, 2, 3, and 4 h after incubation start), the medium from a set of 3 dishes was removed. To collect the membrane-bound radioactivity, the cells were treated with 0.2 M glycine buffer containing 4 M urea, pH 2.5, for 5 min on ice. To collect cells containing internalized radioactivity, a treatment with 1 M NaOH at 37°C for 0.5 h was used. A percentage of internalized radioactivity was calculated for each time point.
To quantify the IGF-1R expression in the DU-145 cell line, the cells were incubated for 4 h at 4°C with the radiolabeled Affibody molecule at concentrations in the range of 0.08–27 nM in complete medium. For each data point, 4 dishes were used, including 1 presaturated with unlabeled DOTA-ZIGF1R:4551 at a 3.7-μM concentration to determine specific binding. After incubation, the medium was aspirated, and the cells were detached by trypsin-EDTA solution and counted. Radioactivity of the samples was measured. The data were analyzed using Prism (version 5.04 [GraphPad Software] for Windows [Microsoft]).
In Vivo Studies
All animal experiments were planned and performed in accordance with national legislation on laboratory animals’ protection and were approved by the Ethics Committee for Animal Research in Uppsala.
Animal Model Validation: Biodistribution in Normal NMRI Mice
Subsets of cells in colon, lung, pancreas, salivary gland, and stomach express IGF-1R (http://www.proteinatlas.org/index.php). To determine the cross-reactivity of 111In-DOTA-ZIGF1R:4551 with murine IGF-1R in healthy tissues, NMRI mice (average weight, 32.9 ± 1.5 g) were intravenously injected (tail) with 25 kBq of 111In-DOTA-ZIGF1R:4551 in 100 μL of phosphate-buffered saline (PBS). The injected protein dose was adjusted by dilution with nonlabeled DOTA-ZIGF1R:4551 to 0.03, 1, or 10 μg per mouse. A group of 4 mice was used for each protein dose. At 4 h after injection, mice were sacrificed by injection of a lethal dose of anesthesia (20 μL of Ketalar–Rompun per gram of body weight: Ketalar [50 mg/mL; Pfizer], 10 mg/mL; Rompun [20 mg/mL; Bayer]), followed by heart puncture and exsanguination with a 1-mL syringe rinsed with heparin (5,000 IE/mL; Leo Pharma). Samples of blood, colon, lung, liver, spleen, pancreas, salivary gland, stomach, kidney, muscle, and bone were collected and weighed, and their radioactivity was measured. Tissue uptake was calculated as percentage of injected radioactivity per gram.
Biodistribution in NMRI nu/nu Mice Bearing DU-145 Prostate Cancer Xenografts: Influence of Protein Dose on Uptake in Tumors and Normal Tissues
For xenografting, 5 × 106 DU-145 cells (in Matrigel [BD Biosciences]) were subcutaneously implanted in the right hind leg. At the time of the experiment, the average animal weight was 32.7 ± 1.6 g, and the average tumor size was 0.44 ± 0.19 g. The mice were randomly distributed into 4 groups and injected with 25 kBq of 111In-DOTA-ZIGF1R:4551 in 100 μL of PBS. The injected protein dose was adjusted by dilution with nonlabeled DOTA-ZIGF1R:4551 to 0.1, 0.3, 1, or 300 μg per mouse. A group of 4 mice was used for each protein dose. At 4 h after injection, mice were sacrificed and analyzed as described.
Biodistribution of 111In-DOTA-ZIGF1R:4551 in NMRI nu/nu Mice Bearing DU-145 Prostate Cancer Xenografts at Different Time Points
DU-145 xenografts were implanted in male NMRI nu/nu mice as described. At the time of the experiment, the average animal weight was 32.6 ± 2.3 g, and average tumor size was 0.55 ± 0.22 g. The mice were randomly distributed into 5 groups and injected with 25 kBq (1 μg) of 111In-DOTA-ZIGF1R:4551 in 100 μL of PBS. At 1, 2, 4, 8, and 24 h after injection, mice were sacrificed and analyzed as described.
γ-Camera Imaging
The imaging experiment was performed 8 h after injection. Three mice with DU-145 xenografts (0.9–1.0 cm3) were injected with 111In-DOTA-ZIGF1R:4551 (1 MBq, 1 μg, in 100 μL of PBS). Immediately before imaging, the animals were sacrificed by overdosing Ketalar–Rompun. After euthanasia, the urine bladders were excised. The imaging experiment was performed using an Infinia γ-camera (GE Healthcare) equipped with a medium-energy general purpose collimator. Static images (30 min) were obtained with a zoom factor of 2 in a 256 × 256 matrix. The evaluation of the images was performed using Osiris 4.19 software (University Hospital of Geneva, Switzerland). In each animal, a region of interest was drawn around the tumor. The same region was copied to a contralateral thigh. Calculation of tumor–to–contralateral thigh ratios was based on average counts in the regions of interest.
Statistics
Data on cellular uptake and biodistribution were analyzed by unpaired, 2-tailed t test using Prism to determine any significant differences (P < 0.05).
RESULTS
Characterization of Affibody Molecule ZIGF1R:4551 and Conjugation with DOTA
The probe to be used for imaging of IGF-1R–expressing cells was based on the Affibody molecule ZIGF1R:4551. The affinity for binding to IGF-1R–expressing MCF-7 cells was 500 ± 79 pM by flow cytometry (Fig. 1A). The melting point of the protein was 62°C. To prepare the imaging probe, MMA-DOTA was coupled to ZIGF1R:4551. After high-performance liquid chromatography purification of DOTA-ZIGF1R:4551, no unconjugated ZIGF1R:4551 could be detected.
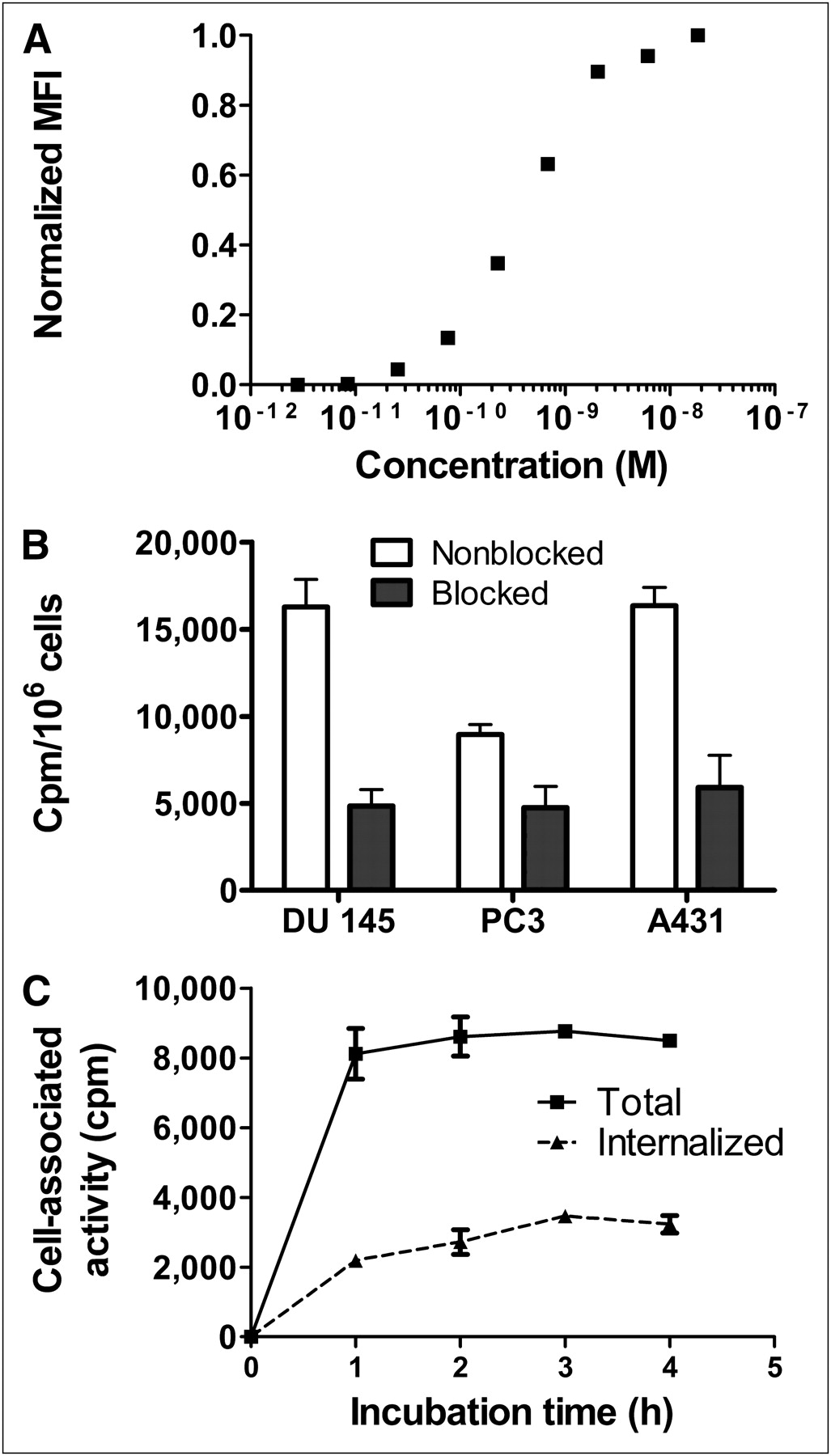
In vitro characterization of ZIGF1R:4551. (A) Affinity determination of ZIGF1R:4551 binding to MCF-7 cells. Cells were stained with different concentrations of ZIGF1R:4551-OG, followed by analysis by flow cytometry. Mean fluorescence intensity (MFI) after background subtraction was recalculated on scale from 0 (only background intensity) to 1 (maximum difference between MFI and background) and was plotted as a function of concentration of ZIGF1R:4551. The experiment was repeated 3 times, and a representative curve is shown. (B) Binding specificity of 111In-DOTA-ZIGF1R:4551 Affibody molecule to IGF-1R–expressing cells. For presaturation of IGF-1R, a 100-fold molar excess of nonradioactive IGF-1R–binding Affibody molecule was added. Data are mean values from 3 cell dishes ± SD. (C) Binding and internalization of 111In-DOTA-ZIGF1R:4551 by prostate cancer DU-145 cell line. Cells were incubated with 111In-DOTA-ZIGF1R:4551 at 37°C. Data are mean values from 3 cell dishes ± SD; some error bars are not seen because they are smaller than the symbols.
Labeling and In Vitro Evaluation of 111In-DOTA-ZIGF1R:4551
Labeling provided a radiochemical purity of 98.9% ± 0.2% (at 50°C) or 99.7% ± 0.1% (at 90°C), which excluded the need for additional purification. For biologic experiments, the conjugates were diluted with PBS. Challenge with a 1,000-fold molar excess of EDTA did not cause any detectable release of 111In, showing that the label was stable.
Binding specificity tests were performed to assess that the binding of 111In-DOTA-ZIGF1R:4551 to living IGF-1R–expressing cells was receptor-mediated. Saturation of the receptors by preincubation with nonlabeled DOTA-ZIGF1R:4551 significantly (P < 0.05) decreased the binding of the radiolabeled Affibody molecule, suggesting that the binding was specific (Fig. 1B). Importantly, binding was receptor-specific also after labeling at 90°C, extending the range of methods that might be used for labeling of ZIGF1R:4551. According to the saturation assay, the DU-145 cells expressed 25,000 ± 3,000 receptors per cells.
Data concerning binding and internalization of 111In-DOTA-ZIGF1R:4551 by the prostate cancer DU-145 cell line are presented in Figure 1C. The binding was rapid, reaching a plateau after 1 h of incubation. The internalization of 111In-DOTA-ZIGF1R:4551 was relatively inefficient, with a slow increase of the internalized fraction from 27% ± 2% of the total cell-associated radioactivity after 1 h incubation to 38% ± 3% after 4 h.
In Vivo Studies
Biodistribution in Normal NMRI Mice
Biodistribution of 111In-DOTA-ZIGF1R:4551 in normal NMRI mice was performed to investigate whether the animal model would reflect the situation in humans, in whom the target receptor is expressed in several tissues. Data obtained at 4 h after injection showed that 111In-DOTA-ZIGF1R:4551 was rapidly cleared from the blood irrespective of the amount of added cold tracer (Fig. 2). The clearance was predominantly renal, as radioactivity in intestines with content was below 2% at this time point. Renal clearance was accompanied by an appreciable reabsorption of radioactivity in the kidneys. The accumulation of radioactivity in the pancreas, stomach, lung, and salivary gland was lower at higher injected protein doses (P < 0.05 for uptake values after injection of 0.03 and 10 μg per animal). This finding demonstrates that the uptake in these IGF-1R–expressing tissues is saturable, indicating that it is, at least partially, receptor-mediated. In contrast, the amount of radioactivity in the liver was independent of the protein dose.
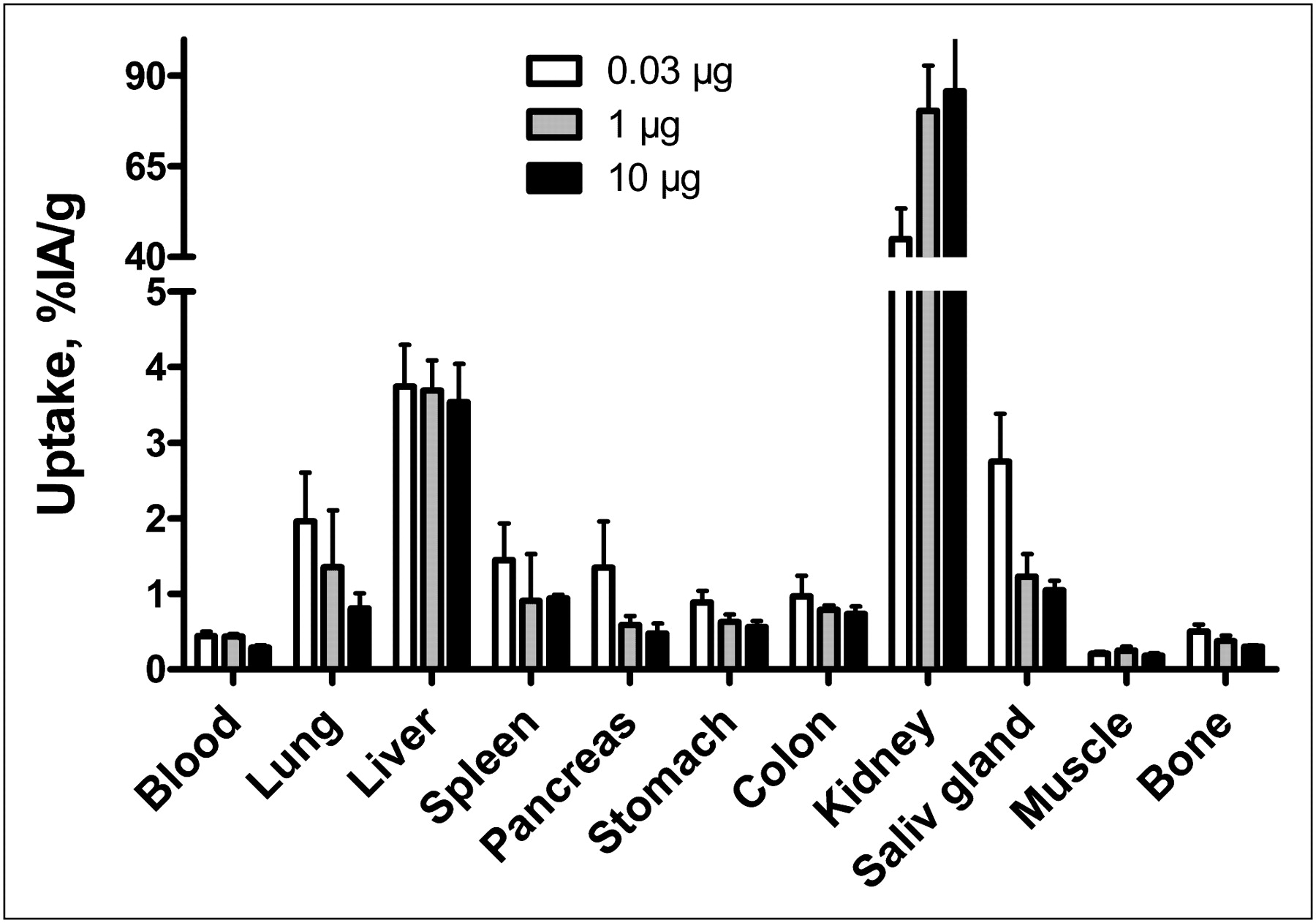
Biodistribution of 111In-DOTA-ZIGF1R:4551 in NMRI mice at 4 h after injection. Values are presented as mean percentage of injected radioactivity per gram of tissue (%IA/g) ± SD (n = 4). Saliv = salivary.
Biodistribution in NMRI nu/nu Mice Bearing DU-145 Prostate Cancer Xenografts: Influence of Protein Dose on Uptake in Tumors and Normal Tissues
The results of the previous experiment suggest that uptake of 111In-DOTA-ZIGF1R:4551 is receptor-mediated for several tissues. Our previous studies with targeting of epidermal growth factor receptor (29) showed that it was possible to partially saturate receptors in normal tissues to a higher extent than in tumors, leading to an increase in tumor-to-organ uptake ratio. To evaluate whether this was possible for IGF-1R, the biodistribution as a function of injected dose at 4 h after injection in male outbreed NMRI nu/nu mice bearing DU-145 prostate cancer xenografts was studied (Fig. 3). In agreement with the study in non–tumor-bearing mice, the accumulation of radioactivity in the lung, stomach, pancreas, and salivary gland significantly decreased (P < 0.05) with increasing protein dose, supporting the hypothesis that saturation occurred in these tissues. In contrast, no significant difference (P < 0.05) was seen in blood, spleen, liver, muscle, or bone at different injected protein doses, suggesting that ligand–receptor interaction did not play any significant role in accumulation of radioactivity in those organs. The lungs showed a separate pattern of radioactivity accumulation. A large decrease was observed when the protein dose was increased from 1 to 300 μg (P < 0.05), whereas no significant differences could be seen between the lowest 3 doses. There was no significant difference in the tumor uptake after injection of 0.1–1 μg of protein per mouse; however, an injection of 300 μg caused a highly significant (P < 0.0005) decrease of radioactivity accumulation in tumors.

Dose-escalation study of 111In-DOTA-ZIGF1R:4551 in male NMRI nu/nu mice with subcutaneous DU-145 xenografts at 4 h after injection. Values are presented as mean percentage of injected radioactivity per gram of tissue (%IA/g) ± SD (n = 4). Saliv = salivary.
The influence of injected protein dose on tumor-to-organ ratios is presented in Figure 4. An increase of the injected protein dose from 0.3 to 1 μg resulted in an increase of the tumor-to-pancreas ratio by 1.8-fold and of the tumor–to–salivary gland ratio by 1.5-fold (P < 0.005 in both cases). In contrast, the tumor-to-liver ratio decreased when the dose was increased. With most organs and tissues, no significant difference in tumor-to-organ ratios could be seen—that is, the dependence on the amount of injected protein was the same as for the tumor.
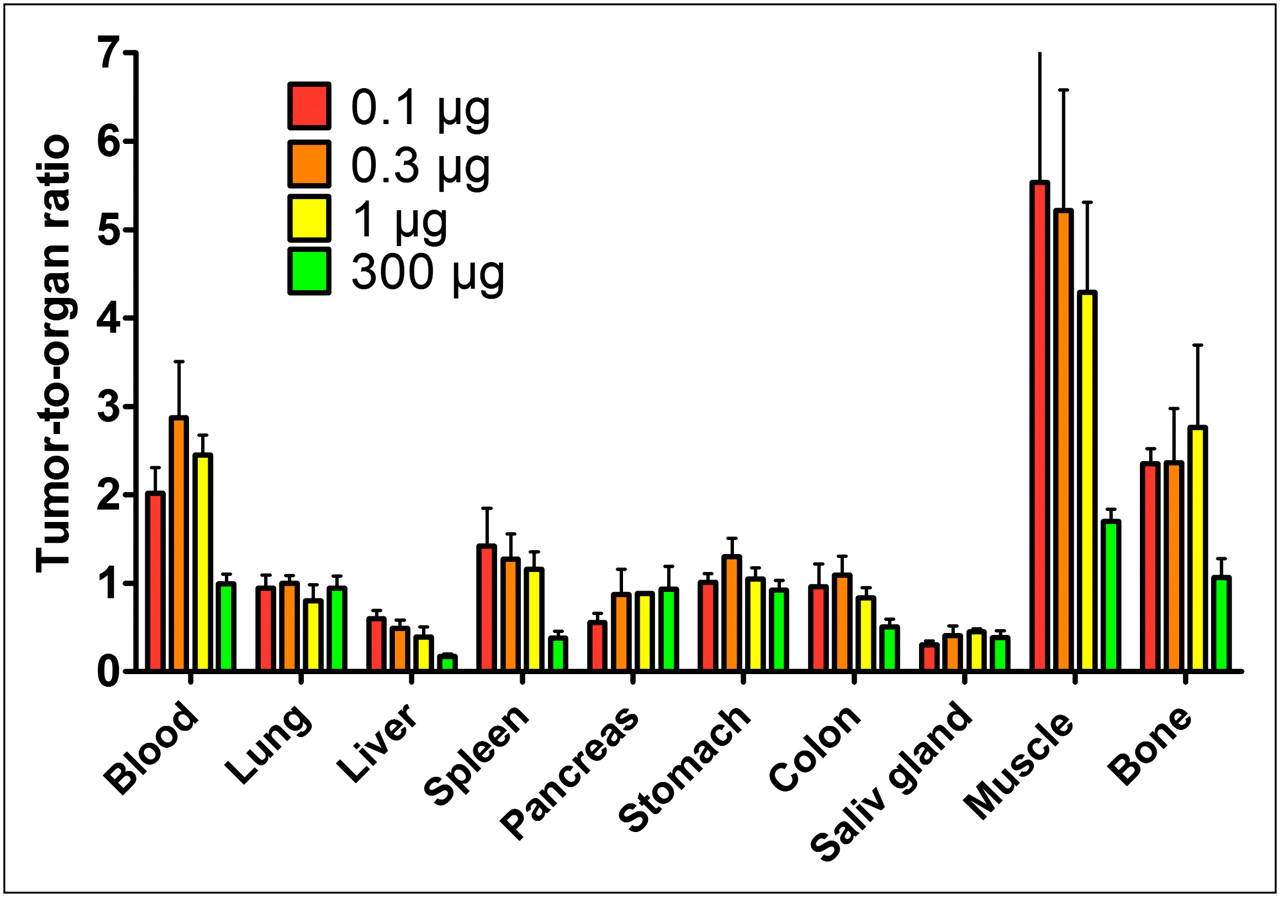
Tumor-to-organ ratios for 111In-DOTA-ZIGF1R:4551 in male NMRI nu/nu mice with subcutaneous DU-145 xenografts at 4 h after injection. Values are presented as mean value ± SD (n = 4). Saliv = salivary.
Biodistribution in NMRI nu/nu Mice Bearing DU-145 Prostate Cancer Xenografts: Results for Different Time Points
The biodistribution as a function of time was also investigated, and the results are presented in Figure 5. The results demonstrate a rapid clearance from blood and lungs. The concentration of radioactivity in liver, salivary gland, and spleen was rather constant over the course of the experiment. Similarly, no significant difference was observed in muscles and bones over the course of the experiment (except that the level at 1 h after injection was higher than at 8 and 24 h). There was slow washout of radioactivity from tumors and normal IGF-1R–expressing organs (colon, lung, pancreas, salivary gland, and stomach). The washout rate was somewhat slower from the tumor than from tissues, which increased the tumor-to-organ ratios for blood, lung, and pancreas with time (Fig. 6). The tumor-to-bone ratio reached its maximum at 8 h after injection.
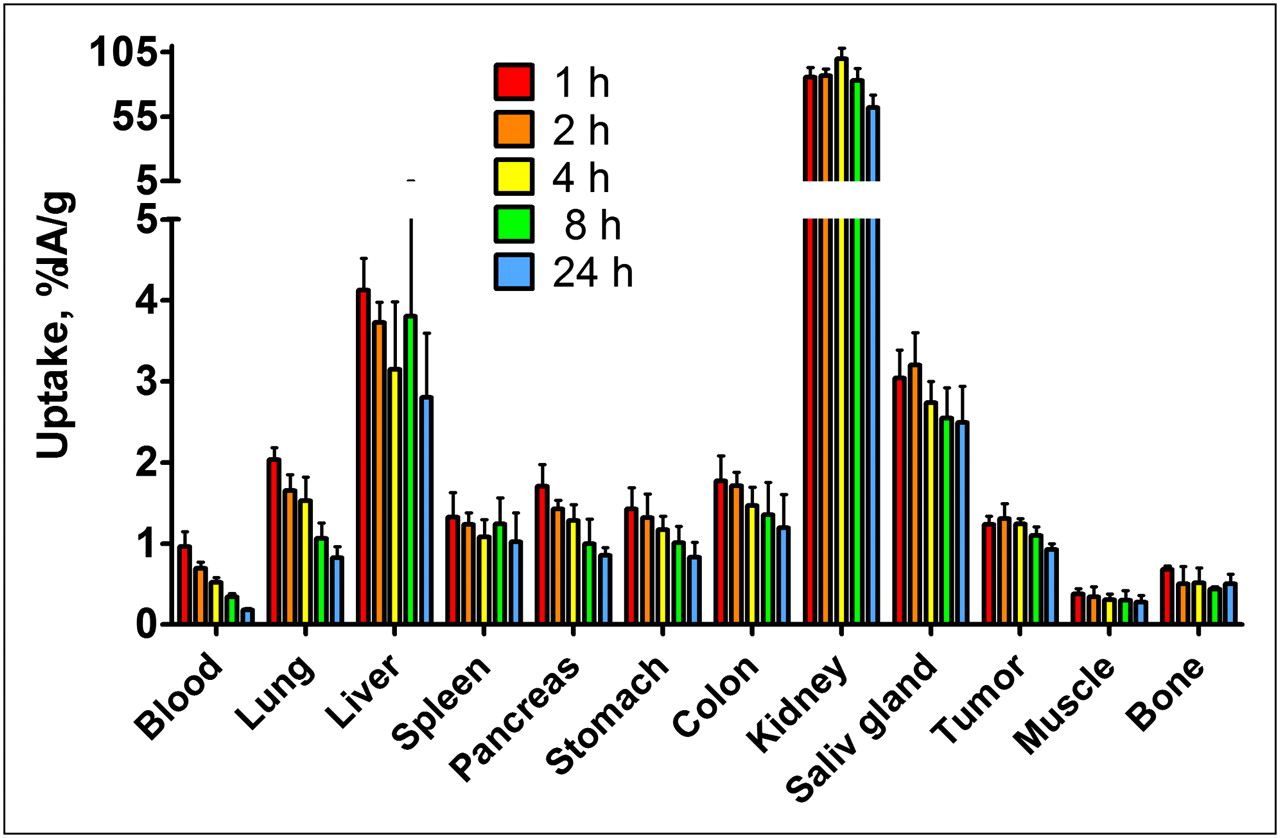
Biodistribution of 111In-DOTA-ZIGF1R:4551 (injected dose, 1 μg) in male NMRI nu/nu mice with subcutaneous DU-145 xenografts at different time points. Values are presented as mean percentage of injected radioactivity per gram of tissue (%IA/g) ± SD (n = 4). Saliv = salivary.

Tumor-to-organ ratios for 111In-DOTA-ZIGF1R:4551 (injected dose, 1 μg) in male NMRI nu/nu mice with subcutaneous DU-145 xenografts at different time points. Values are presented as mean value ± SD (n = 4).
γ-Camera Imaging
Images acquired at 8 h after the administration of 111In-DOTA-ZIGF1R:4551 to immunodeficient mice bearing subcutaneous DU-145 are presented in Figure 7. The tumor xenografts were visualized with appreciably higher accumulation of radioactivity than in contralateral sites. The tumor–to–contralateral site ratio was 2.9 ± 0.1 (average ± SD). Also, as predicted from the biodistribution studies, there was substantial accumulation of radioactivity in the kidneys, liver, and salivary gland, which were thereby clearly visualized.

Imaging of IGF-1R expression in DU-145 prostate cancer xenografts in NMRI nu/nu mice using111In-DOTA-ZIGF1R:4551. Planar γ-camera images were acquired at 8 h after injection. Arrows point to tumor (T), kidney (K), liver (L), and salivary gland (SG).
DISCUSSION
In this work, we have investigated the feasibility of using Affibody molecules for radionuclide molecular imaging of IGF-1R–expressing tumors in vivo. Imaging of IGF-1R can potentially provide essential information influencing management of several different tumor types. For example, this method has a potential to allow for stratification of patients with androgen-independent prostate cancer for IGR-1R–targeting therapy and for monitoring the response to such therapy. In principle, several classes of radiolabeled molecular targeting probes can be considered for imaging of IGF-1R overexpression—such as anti–IGF-1R monoclonal antibodies or fragments thereof; a natural ligand, IGF-1, or its mutated variants (30–33); or scaffold proteins such as Affibody molecules.
An issue with monoclonal antibodies for radionuclide molecular imaging is their large size, resulting in slow blood clearance and slow tumor accumulation. Thus, imaging of molecular targets is possible only several days after injection, which is an essential clinical disadvantage. The applicability of radiolabeled antibodies for molecular imaging is also limited by the unspecific uptake of proteins with a molecular weight of more than 45 kDa in tumors, the so-called enhanced permeability and retention effect (34), because this can lead to false-positive findings. A possible advantage of monoclonal antibodies is that a therapeutic antibody with known targeting efficiency and safety profile can be radiolabeled, providing a facile way to develop the imaging agent. Recently, the use of radiolabeled anti–IGF-1R R1507 antibody for immunoSPECT and immunoPET of triple-negative breast cancer xenografts was reported, demonstrating the feasibility of in vivo IGF-1R detection (30). As expected, a good imaging contrast was not obtained until 3 d after the administration.
The use of IGF-1 and its mutated variants (31–33) for in vivo targeting of IGF-1R–overexpressing or IGF-1R–transfected tumor xenografts has shown promising results. The major issue with IGF-1–based probes is their agonistic action, potentially eliciting a broad range of physiologic responses. Scaffold-based affinity proteins, such as Affibody molecules, may have agonistic, antagonistic, or no effect on their target receptor, much like antibodies. In fact, the parental IGF-1R–binding Affibody molecule Z4:40 is an IGF-1R antagonist (26).
In this study, we have for the first time evaluated the use of Affibody molecules for in vivo imaging of IGF-1R expression in a prostate cancer model. We have shown that conjugation of DOTA to ZIGF1R:4551 and subsequent labeling with 111In provides a conjugate with preserved binding specificity (Fig. 1B). Biodistribution in normal mice demonstrated specific (dose-dependent) binding of 111In-DOTA-ZIGF1R:4551 to IGF-1R–expressing organs and tissues (Fig. 2). This finding suggests that 111In-DOTA-ZIGF1R:4551 possesses cross-reactivity to murine IGF-1R, making a mouse model adequate for evaluation of IGF-1R targeting in vivo because a high expression of the target in normal organs might appreciably influence the imaging results, especially when the target level in the tumor is low. Injection of a high 111In-DOTA-ZIGF1R:4551 protein dose caused a significant decrease of uptake in tumors, suggesting a saturability of IGF-1R targeting in vivo—a strong indication of targeting specificity. Further, after optimization of the injected dose, an increase in tumor-to-blood, tumor–to–salivary gland, and tumor-to-pancreas ratio could be achieved. This study suggests that the optimal time point for imaging aquisition is between 8 and 24 h after injection. Experimental γ-camera imaging confirmed the feasibility of visualization of IGF-1R–expressing prostate cancer xenografts. A limitation of the tested conjugate is the high accumulation of radioactivity in the liver and kidneys. Several approaches can be explored to overcome this limitation. For example, the probe investigated in this study contained a hexahistidine tag at the N-terminus to facilitate immobilized metal ion affinity chromatographic purification of the construct, as we have recently shown to be associated with elevated liver uptake of radioactivity (27,35). Using alternative purification schemes that do not require the use of hexahistidine tags might reduce liver uptake of IGF-1R–targeting Affibody molecules. The use of a nonresidualizing radiohalogen label is a complementary approach to appreciably reduce renal retention of radioactivity in comparison with radiometal labels (36). Our recent data have also demonstrated that low renal radioactivity retention can be obtained using a cysteine-containing peptide-based GGGC chelator for 99mTc, which may be considered as a chelator in future studies of IGF-1R imaging (37).
The possibility of visualizing IGF-1R expression in vivo is not only of value for treatment optimization of prostate cancer. There is strong evidence that expression of IGF-1R and cross-talk between HER2 and IGF-1R signaling is a potential reason for trastuzumab resistance in disseminated HER2-positive breast cancer (4,38). Thus, determination of IGF-1R status in those tumors could also be of value in the clinic. Furthermore, IGF-1R signaling has been suggested to be involved in development of gefitinib resistance in non–small cell lung cancer (39,40). Thus, further research regarding applications of the anti–IGF-1R Affibody molecule is warranted.
CONCLUSION
This study has for the first time demonstrated the feasibility of in vivo targeting of IGF-1R–expressing prostate cancer xenografts using Affibody molecules. Further development of the radiolabeled probe might result in a useful clinical tool for stratification of patients with prostate cancer for IGF-1R–targeting therapy.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This research was financially supported by grants from the Swedish Cancer Society (Cancerfonden), the Swedish Research Council (Vetenskapsrådet), and O.E. and Edla Johansson Foundation. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Dec. 15, 2011.
- © 2012 by the Society of Nuclear Medicine, Inc.
REFERENCES
- Received for publication March 20, 2011.
- Accepted for publication August 17, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}