


Abstract
Panitumumab, a human monoclonal antibody that binds to the epidermal growth factor receptor (HER1), was approved by the Food and Drug Administration in 2006 for the treatment of patients with HER1-expressing carcinoma. In this article, we describe the preclinical development of 86Y-CHX-A″-diethylenetriaminepentaacetic acid (DTPA)-panitumumab for quantitative PET of HER1-expressing carcinoma. Panitumumab was conjugated to CHX-A″-DTPA and radiolabeled with 86Y. In vivo biodistribution, PET, blood clearance, area under the curve, area under the moment curve, and mean residence time were determined for mice bearing HER1-expressing human colorectal (LS-174T), prostate (PC-3), and epidermoid (A431) tumor xenografts. Receptor specificity was demonstrated by coinjection of 0.1 mg of panitumumab with the radioimmunoconjugate. Results: 86Y-CHX-A″-DTPA-panitumumab was routinely prepared with a specific activity exceeding 2 GBq/mg. Biodistribution and PET studies demonstrated a high HER1-specific tumor uptake of the radioimmunoconjugate. In mice bearing LS-174T, PC-3, or A431 tumors, the tumor uptake at 3 d was 34.6 ± 5.9, 22.1 ± 1.9, and 22.7 ± 1.7 percentage injected dose per gram (%ID/g), respectively. The corresponding tumor uptake in mice coinjected with 0.1 mg of panitumumab was 9.3 ± 1.5, 8.8 ± 0.9, and 10.0 ± 1.3 %ID/g, respectively, at the same time point, demonstrating specific blockage of the receptor. Normal organ and tumor uptake quantified by PET was closely related (r2 = 0.95) to values determined by biodistribution studies. The LS-174T tumor had the highest area under the curve (96.8 ± 5.6 %ID·d·g−1) and area under the moment curve (262.5 ± 14.9 %ID·d2·g−1); however, the tumor mean residence times were identical for all 3 tumors (2.7–2.8 d). Conclusion: This study demonstrates the potential of 86Y-CHX-A″-DTPA-panitumumab for quantitative noninvasive PET of HER1-expressing tumors and represents the first step toward clinical translation.
The epidermal growth factor receptor (HER1 or Erb1) is a transmembrane glycoprotein belonging to subclass I of the tyrosine kinase receptor superfamily (1,2). HER1 signaling is firmly regulated in normal cells. The receptor is overexpressed in cancers ranging from lung to colorectal because of HER1 gene amplification and anomalous expression and signaling in cancer cells. Overexpression of the receptor is associated with poor survival, disease progression, and resistance to conventional chemotherapy (1,2). To overcome resistance to chemotherapy and improve outcomes, HER1-targeted therapies are actively being developed. Two major classes of clinical therapies have been explored in the treatment of HER1-expressing cancer, with moderate success (3,4). These are anti-HER1 monoclonal antibodies (mAbs) and HER1-specific tyrosine kinase inhibitors. Currently, 2 mAbs, cetuximab (Erbitux; ImClone and Bristol-Myers Squibb) and panitumumab (Vectibix; Amgen), are approved by the Food and Drug Administration.
Cetuximab is a chimeric anti-HER1 IgG1-isotype mAb indicated for use in patients with HER1-expressing metastatic colorectal cancer as a single-agent immunotherapy or in combination with irinotecan-based chemotherapy (3,4). The antitumor activity of cetuximab, however, requires high doses, and adverse reactions related to immune response and hypersensitivity to the antibody have been reported in approximately 19% of patients, with 3% of the patients experiencing severe reactions (4–6). The fully human anti-HER1 mAb panitumumab was developed using XenoMouse technology to improve therapeutic efficacy and decrease the potential of eliciting immune responses in patients (7). Panitumumab binds to the ligand-binding domain (domain III) of HER1 and is rapidly internalized, leading to downregulation of cell surface HER1 in vitro and in vivo (7,8). In addition to inhibiting phosphorylation of HER1 and mitogen-activated protein kinases or Akt, panitumumab also causes cell cycle arrest and inhibits tumor growth by suppressing the production of proangiogenic factors such as vascular endothelial growth factor and IL-8 by tumor cells (7,8).
Panitumumab was approved by the Food and Drug Administration in 2006 for the treatment of patients with HER1-expressing, metastatic colorectal carcinoma with disease progression during or after fluoropyrimidine-, oxiplatin-, and irinotecan-containing chemotherapy regimens (9–11). Panitumumab therapy is well tolerated in patients (12,13). A phase III trial of 463 patients with refractory metastatic colorectal cancer compared panitumumab plus best supportive care versus best supportive care alone. In the panitumumab group, as compared with best supportive care alone, a 46% reduction in the tumor-progression rate was reported (14). Panitumumab also significantly improved progression-free survival with manageable toxicity and was efficient in time-related endpoints. The clinical efficacy of panitumumab is currently being evaluated in patients with other types of cancers such as lung, breast, renal, head and neck, and ovarian (10).
A critical factor in screening patients for targeted therapy is evaluating the presence and amount of the specific target in the tumor and its relevance to the disease state. Initial clinical experience with both cetuximab and panitumumab therapy revealed that HER1 levels detected by immunohistochemistry did not correlate with response to anti-HER1 immunotherapy (15,16). Along with other pathologic procedures and tests, noninvasive nuclear imaging is often used to assess the status of the specific target. For instance, to assess the status of HER1 expression and cetuximab distribution, cetuximab has been radiolabeled with radionuclides such as 99mTc and 111In for SPECT (17–20), and cetuximab radiolabeled with 64Cu and 89Zr has been explored for PET (21–24). Panitumumab and cetuximab bind to different epitopes of HER1; therefore, a need to develop a panitumumab-specific imaging agent exists. Panitumumab conjugated with CHX-A″-diethylenetriaminepentaacetic acid (DTPA) and radiolabeled with 111In has been extensively evaluated preclinically in this laboratory (25). In the report from that study, conjugating 1–2 molecules of CHX-A″-DTPA to panitumumab did not alter the binding affinity of panitumumab. Panitumumab was found to retain reactivity with HER1 after modification with the CHX-A″-DTPA ligand and when radiolabeled with 111In. Excellent tumor targeting by 111In-CHX-A″-DTPA-panitumumab was demonstrated in vivo by direct quantitation of tumors and normal tissues in 5 tumor xenograft models. Considering the superiority of PET over single-photon scintigraphy, the development of a panitumumab-specific PET radioimmunoconjugate was deemed a worthwhile pursuit.
Of the numerous longer-lived positron-emitting radionuclides available for radioimmunoimaging—such as 124I, 86Y, 64Cu, and 89Zr—we selected 86Y because of its appropriate half-life (14.7 h), suitability for internalizing mAbs, well-established chemistry, and availability (26,27). In addition to these attractive features, 86Y can also serve as a surrogate marker for 90Y-based radioimmunotherapy and peptide receptor radionuclide therapy (28,29). In a study describing 64Cu-DOTA-panitumumab, the radioimmunoconjugate was successfully used to image HER1-expressing head and neck squamous cell tumor xenografts in mice (30).
In the present study, the preparation and evaluation of 86Y-CHX-A″-DTPA-panitumumab for potential use in risk stratification and quantitative noninvasive imaging of HER1 and assessment of panitumumab uptake in preclinical cancer models are described. To achieve this objective, 86Y-CHX-A″-DTPA-panitumumab was assessed by in vivo biodistribution; PET and quantification; blood pharmacokinetics; and detailed analysis of area under the curve (AUC), area under the moment curve (AUMC), and mean residence time (MRT) for mice bearing HER1-expressing human colorectal (LS-174T), prostate (PC-3), and epidermoid (A431) tumor xenografts.
MATERIALS AND METHODS
Preparation of 86Y-CHX-A″-DTPA-Panitumumab
Production and Purification of 86Y
86Y was produced by the previously described 86Sr(p,n)86Y reaction, with minor modifications in the postirradiation processing of the SrCO3 target (27). Briefly, the postirradiated SrCO3 target was dissolved in 500 μL of 3 M ultrapure-grade nitric acid and heated to dryness twice; 300 μL of 8 M ultrapure-grade nitric acid were added to the residue to dissolve 86Y. The mixture was allowed to cool in an ice bath to precipitate the strontium. The supernatant containing 86Y was separated and heated to dryness. The 86Y was extracted with 2 × 300 μL of 3 M nitric acid, loaded onto a preequilibrated 0.5-mL bed volume of strontium resin (EiChrom Industries), and eluted with 3 M nitric acid. The eluted 86Y solution was heated to dryness, and the resultant 86Y residue was extracted with 3 × 200 μL of 2 M nitric acid and loaded onto a preconditioned yttrium-selective RE-Spec resin column (EiChrom Industries). The 86Y was eluted with 2 M nitric acid. The eluate containing 86Y was heated to dryness, and the 86Y residue was dissolved in 0.1 M nitric acid for radiolabeling procedures.
Radiolabeling CHX-A″-DTPA-Panitumumab with 86Y
The bifunctional chelate, CHX-A″-DTPA, was conjugated to panitumumab as previously described (25). The chelate-to-protein ratio was determined using the Y(III)–Arsenazo(III) complex assay (25,31). For radiolabeling, a freshly prepared solution of ascorbic acid (50 μL, 220 μg/μL) was first added to the 86Y solution (140–170 MBq in 0.1 M nitric acid, 500 μL) to prevent radiolysis of the mAb. The solution was neutralized to pH 5–6 by the addition of ammonium acetate buffer (50 μL 5 M, pH 7.0). CHX-A″-DTPA-panitumumab (50 μg in 0.15 M ammonium acetate) was added to the mixture, stirred briefly in a vortex mixer, and incubated at room temperature for 30 min. The reaction was quenched by the addition of ethylenediaminetetraacetic acid solution (4 μL, 0.1 M). The radiolabeled product was purified using a PD-10 desalting column (GE Healthcare). Size-exclusion high-performance liquid chromatography using a TSK-3000 column (Toso-Haas) was performed to ascertain the purity of the radioimmunoconjugate using previously described solvent conditions and analysis parameters (27).
Cell Lines
HER1-expressing human LS-174T, PC-3, and A431 carcinoma cells (American Type Culture Collection) were grown as a monolayer at 37°C, in a humidified atmosphere of 5% CO2 and 95% air. LS-174T and A431 cells were cultured in Dulbecco's minimal essential medium containing 10% FetaPLEX (Gemini Bio-Products) and 10 mM glutamine solution. PC-3 cells were cultured in RPMI 1640 medium containing FetaPLEX (10%). Medium and supplements were obtained from Quality Biologicals, Invitrogen, and Lonza.
In Vitro Evaluations
In radioligand cell-binding studies, the immunoreactivity of the 86Y-CHX-A″-DTPA-panitumumab was determined using a HER1-positive human A431 fixed-cell radioimmunoassay (25).
Animal and Tumor Models
Female athymic nu/nu mice (Charles River Laboratory) were injected subcutaneously with 2 × 106 cells of each cell line (200 μL of medium containing 20% Matrigel [BD Biosciences]). In vivo experiments were performed when the tumor diameter reached 0.5–0.7 cm. Tumor necrosis was examined by hematoxylin and eosin staining, and HER1 expression was confirmed by immunohistochemistry on selected formalin-fixed, paraffin-embedded tumors. Tumors were carefully removed and fixed in 4% buffered paraformaldehyde solution and incubated overnight at 4°C. After the incubation period, the fixed tumor was washed with phosphate-buffered saline solution and stored in 70% ethanol solution at 4°C before paraffin-embedding. For immunohistochemistry experiments, panitumumab was used as the primary antibody to determine the available binding sites on the tumor sections. Immunohistochemistry was performed and analyzed at Histoserv, Inc. All animal studies were performed in accordance with the National Institutes of Health guidelines for the humane use of animals, and all procedures were reviewed and approved by the National Cancer Institute Animal Care and Use Committee.
In Vivo Evaluations
Biodistribution and Pharmacokinetic Studies
Female athymic mice bearing human LS-174T, PC-3, or A431 tumor xenografts were injected intravenously via the tail with 0.4–0.6 MBq (less than 5 μg) of 86Y-CHX-A″-DTPA-panitumumab. To determine HER1 specificity, panitumumab (0.1 mg) was coinjected with the radiotracer in an additional set of mice bearing each of the tumor xenografts. At the desired times, the animals were sacrificed by CO2 inhalation. Tumor, blood, and selected normal organs were harvested and wet-weighed, and the radioactivity was measured in a Wizard 1480 γ-counter (PerkinElmer). The percentage injected dose per gram (%ID/g) of tissue was calculated by comparison with standards representing 10% of the injected dose per animal.
Blood pharmacokinetics of the 86Y-CHX-A″-DTPA-panitumumab was determined as previously described (27). The percentage injected dose per milliliter of blood was calculated for each of the samples and clearance determined by biphasic nonlinear regression analysis using GraphPad Prism (version 5; GraphPad Software). Noncompartmental pharmacokinetics were determined for AUC, AUMC, and MRT using trapezoidal integration analysis (32).
PET Studies
Small-animal PET studies were performed using the Advanced Technology Laboratory Animal Scanner at the National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health. Whole-body imaging studies (6 bed positions, total acquisition time of 1 h per mouse) were performed on mice anesthetized with 1.5%−1.7% isoflurane on a temperature-controlled bed. Female athymic mice bearing LS-174T, A431, and PC-3 tumor xenografts (n = 3) were injected intravenously with 1.8–2.0 MBq of 86Y-CHX-A″-DTPA-panitumumab. To determine HER1 specificity, excess panitumumab (0.1 mg) was coinjected with the radiotracer. 86Y cylinder phantoms were imaged during each imaging session for normalization and quantitative analysis. The energy window for PET acquisition of 86Y was set between 400 and 700 keV. The imaging data were reconstructed using a 2-dimensional Fourier rebinned ordered-subsets expectation maximization method with scatter correction (linear background subtraction). Additional dead time, decay, and background corrections were applied for quantitative analysis. The reconstructed images were processed and analyzed using the AMIDE: A Medical Image Data Examiner software program. To minimize spillover effects, regions of interest were drawn to enclose approximately 80%−90% of the organ of interest to avoid the edges. To minimize partial-volume effects caused by nonuniform distribution of the radioactivity in the containing volume, smaller regions of interest were consistently drawn to enclose the organ. After the imaging studies, the mice were euthanized and in vivo biodistribution studies were performed to determine the correlation between %ID/cm3 as assessed by PET and %ID/g obtained from biodistribution studies.
Statistical Analysis
All numeric data were expressed as the mean of the values ± SEM. GraphPad Prism, version 5, was used for statistical analysis. A P value of less than 0.05 was considered statistically significant.
RESULTS
Radiochemistry and In Vitro Evaluations
Panitumumab was modified with the acyclic ligand CHX-A″-DTPA at a 10:1 molar excess of chelate to protein, yielding a final chelate-to-protein ratio of 1.6 chelate molecules per protein molecule. The 86Y-CHX-A″-DTPA-panitumumab conjugate was successfully prepared, with the radiochemical yields ranging from 60% to 75% and specific activity exceeding 2 GBq/mg. The 86Y-CHX-A″-DTPA-panitumumab conjugate demonstrated excellent in vitro receptor specificity, as exhibited by an immunoreactivity (%) of 73.51 ± 4.76 and nonspecific binding (%) of 3.79 ± 1.69 (n = 4) on fixed A431 cells. On high-performance liquid chromatography analysis, the radioimmunoconjugate exhibited excellent stability after storage in the refrigerator at 4°C for 1 d and retained the immunoreactivity (Supplemental Fig. 1; supplemental materials are available online only at http://jnm.snmjournals.org).
In Vivo Evaluations
Biodistribution Studies
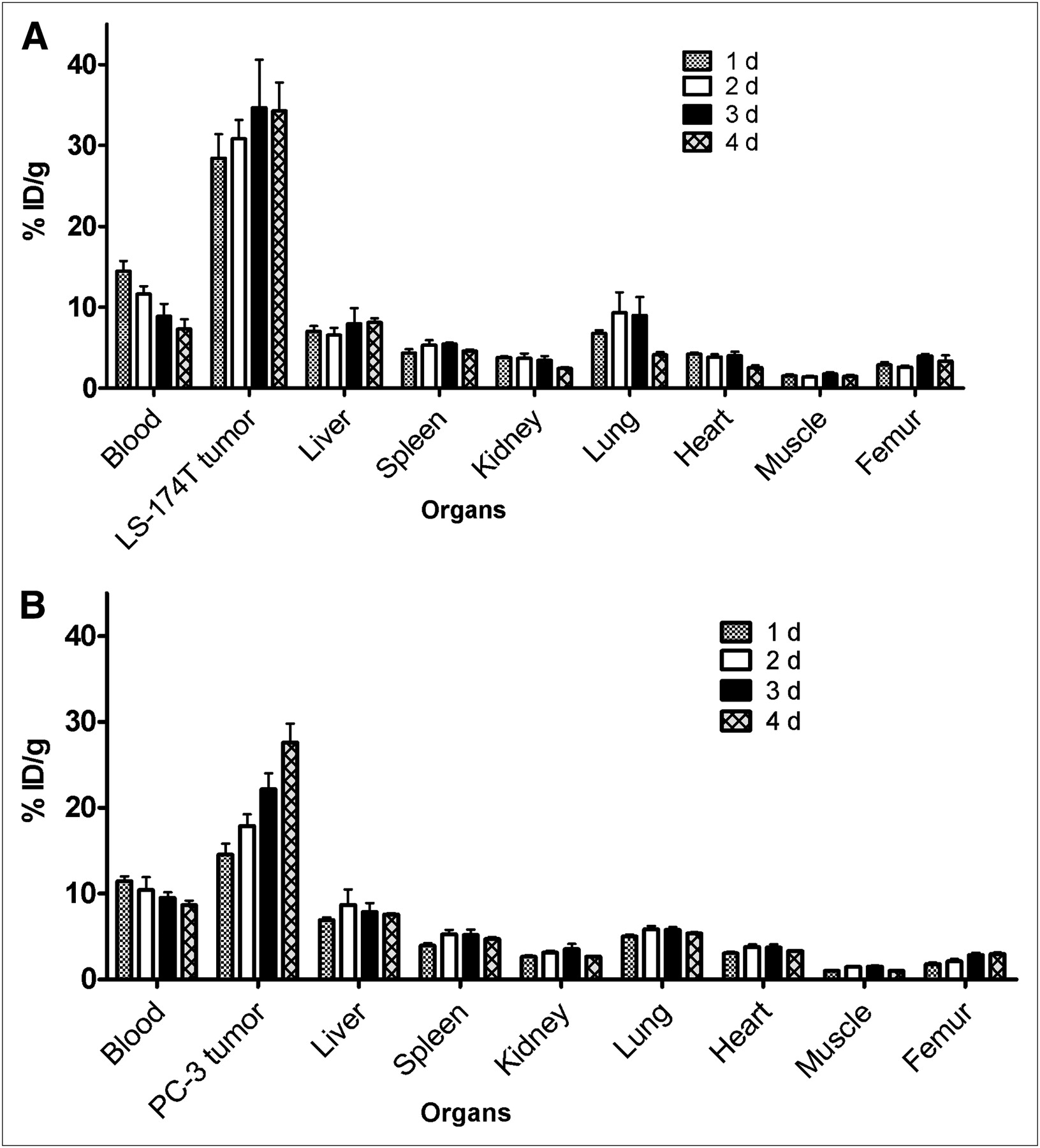
In mice bearing LS-174T tumor xenografts, an approximately 50% decrease in the blood-pool activity was observed over 4 d (14.47 ± 1.28 %ID/g at 1 d to 7.30 ± 1.21 %ID/g at 4 d) (Fig. 1A). An opposite trend was observed in tumor uptake, with the %ID/g of 28.43 ± 2.93 observed at 1 d increasing to 34.30 ± 3.47 %ID/g at 4 d after injection (Fig. 1A). The tumor-to-blood ratio increased more than 2-fold from 2.0 at 1 d to 4.7 at 4 d after injection. In mice bearing PC-3 xenografts, the blood-pool activity of the radiotracer decreased from 11.45 ± 0.56 %ID/g at 1 d to 8.66 ± 0.52 %ID/g at 4 d after injection (Fig. 1B). In contrast, an approximately 50% increase in the tumor uptake was observed from a 1- to 4-d period (14.53 ± 1.29 %ID/g at 1 d to 27.61 ± 2.81 %ID/g at 4 d after injection). The tumor-to-blood ratios in mice bearing PC-3 xenografts were relatively lower than those observed in mice bearing LS-174T xenografts (1.27 at 1 d after injection to 3.18 at 4 d after injection) because of the slower localization of the radiotracer in PC-3 xenografts than in the LS-174T xenografts.
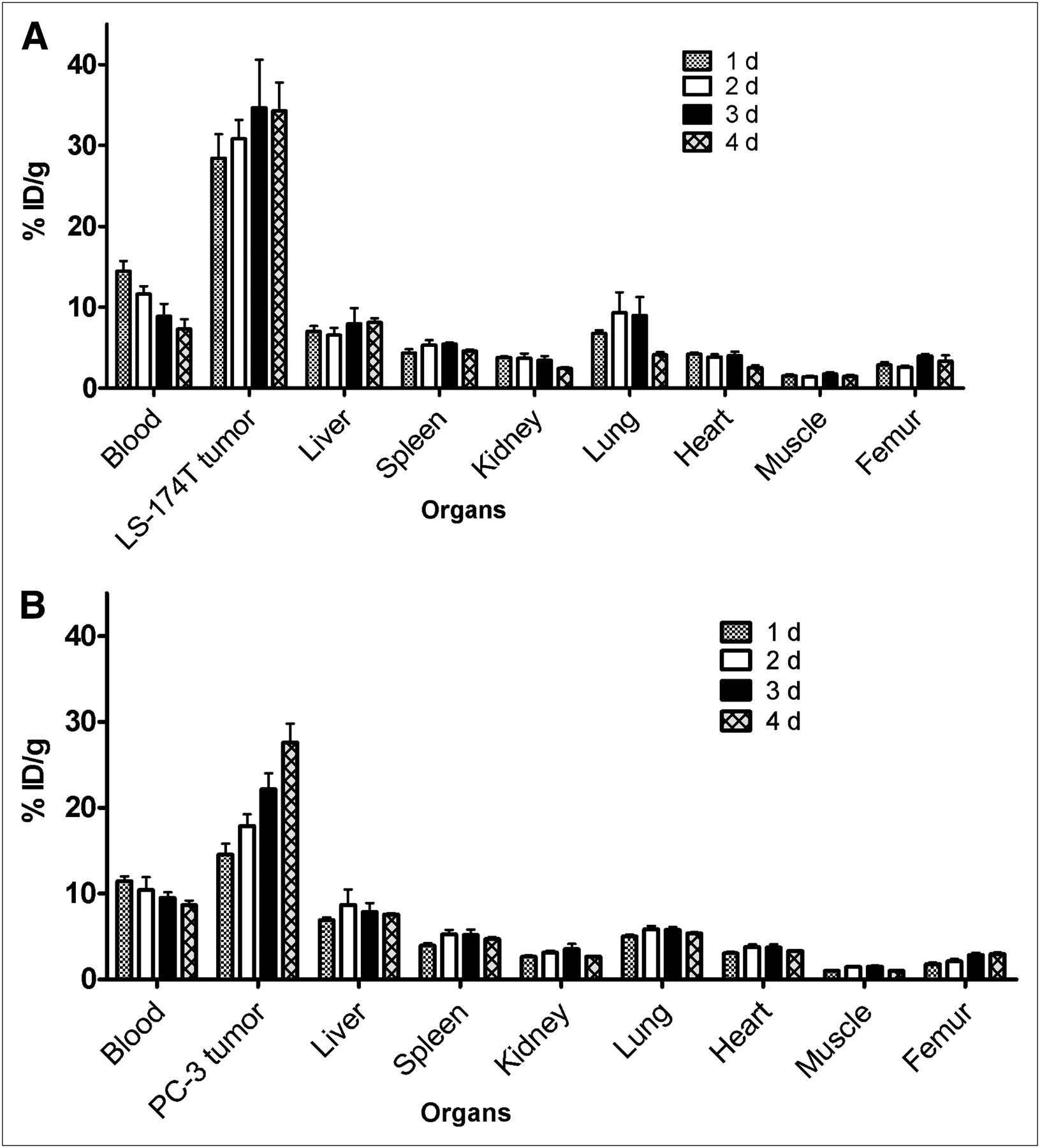
Biodistribution of 86Y-CHX-A″-DTPA-panitumumab in selected organs of female athymic (NCr) nu/nu mice bearing human LS-174T (A) and PC-3 (B) tumor xenografts. Biodistribution data were obtained at 1, 2, 3, and 4 d after intravenous injection of 86Y-CHX-A″-DTPA-panitumumab. All values are expressed as %ID/g. Data represent mean value ± SEM from at least 4 determinations.
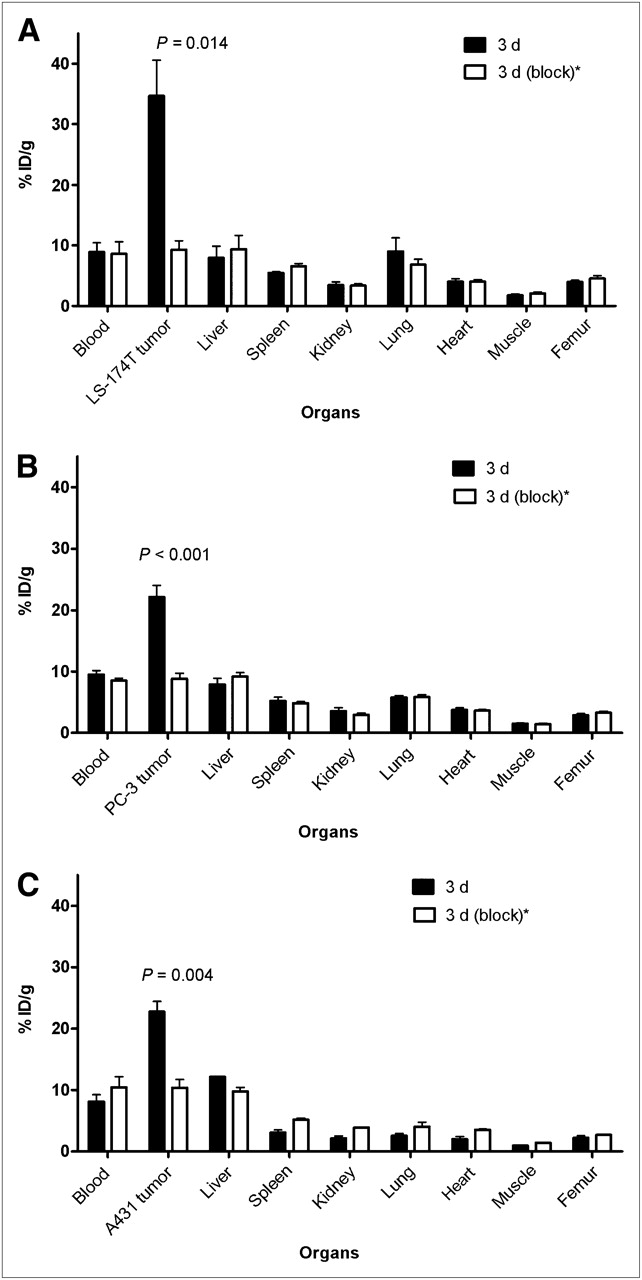
The 86Y-CHX-A″-DTPA-panitumumab uptake in all the tumor models was HER1-mediated, as demonstrated by the receptor-blocking experiments performed by coinjecting 0.1 mg of panitumumab (Fig. 2). In mice bearing LS-174T (Fig. 2A), PC-3 (Fig. 2B), or A431 (Fig. 2C) tumors, the tumor %ID/g at 3 d was 34.65 ± 5.9, 22.1 ± 1.9, and 22.74 ± 1.7, respectively. The corresponding tumor %ID/g in mice coinjected with 0.1 mg of panitumumab was 9.28 ± 1.5, 8.80 ± 0.9, and 10.04 ± 1.3, respectively, at the same time, thus demonstrating the specificity of the radioimmunoconjugate. Immunohistochemistry revealed varied levels of HER1 expression in all tumors. A431 tumors had the highest expression levels of HER1 (Table 1). LS-174T tumor sections had the weakest HER1 staining patterns (+), A431 tumor sections had the strongest HER1 staining patterns (+++), and PC-3 tumor sections demonstrated either a weak (+) or a moderate (++) HER1 staining pattern.
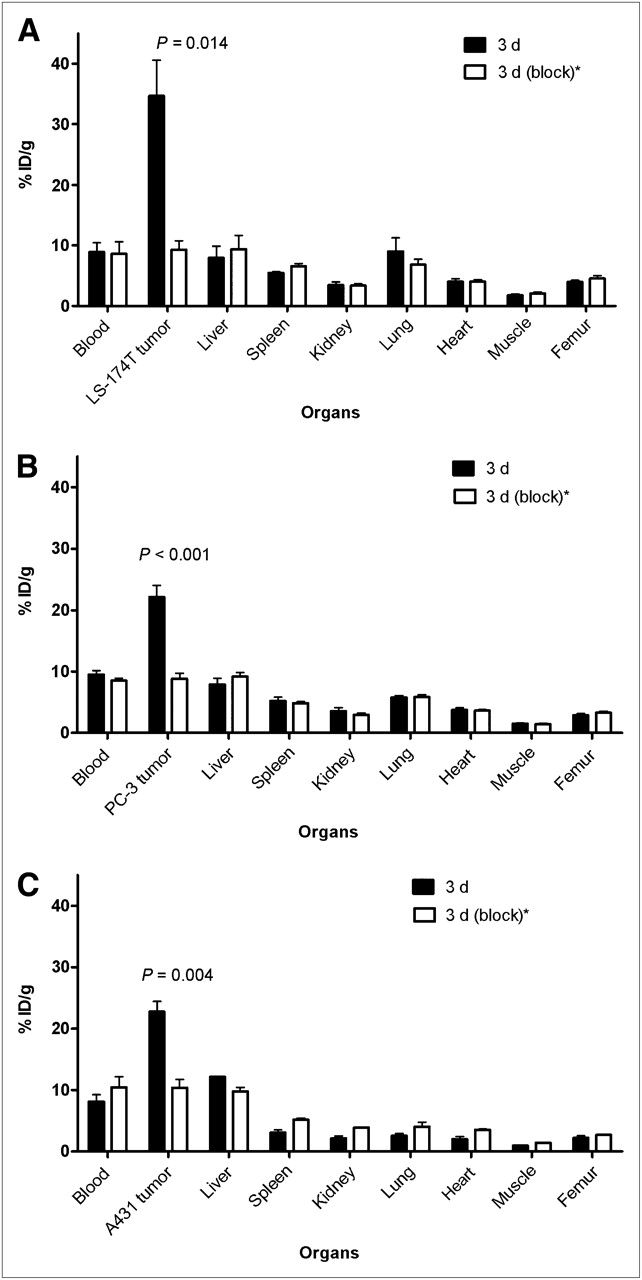
Receptor-meditated uptake of 86Y-CHX-A″-DTPA-panitumumab in selected organs of female athymic (NCr) nu/nu mice bearing human LS-174T (A), PC-3 (B), and A431 (C) tumor xenografts. Biodistribution data were obtained at 3 d after injection. All values are expressed as %ID/g. Data represent mean value ± SEM from at least 3 determinations. *Receptor blocking studies were performed by coinjecting 0.1 mg of panitumumab with radiotracer.
Pharmacokinetic Characteristics of 86Y-CHX-A″-DTPA-Panitumumab in Female Athymic (NCr) nu/nu Mice
Pharmacokinetic Analysis
From the blood clearance studies, the half-life of the α-phase of the biphasic blood clearance ranged from 2.7 ± 1.2 h for mice bearing PC-3 xenografts to 3.7 ± 1.7 h for mice bearing LS-174T xenografts (Table 1). The half-life of the β-phase was identical for all 3 tumor models. The mice bearing LS-174T tumors had the highest AUC (96.8 ± 5.6 %ID·d·g−1) and AUMC (262.5 ± 14.9 %ID·d2·g−1). The tracer accumulation was significantly higher in LS-174T tumors (P < 0.05) than in A431 and PC-3 tumors, as shown in Table 1. However, the tumor MRTs were identical for all 3 tumors (2.7–2.8 d). The LS-174T tumor AUC[0→4 d]:blood AUC[0→4 d] ratio of 3:1 was nearly 1.5 times greater than the PC-3 and A431 tumor AUC[0→4 d]:blood AUC[0→4 d] ratio of 2.0 (Table 1).
PET Studies
The linearity of the PET-assessed concentration versus the radioactivity concentration measured in a Capintec CRC-127R dose calibrator was r2 = 0.99 in the radioactivity range of 0.03–3.63 MBq/mL of 86Y solution from cylindric phantom studies.
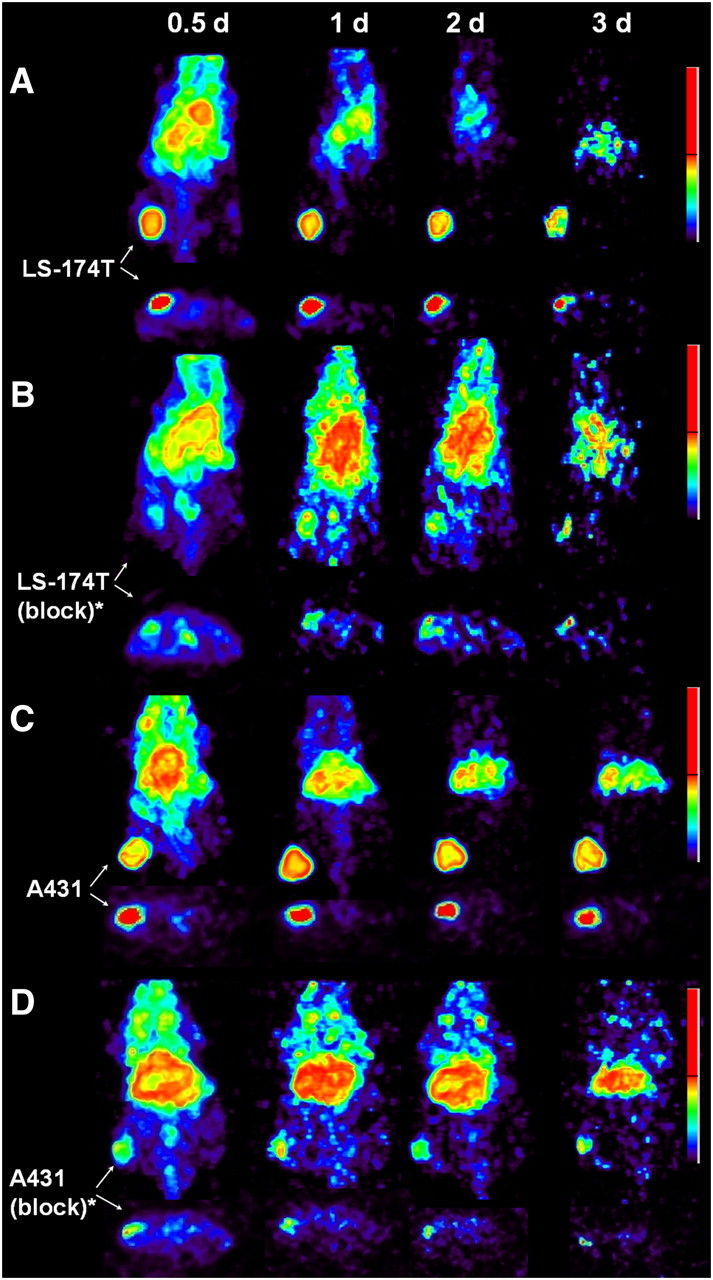
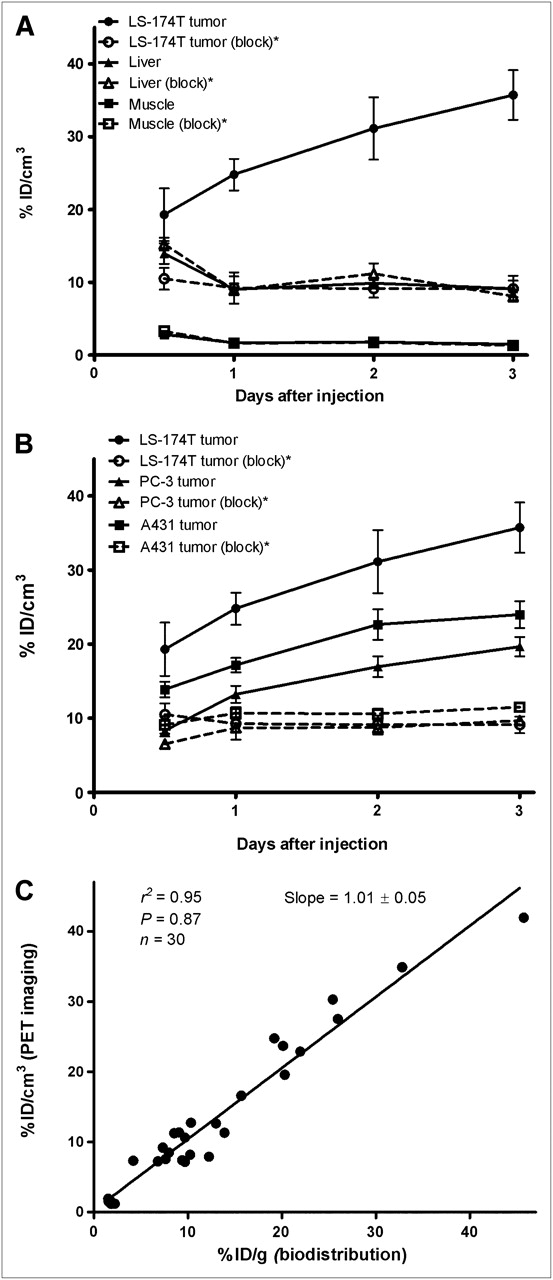
Small-animal PET studies were performed in female athymic mice bearing LS-174T (Figs. 3A−3B), A431 (Figs. 3C−3D), and PC-3 (Supplemental Fig. 2) xenografts injected with 1.8–2.0 MBq of 86Y-CHX-A″-DTPA-panitumumab or 86Y-CHX-A″-DTPA-panitumumab coinjected with 0.1 mg of panitumumab. The LS-174T (Fig. 3A) and A431 (Fig. 3C) tumors were clearly visualized in maximum-intensity projections (top) and transverse slices (bottom) of mice imaged from 0.5 to 3 d after injection of the radioimmunoconjugate. The tumor-to-background ratios improved over the period, mostly because of the decrease of radioactivity in blood, liver, and background, whereas the tumor uptake increased. In contrast, when 0.1 mg of panitumumab was coinjected with the radiotracer, the tumors were poorly visualized because of receptor-specific blockage, demonstrating the HER1-specificity of 86Y-CHX-A″-DTPA-panitumumab (Figs. 3B and 3D). No significant differences were found in the liver and muscle uptake of mice injected with 86Y-CHX-A″-DTPA-panitumumab and mice coinjected with 0.1 mg of cold panitumumab (Fig. 4A). As shown in Figure 4B, the quantitated tumor uptake of mice injected with 86Y-CHX-A″-DTPA-panitumumab and mice injected with 86Y-CHX-A″-DTPA-panitumumab plus 0.1 mg of cold panitumumab was significantly different at 1, 2, and 3 d after injection. However, the tumor uptake was not significantly different at 0.5 d after injection (P = 0.08 for LS-174T, P = 0.10 for A431, and P = 0.09 for PC-3 tumors). For mice bearing LS-174T tumors, the PET-assessed tumor AUC[0→3 d] of mice injected with 86Y-CHX-A″-DTPA-panitumumab was 3.1 times greater than that of mice coinjected with 0.1 mg of panitumumab (Table 1). The PET-assessed tumor AUC[0→3 d] of mice bearing PC-3 and A431 tumors injected with 86Y-CHX-A″-DTPA-panitumumab was 1.6 and 1.9 times, respectively, greater than that of groups coinjected with 0.1 mg of panitumumab. In fact, a statistically significant difference was observed in tumor-bearing mice injected with 86Y-CHX-A″-DTPA-panitumumab alone and 86Y-CHX-A″-DTPA-panitumumab coinjected with 0.1 mg of panitumumab. The liver, tumor, and muscle uptake quantified by PET at all times was closely related (r2 = 0.95, P = 0.87, n = 30) to values determined by in vivo biodistribution studies (Fig. 4).
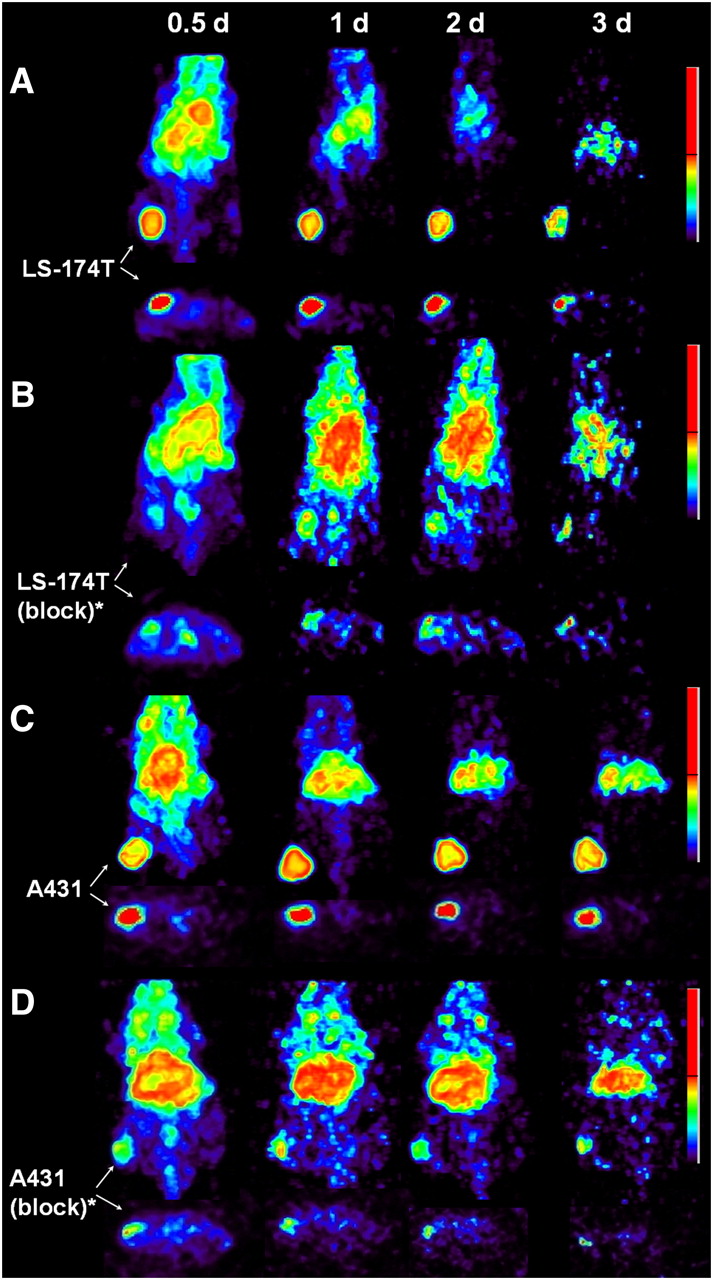
Representative reconstructed and processed maximum-intensity projections (top) and transverse slices (bottom) of female athymic (NCr) nu/nu mouse bearing human LS-174T (A and B) and A431 (C and D) tumor xenografts. Mice in A and C were injected intravenously via tail vein with 1.8–2.0 MBq of 86Y-CHX-A″-DTPA-panitumumab, and mice in B and D were coinjected intravenously via tail vein with 1.8–2.0 MBq of 86Y-CHX-A″-DTPA-panitumumab and 0.1 mg of panitumumab for blocking HER1. Tumors are indicated with white arrows. Scale represents percentage of maximum and minimum threshold intensity. *Receptor blocking studies were performed by coinjecting 0.1 mg of panitumumab with radiotracer.
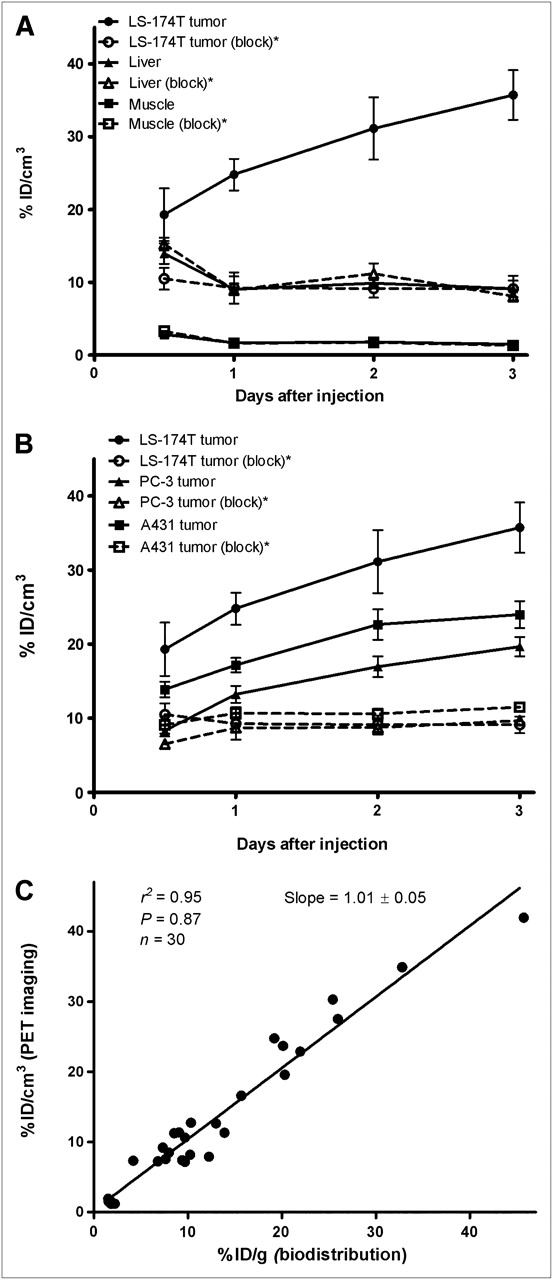
(A) Time–activity curve and uptake values of 86Y-CHX-A″-DTPA-panitumumab in selected organs of female athymic (NCr) nu/nu mice bearing human LS-174T xenografts assessed through quantitative small-animal PET. (B) Comparative time–activity curves of 86Y-CHX-A″-DTPA-panitumumab in female athymic (NCr) nu/nu mice bearing LS-174T, A431, and PC-3 tumor xenografts. (C) Correlation between organ %ID/g values assessed through in vivo biodistribution studies and quantitative small-animal PET. All uptake values derived from PET studies are expressed as %ID/cm3. Data represent mean value ± SEM from 3 determinations. *Receptor blocking studies performed by coinjecting 0.1 mg of panitumumab with radiotracer.
DISCUSSION
Advances in genomics and proteomics are revolutionizing cancer therapy. Significant progress has been made in the development of targeted cancer therapy, wherein the drug specifically targets a unique protein or gene product overexpressed in tumors. Traditionally, in vitro assays of tumor biopsy material are used to evaluate the expression of the tumor biomarker that is vital for selecting patients for targeted therapy. Complementary to biopsy assays, molecular imaging has been used to measure regional tumor target expression and therefore select patients for appropriate cancer therapy and to evaluate treatment response (33,34).
Toward this end, 86Y-CHX-A″-DTPA-panitumumab was explored as a noninvasive molecular imaging tool for selecting patients for HER1-targeted panitumumab therapy and for dosimetry assessment for possible targeted 90Y therapy.
86Y-CHX-A″-DTPA-panitumumab was routinely prepared with a specific activity exceeding 2 GBq/mg (0.3 GBq/nmol) and radiochemical yields exceeding 60%. When compared with 64Cu-DOTA mAb and 89Zr-desferrioxamine-mAb, 86Y-CHX-A″-DTPA-mAb offers a viable alternative because of its greater in vivo stability, greater tumor-to-background ratios, and significant ease of preparation, as demonstrated by this study.
The biodistribution, noncompartmental pharmacokinetics, and imaging data reveal HER1-mediated uptake and accumulation in HER1-expressing tumor xenografts (Figs. 1–4⇑⇑⇑; Table 1). 86Y-CHX-A″-DTPA-panitumumab had a relatively longer half-life and slower blood clearance than did radiolabeled cetuximab (20). The biodistribution and blood pharmacokinetics of 86Y-CHX-A″-DTPA-panitumumab were similar to those of 111In-CHX-A″-DTPA-panitumumab, except for lung and femur uptake. These minor differences in 111In- and 86Y-labeled mAb may be attributed to radiometabolites, as previously observed (27). Data on blood clearance and tumor residence time as obtained in this study should prove useful for dosing in panitumumab-related therapies.
Studies have concluded that 64Cu-DOTA-cetuximab could be used to detect and quantify HER1 expression and therefore monitor therapeutic response (22,23). However, no correlation among relative in vitro expression determined by flow cytometry, relative ex vivo expression determined by immunohistochemistry, and tumor uptake and accumulation (Table 1) was found in the studies presented here. In fact, the cell line demonstrating the lowest HER1 expression (LS-174T) resulted in the highest tumor uptake and accumulation (Table 1; Figs. 1–4⇑⇑⇑). A recent study performed with 89Zr-labeled cetuximab also found no correlation between ex vivo expression of HER1 and radioimmunoconjugate uptake (21). The report describing 64Cu-DOTA-panitumumab also described discrepancies in tumor uptake and ex vivo HER1 expression levels determined by immunohistochemistry (30). This apparent dichotomy can be explained by the fact that in vivo accretion in tumor is actually dependent on many physiologic factors including tumor vasculature, blood flow, tumor interstitial pressure, and antigen shedding. Above all else, there are clearly obvious differences between the in vivo and the in vitro milieu of the surrounding environment ranging from cell-to-cell interactions to growth factors and such (35). Therefore, to correlate HER1 expression and tumor uptake, further studies are warranted. In vivo determination of the distribution of the targeted biomarker and simultaneous determination of its true availability to the drug for therapy provide an extremely important metric that conveys the suitability of the patient for therapy and profoundly personalizes the treatment to the patient. The added advantage is that this information is obtained noninvasively. PET with 86Y-CHX-A″-DTPA-panitumumab may have an extremely useful role in the selection of patients for panitumumab-related therapy because it would indicate HER1 accessibility to the antibody. However, it is also possible that 86Y-CHX-A″-DTPA-panitumumab imaging by itself may not predict the response to therapy but it indicates only how much panitumumab reaches the tumor. 86Y-CHX-A″-DTPA-panitumumab imaging does not reveal the status of v-kis-ras2 Kirsten rat sarcoma viral oncogene mutations, which is critical for response to HER1 immunotherapy (36–38). Thus, the role of panitumumab imaging may be complementary and best used together with assays to determine v-kis-ras2 Kirsten rat sarcoma viral oncogene mutations and HER1 gene amplification and polymorphism (36–38).
The available radionuclides for PET radioimmunoimaging are 124I, 64Cu, 89Zr, and 86Y. Each of these radionuclides has its own specific drawback (26). Panitumumab is rapidly internalized; therefore, we anticipate that 124I will be dehalogenated rapidly in vivo and result in poor tumor-to-background ratio. 64Cu-1,4,8,11-tetraazacyclotetradecane-N,N′,N″,N′″-tetraacetic acid-1A3 has previously been reported for clinical PET of metastatic colorectal cancer (39,40). Although all 17 primary and recurrent sites were clearly visualized in patients, only 23 of 39 metastatic sites (59%) were detected (40). The detection of lung and liver metastasis was seriously hindered by nonspecific uptake in the liver and the blood because of dissociation of the 64Cu from the currently used chelates for radiolabeling mAbs. 89Zr is an attractive positron emitter because of its longer half-life; however, preparation of 89Zr-labeled mAbs is a multistep, tedious process and 89Zr has been shown to dissociate from the currently used chelates and to localize in the bone thereafter (24). On the other hand, the chelation chemistry and the preparation of yttrium-labeled mAbs for clinical use are well established. Although the half-life of 86Y is slightly longer than that of 64Cu, the abundance of positrons is also almost twice that of 64Cu. With these advantages over 64Cu, we anticipate much lower amounts of 86Y will be required for quantitative immunoPET at 2 d after injection. On the basis of those previous studies performed with 64Cu-labeled mAb (39,40), we anticipate that injection of between 0.18 and 0.37 GBq of the radioimmunoconjugate will result in useful quantitative images up to 2–3 d after injection.
We are currently performing radioimmunotherapy of HER1-expressing solid tumors with 90Y-CHX-A″-DTPA-panitumumab. Therefore, 86Y-CHX-A″-DTPA-panitumumab serves as a surrogate PET marker for dosimetry and selection of subjects for 90Y CHX-A″-DTPA-panitumumab radioimmunotherapy of HER1-expressing carcinoma. To achieve the long-term goal of clinical translation of 86Y-CHX-A″-DTPA-panitumumab, PET/CT and MRI studies are currently being performed with mice bearing orthotopic and disseminated tumors.
CONCLUSION
86Y-CHX-A″-DTPA-panitumumab has been prepared with high specific activity. The utility of the radioimmunoconjugate for noninvasive PET of HER1-expressing tumors in preclinical models has been demonstrated. 86Y-CHX-A″-DTPA-panitumumab as a radiotracer may be used for the assessment of panitumumab uptake, which may be important for risk stratification, patient screening, and appropriate dosage selection. This preclinical study elucidating the biologic and pharmacokinetic characteristics of 86Y-CHX-A″-DTPA-panitumumab represents the first step toward the clinical translation.
Acknowledgments
We thank Jurgen Seidel (National Cancer Institute, National Institutes of Health) for technical input on the operations of the NIH ATLAS small-animal PET scanner. This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and the U.S. Department of Health and Human Services.
Footnotes
-
COPYRIGHT © 2010 by the Society of Nuclear Medicine, Inc.
References
- Received for publication October 2, 2009.
- Accepted for publication February 17, 2010.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Antibody Positron Emission Tomography Imaging in Anticancer Drug Development
- Glypican-3-Targeted 89Zr PET Imaging of Hepatocellular Carcinoma
- Development of a Carbon-14 Labeling Approach to Support Disposition Studies with a Pegylated Biologic
- PET and MRI of Metastatic Peritoneal and Pulmonary Colorectal Cancer in Mice with Human Epidermal Growth Factor Receptor 1-Targeted 89Zr-Labeled Panitumumab