


Abstract
Chemokine receptor 4 (CXCR4)–chemokine ligand 12 (CXCL12) interactions have been shown to play key roles in cancer cell survival, proliferation, chemotaxis, homing, adhesion, tumor angiogenesis, and resistance to conventional and targeted therapies. Given its extensive involvement in cancer progression, the CXCR4–CXCL12 axis has been considered a therapeutic target. Several inhibitors blocking this signaling cascade are in phase I trials. Because CXCR4 is constitutively expressed in a wide variety of normal tissues, patient stratification and noninvasive monitoring would improve therapeutic outcome and reduce unnecessary toxicities. This review focuses on recent developments in CXCR4-based imaging agents and their potential role in the molecular diagnosis and treatment of cancer.
Most deaths from cancer stem from metastasis, and chemokine receptors are emerging as key elements in this process. Chemokine receptors are 7-transmembrane–spanning proteins of the G-protein–coupled receptor superfamily. The 19 known chemokine receptors, in association with their ligands (48 chemokines), direct immune cell and hematopoietic stem cell trafficking. Chemokines are small proteins (8–12 kDa) that are classified into 4 groups (CXC, CX3C, CC, and C) based on the arrangement of highly conserved cysteine residues at the N terminus (1). Several of the chemokines and receptors interact with more than one receptor or ligand. Of the six receptors that are known to bind a single ligand, the chemokine receptor 4 (CXCR4)–chemokine ligand 12 (CXCL12) (also known as stromal cell–derived factor 1) pair has been gaining significant attention because of its role as a putative coreceptor for HIV entry, its role in promoting metastasis in tumors, its involvement in cell trafficking in autoimmune disease and inflammatory conditions, and its role in stem cell maintenance. This review will focus on the role and the detection of CXCR4 in cancer. Excellent reviews on the role of CXCR4 in other diseases can be found elsewhere (2–5).
CXCR4 EXPRESSION, REGULATION, AND SIGNALING IN CANCER
CXCR4 is overexpressed in more than 23 human cancer types including breast, brain, ovary, and prostate cancer and melanoma. CXCR4 expression in normal tissues is markedly lower than in tumors (6). Increased CXCR4 expression in tumors is associated with an aggressive phenotype (2,7–9). CXCR4 expression enables tumor cells to home to organs expressing abundant levels of CXCL12, such as lungs, bone, liver, brain, and bone marrow, leading to establishment of metastases (Fig. 1A). Accordingly, overexpression of CXCR4 in primary tumors is directly correlated to increased risk for local recurrence, distant metastasis, and poor survival rates in breast, colon, and several other cancers (2,6,8–10).
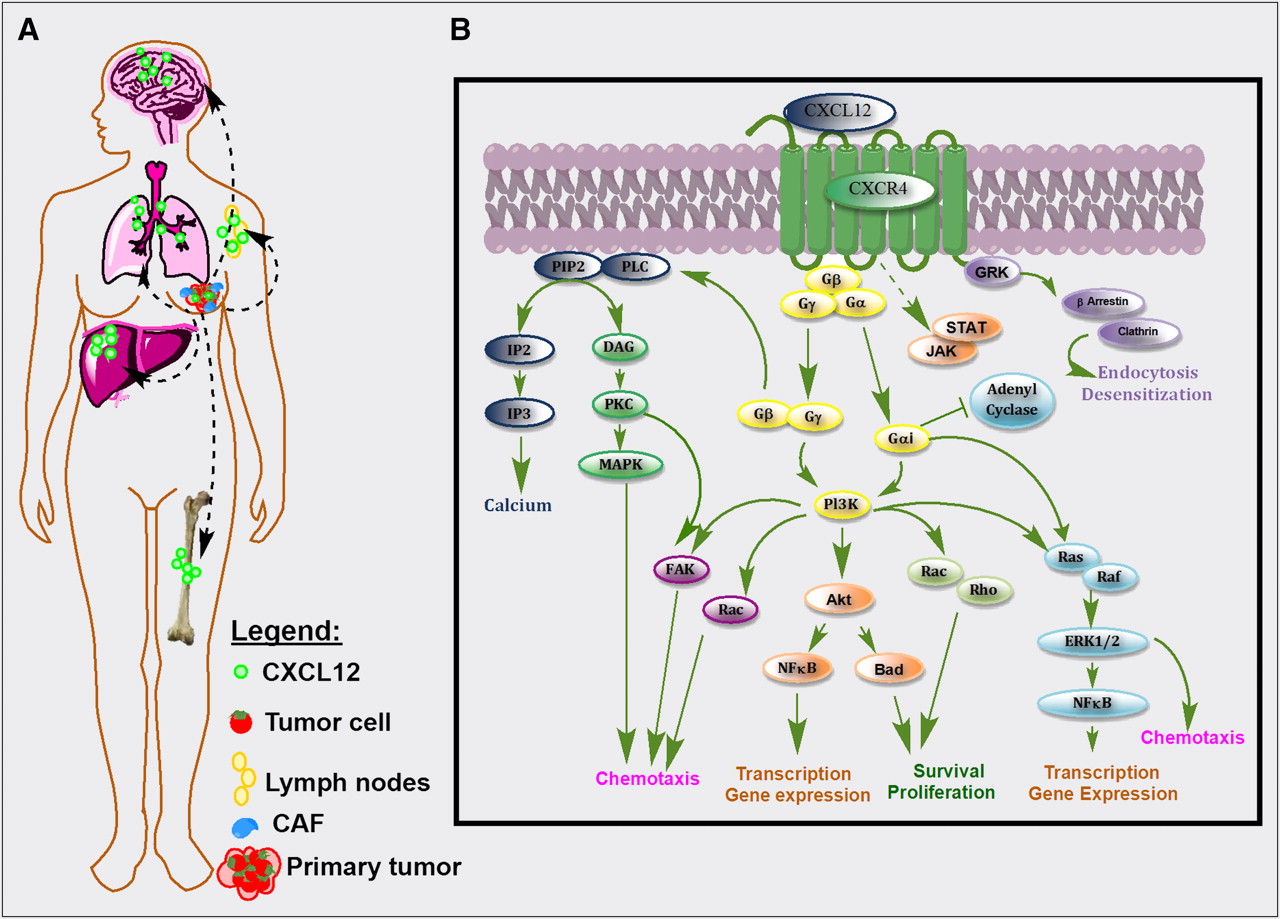
(A) CXCL12 is abundantly expressed in normal tissues such as lungs, liver, and bone marrow and is also secreted by tumor and stromal cells. CXCR4–CXCL12 interactions in tumor induce release of vascular endothelial growth factor, increase vascular permeability, and promote tumor angiogenesis and recruitment of tumor-associated macrophages. The resulting increased proliferative, migratory, and invasive properties of tumor cells enable their escape from primary tumors. Tumor cells that overexpress CXCR4 migrate toward the chemoattractant gradient, like leukocytes, and home to organs that release CXCL12. (B) Activation of CXCR4 by CXCL12 in primary tumor and in metastases leads to G-protein–coupled signaling through IP3, PI3K/Akt, and MAPK pathways, promoting cell survival, proliferation, and chemotaxis. Akt = serine/threonine protein kinase Akt (or protein kinase B, PKB); Bad = Bcl-2–associated death promoter; CAF = carcinoma-associated fibroblast; DAG = diacylglycerol; GRK = G protein–coupled receptor kinases; FAK = focal adhesion kinase; IP2 = inositol (1,4)-bisphosphate; IP3 = inositol (1,4,5) trisphosphate; PIP2 = phosphatidylinositol 4,5-bisphosphate; Pl3K = phosphoinositide 3-kinase; PKC = protein kinase C; PLC = phospholipase C; Ras = rat sarcoma protein family; Rho = Rho family of GTPases.
In addition to primary tumors, metastases frequently exhibit increased CXCR4 expression, which may offer a new strategy for their early detection (9,11). Neutralization of CXCR4 chemotaxis by use of low-molecular-weight agents, peptides, antibodies, or biologic agents such as small interfering RNA significantly reduces metastatic burden in preclinical models of various cancers (8,12,13).
Binding of CXCL12 to CXCR4 leads to the formation of a complex with the Gαi subunit G protein, resulting in the inhibition of adenylyl cyclase–mediated cyclic adenosine monophosphate production and mobilization of intracellular calcium. Dissociation of the Gαi subunit from Gβγ leads to activation of multiple downstream targets, including Rho, extracellular signal-regulated kinases (ERK1/2), mitogen-activated protein kinase (MAPK), and AKT effectors, as shown in Figure 1B, leading to cell survival, proliferation, and chemotaxis (2,8,14). Partially independent of G protein, the Janus-activated kinase/signal transducers and activators of the transcription JAK/STAT pathway are also activated through CXCR4 (8). In addition to those signaling cascades, the CXCR4–CXCL12 axis is also known to transactivate HER2 receptor (8,15) and mediate estrogen-independent tumorigenesis, metastasis, and resistance to endocrine therapy (10). Also, activation of the CXCR4–CXCL12 axis results in tumor resistance to conventional and targeted therapies by directly promoting cancer cell survival, invasion, and the cancer stem or tumor-initiating cell phenotype; recruiting myeloid bone marrow–derived cells to facilitate tumor recurrence and metastasis indirectly; promoting angiogenesis directly or in a paracrine manner; and providing a metastatic niche for cancer cells in the bone marrow (6,16). Although the role of CXCR4 in cancer cells is well established, recent studies have also identified increased expression of CXCR4 in cancer-associated fibroblasts (17). Cancer-associated fibroblasts play an important role in tumorigenesis and have been implicated in neoplastic progression, tumor growth, angiogenesis, and metastasis (17). Data from a recent study by Eck et al. suggest that soluble breast cancer factors initiate the transdifferentiation of normal human mammary fibroblasts to tumor-promoting cancer-associated fibroblasts through the induction of matrix metalloproteinase-1 and CXCR4 expression (18). Furthermore, the role of another chemokine receptor, CXCR7, in modulating the CXCR4 signaling cascade is emerging. CXCR7 is expressed on tumor cells, binds to CXCL12, and forms heterodimers with CXCR4 (19,20). Recent studies show that the CXCR4/CXCR7 complex impairs G protein–coupled signaling and constitutively recruits β-arrestin–dependent signal transduction pathways including MAPK, stress-activated protein kinase, and ERK1/2 activation, leading to increased cell migration of CXCR4-expressing breast cancer cells (21). These findings suggest a possible role for CXCR7 binding agents in modulating CXCR4 signaling through the formation of CXCR4/CXCR7 heterodimers.
Accumulating evidence substantiates the CXCR4–CXCL12 axis as a therapeutic target in cancer. In addition to reduced metastatic burden, CXCR4 inhibition can also synergize with standard chemotherapy in various tumor models (22,23). Furthermore, expression of CXCR4 in many tumors is regulated at the transcriptional level by hypoxia, nuclear factor κB, and Yin Yang 1; at the translational level by HER2; and at the posttranslational level by E3 ubiquitin ligase and HER2, which may have implications for combination therapies (8). Recent clinical trials showed that short-term treatment with CXCR4 antagonists is safe, supporting the use of CXCR4-targeted agents as adjuvants to currently available therapies (24). Accordingly, several CXCR4 inhibitors are in phase I trials (e.g., NCT01120457 [clinical trials.gov]). Considering the large number of normal functions that are affected by the CXCR4–CXCL12 axis (2,6,8), long-term inhibition of CXCR4 may need continuous monitoring. Therefore, development of CXCR4-based imaging agents would be beneficial for the translation of CXCR4 inhibitors to evaluate primary tumors for elevated CXCR4 expression and therapeutic intervention, to screen for secondary metastatic spread to both local and distant sites, and to allow for therapeutic monitoring.
CXCR4-BASED IMAGING AGENTS
CXCR4, unlike other chemokine receptors, is characterized by a strong negatively charged extracellular surface. Not surprisingly, CXCL12 and most of the CXCR4-binding agents, including the peptide-based inhibitors T22, T140, and several small-molecule-antagonist scaffolds such as cyclams, are highly basic and positively charged. A detailed overview of the available CXCR4-binding agents has been previously published (25). Most of the CXCR4-targeted imaging agents to date have originated from those scaffolds.
Small Molecules
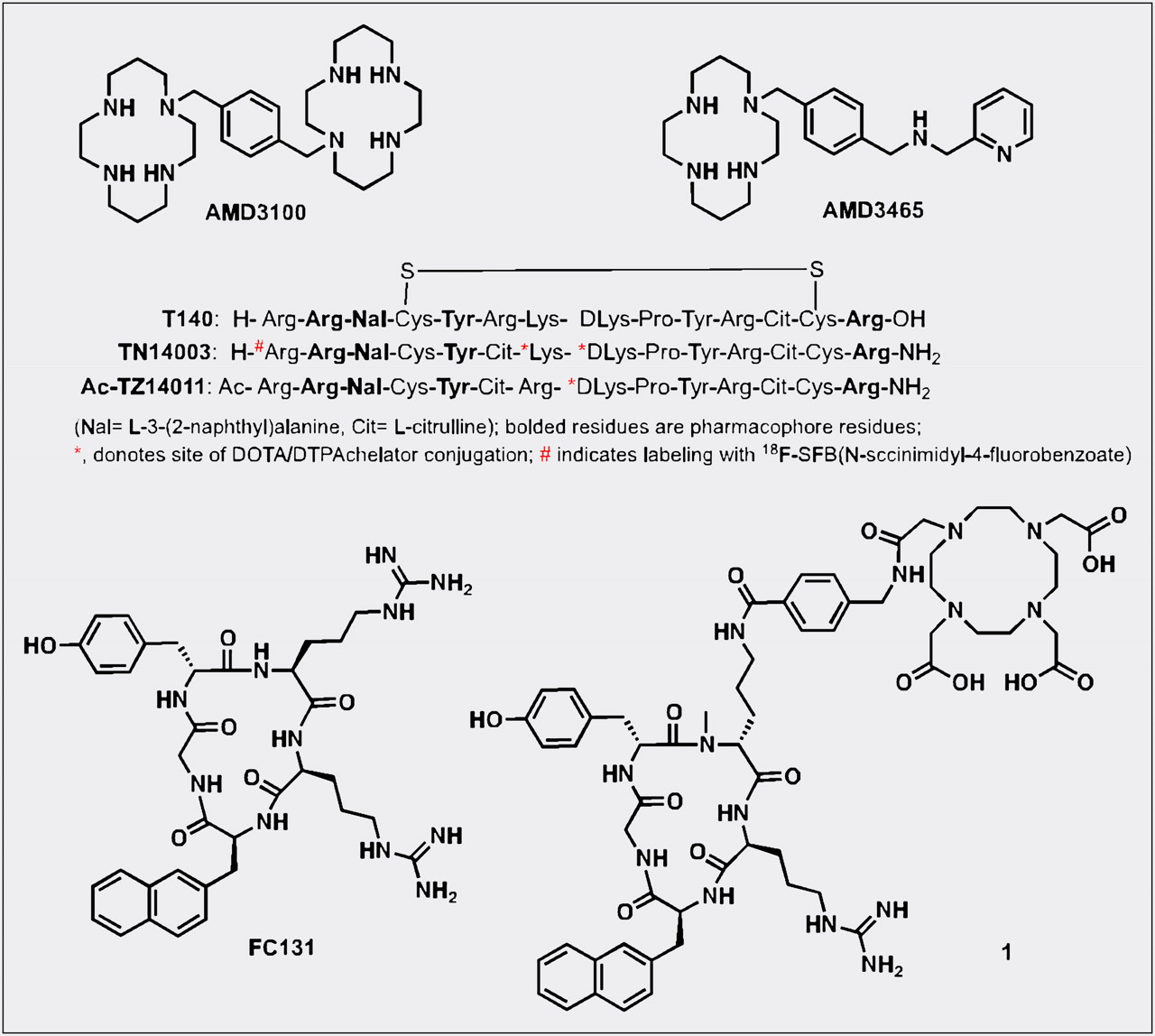
The bicyclam AMD3100 (1,1′-[1,4-phenylenebis(methylene)]bis [1,4,8,11-tetraazacyclotetradecane]; Fig. 2) was the first nonpeptide CXCR4 inhibitor to enter clinical trials and is now used for stem cell mobilization (3). Cyclams have the ability to form strong complexes with transition metals such as copper and zinc, opening the door for a radiolabeled analog of AMD3100 and CXCR4 expression imaging. Fortuitously, the affinity of AMD3100 increases by 7-fold when chelated to copper (11). Jacobson et al. radiolabeled AMD3100 using 64Cu and noted that, in immune-competent mice, uptake of radioactivity was high in the liver and lymphoid organs (26). Applicability of that agent for imaging-graded levels of CXCR4 expression was later demonstrated by Nimmagadda et al. using subcutaneous U87 brain tumors (∼2% CXCR4+), U87 tumors stably expressing CXCR4 (>95%, U87-stb-CXCR4), and orthotopic MDA-MB-231 (∼10%) and DU4475 (∼90%) breast tumor xenografts. PET imaging with 64Cu-AMD3100 demonstrated accumulation of radioactivity in the U87-stb-CXCR4 and DU4475 tumors at 90 min after injection of the radiotracer (11). Similar to Jacobson et al., Nimmagadda et al observed considerable uptake in the liver and lymphoid organs. Given that CXCR4 is expressed on leukocytes, on monocytes, and in the liver, radioactivity accumulation in these organs, except for the majority of nonspecific uptake in the liver, is due to target-specific binding. Those results were later confirmed by blocking studies and other groups (11,27).

Common structural motifs of CXCR4 imaging agents.
Because metastases often have elevated levels of CXCR4 expression, using an experimental model of lung metastasis derived from breast cancer cells, Nimmagadda et al. demonstrated that 64Cu-AMD3100-PET enables noninvasive, in vivo visualization of metastases (Fig. 3). Successive ex vivo biodistribution and molecular characterization studies confirmed that the radioactivity uptake observed in the lungs was indeed due to elevated CXCR4 expression in the metastases (11).
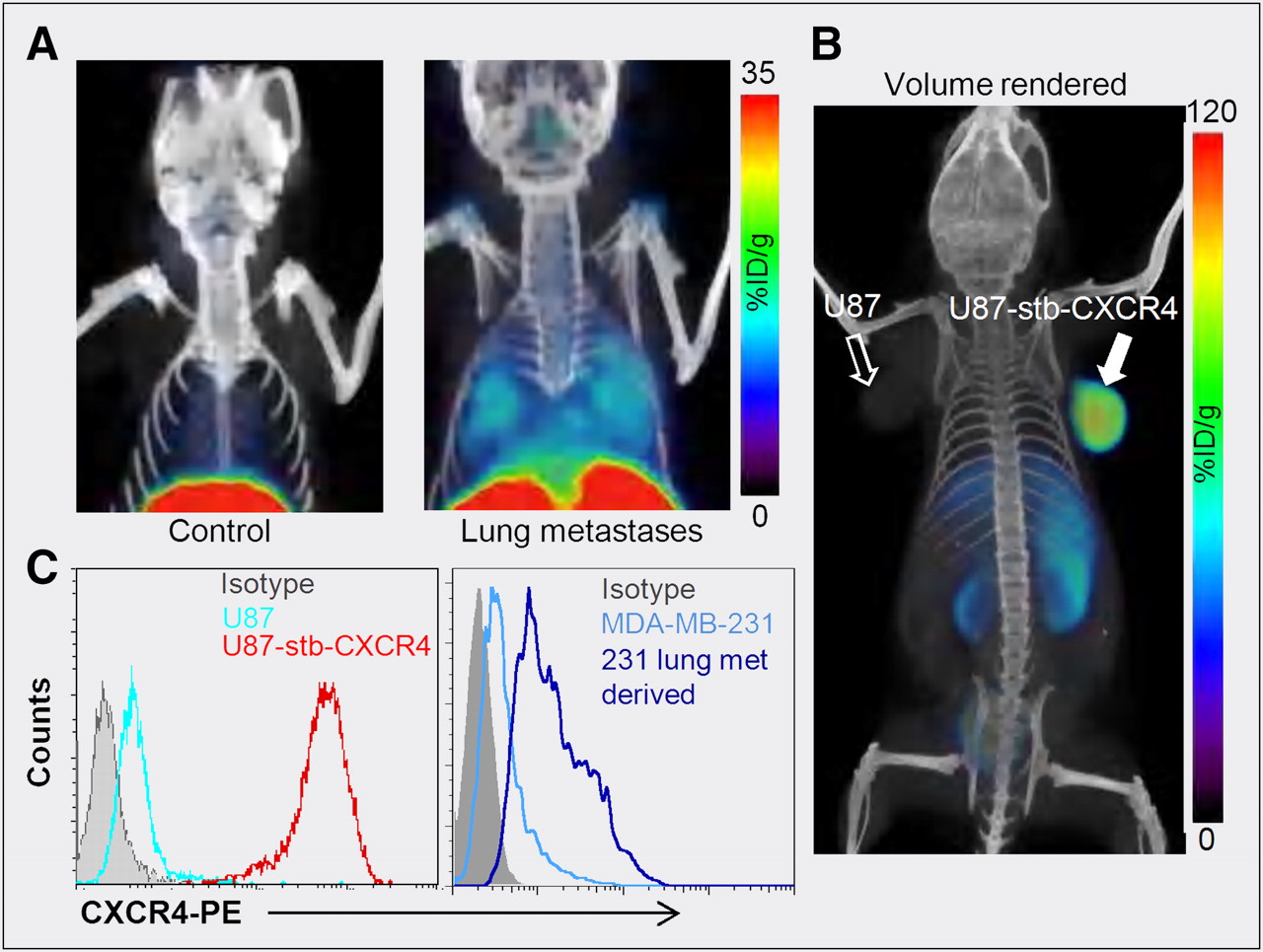
Although AMD3100 shows promise as a PET agent, low affinity for CXCR4 and a scaffold not flexible for the development of 18F-labeled analogs may limit clinical use. A second-generation, monocyclam-based CXCR4 inhibitor, AMD3465 (Fig. 2), has high affinity (41.7 ± 1.2 nM), reduced charge, and is smaller in size compared to AMD3100 (28). Using the U87 and U87-stb-CXCR4 glioblastoma model and a colon xenograft model, De Silva et al. showed that 64Cu-AMD3465-PET has the highest target selectivity reported for this class of agents to date (Fig. 3). More important, the pyridine moiety of AMD3465 may allow structural modification for the synthesis of clinically translatable agents.
Monoclonal Antibodies and Peptides
Monoclonal antibodies are gaining attention as radiopharmaceuticals. Monoclonal CXCR4 antibody clone 12G5, which binds the same extracellular loop as CXCL12, was radiolabeled with 125I and studied by Nimmagadda et al. in a glioblastoma tumor model (29). Imaging data showed clear accumulation of 125I-12G5 in the tumors, compared with isotype-matched 125I-IgG2A control antibody. Even though the highest level of radioactivity was seen in the spleen, those results establish the viability of using radiolabeled monoclonal antibodies for imaging CXCR4 expression.
Polyphemusin-based peptides form the foundation for most of the peptide-based CXCR4 imaging agents. In 1998, Tamamura et al. identified T140 (Fig. 2), a 14-residue peptide with a disulfide bridge, as a potent CXCR4 antagonist. Subsequent studies have shown that Arg2, l-3-(2-naphthyl)alanine (Nal)3, Tyr5, and Arg14 in T140 are critical for CXCR4 binding. Although T140 was found to be unstable in serum, many CXCR4-selective and stable analogs with modifications at each terminus were synthesized (30). First within this category of peptides is Ac-TZ14011, with the carboxyl group protection via amidation for stability in vivo and a single amino group (d-Lys8) distant from the pharmacophore allowing for conjugation of chelates. Even though 111In-diethylenetriaminepentaacetic acid (DTPA) conjugation resulted in a nearly 6-fold decrease in affinity to CXCR4, acceptable accumulation observed within the tumors led to further development of these peptides for imaging. Although 111In-DTPA-Ac-TZ14011 uptake was higher in the tumor than in the muscle or blood, a 15- to 200-fold increase in uptake was observed in the liver, kidneys, and spleen (31). Also, Kuil et al. recently reported the synthesis of a bimodal analog of Ac-TZ14011, radiolabeled with 111In-DTPA for SPECT and Cy5.5 for optical imaging. In vivo results indicate that the fluorescent agent may have applicability for image-guided surgical applications (32).
Another amidated analog of T140, TN14003 (Fig. 2), with N-terminal 4-fluorobenzoyl protection (4-18F-T140), has been labeled with 18F using N-succinimidyl-4-18F-fluorobenzoate and evaluated in Chinese hamster ovarian tumor–bearing mice, with tumors stably expressing CXCR4 (33). With 4-18F-T140, CXCR4-positive tumors were distinguishable from control tumors; however, coinjection of unlabeled 4-F-TN14003 was necessary to see increased radioactivity in the CXCR4-positive tumors. The same peptide was also radiolabeled with 64Cu-DOTA on the lysines (34). Because these agents are receptor-targeted and it is likely that tumor cells often have low receptor density, compared with stably transfected cell lines (11), use of agents of low specific activity may face significant challenges in clinical translation. Similarly, CXCL12 radiolabeled with 99mTc or with near-infrared fluorophores have been shown to have poor imaging characteristics in vivo, limiting routine use (35,36).
The pharmacophore amino acid residues of T140 that interact with CXCR4 were further downsized to a cyclic pentapeptide (FC131), which generated a compound with similar potency to T140 itself (37). All of the imaging agents described above, including the low-molecular-weight agents, have moderate to high liver uptake, limiting their use for imaging lesions within this organ. A recent report by Demmer et al. showed a highly specific FC131-based cyclic 68Ga-DOTA–conjugated peptide (1, Fig. 2) with high whole-body contrast, low liver uptake, and fast clearance from the kidneys, potentially obviating that problem (38).
CLINICAL IMPLICATIONS AND FUTURE CHALLENGES
Since the description of the implication of CXCR4 in the metastatic cascade, much progress has been made in establishing its role in tumor progression. The direct involvement of CXCR4 in the metastatic cascade suggests a role for CXCR4 imaging agents in identifying primary tumors with an aggressive phenotype, and also a new approach for the early detection of metastases. The role of the CXCR4–CXCL12 pathway in resistance to antitumor therapies is emerging and will be important in the design of future combination therapies. CXCR4-based imaging agents may also facilitate the temporal evaluation of changes in expression of CXCR4 in tumors in response to combination therapy. Clinical translation faces several challenges: first, the evaluation of these agents in biologically relevant tumor, metastasis, and therapy models so that feasibility of imaging graded levels of CXCR4 expression, sensitivity, and specificity can be established; second, the consideration for CXCR4 desensitization in some tumors and validation of whether high tumor uptake due to surface CXCR4 expression correlates with prognosis; third, the establishment of a correlation between plasma CXCL12 levels and tumor radioactivity uptake and prognosis; and fourth, the establishment of the need for simultaneous evaluation of CXCR4 and CXCR7 levels in the tumors. Some but not all of these challenges can be addressed in preclinical models in parallel with the translation of a suitable agent. As therapeutic agents that target CXCR4 translate to the clinic, the availability of CXCR4 imaging agents will enable not only detection or staging of cancer but also measurement of the concentration of therapeutic agent reaching its target and allow us to unravel the relationship between tumor progression and CXCR4 expression in the biologic system of greatest relevance.
Acknowledgments
We regret that many important studies that elucidated the role of CXCR4 in cancer could not be cited due to editorial constraints. We gratefully acknowledge grant support from the Elsa U. Pardee Foundation and the Maryland Technology Development Corporation. We also thank Drs. Pomper and Bhujwalla for many helpful discussions. No potential conflict of interest relevant to this article was reported.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication September 30, 2011.
- Accepted for publication October 3, 2011.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Theranostics: Leveraging Molecular Imaging and Therapy to Impact Patient Management and Secure the Future of Nuclear Medicine
- Repopulating hematopoietic stem cells from steady-state blood before and after ex vivo culture are enriched in the CD34+CD133+CXCR4low fraction
- SDF-1 Expression is Associated with Poor Prognosis in Osteosarcoma
- PET Imaging of Leukocytes in Patients With Acute Myocardial Infarction
- CXCR4/SDF-1{alpha}-mediated Chemotaxis in an In Vivo Model of Metastatic Esophageal Carcinoma