


Abstract
The organic anion 99mTc-N-[2-[(3-bromo-2,4,6-trimethylphenyl)-amino]-2-oxoethyl]-N-(carboxymethyl)-glycine (99mTc-mebrofenin) and its analogs are widely used for hepatobiliary imaging. Identification of the mechanisms directing bile canalicular transport of these agents will provide insights into the basis of their hepatic handling for assessing perturbations. Methods: We performed studies in animals, including healthy Fischer 344 rats or rats treated with carbon tetrachloride or intrasplenic cell transplantation and healthy Wistar rats or HsdAMC:TR-Abcc2 mutant rats in Wistar background. Onset of hepatic inflammation was verified by analysis of carbon uptake in Kupffer cells. Hepatic clearance of 99mTc-mebrofenin was studied with dynamic imaging, and fractional retention of peak hepatic mebrofenin activity after 60 min was determined. Changes in the expression of bile canalicular transporters were analyzed by real-time polymerase chain reaction and Western blots. Results: Carbon tetrachloride and cell transplantation produced hepatic inflammation with activation of Kupffer cells, resulting in a rapid decline in the expression of the bile canalicular transporters Abcb4, Abcb11, and Abcc2. Among these transporters, decreased expression of Abcc2 was most prominent, and this decline persisted for 4 wk. Next, we examined 99mTc-mebrofenin excretion in HsdAMC:TR-Abcc2 mutant rats (in which Abcc2 expression is naturally inactivated), compared with their healthy counterparts. In healthy HsdRccHan:WIST rats, only 23% ± 3% of the peak 99mTc-mebrofenin activity was retained after 60 min. By contrast, in HsdAMC:TR-Abcc2 mutant rats, 73% ± 5% of the peak 99mTc-mebrofenin activity was retained (P < 0.001). Moreover, the administration of cyclosporin A markedly inhibited 99mTc-mebrofenin excretion in healthy rats, with no further effect on already impaired 99mTc-mebrofenin excretion in HsdAMC:TR-Abcc2 mutant rats. Hepatic excretion of 99mTc-mebrofenin was largely dependent on Abcc2. This molecular basis of 99mTc-mebrofenin excretion will advance studies of pathophysiologic mechanisms in hepatic Abcc2 pathways.
The imaging agent N-[2-[(3-bromo-2,4,6-trimethylphenyl)-amino]-2-oxoethyl]-N-(carboxymethyl)-glycine conjugated to technetium (99mTc-mebrofenin) and related compounds are extensively used for diagnosing cholestasis, gallbladder function, and bile leaks (1). The avidity of these compounds for hepatocytes (>80% of intravenously administered 99mTc-mebrofenin is cleared from blood within 10 min, with incorporation essentially in the liver, followed by rapid excretion in bile) has been highly attractive for imaging applications (2). The mechanism by which 99mTc-mebrofenin enters hepatocytes is unknown, although as an organic anion, basolateral transporters (e.g., members of the solute carrier family [SLC]—SLCO1B1, SLCO1B3, and possibly others) may be involved (3,4). Subsequently, 99mTc-mebrofenin exits hepatocytes without metabolism or modifications to enter the bile canaliculus; however, the transport mechanism by which this exit is accomplished has been unknown. Many bile canalicular transporters have been identified to date, many belonging to adenosine triphosphate (ATP)-binding cassette (ABC) subfamilies, and their properties have been extensively studied because these transporters serve major roles in health and disease, including the characterization of the genetics, molecular and biochemical activities, and phenotypic effects of specific mutations in several instances (5–7).
Recently, we found that inflammatory cytokines—tumor necrosis factor (TNF)-α and interleukin-6—blocked hepatobiliary transport of 99mTc-mebrofenin (8,9). In this way, hepatic excretion of 99mTc-mebrofenin offered an intracellular reporter pathway for assaying activation of inflammatory cells, induced experimentally either by the administration of the hepatotoxin carbon tetrachloride (CCl4) or by the transplantation of cells in the liver. Rapid and persistent inhibition of 99mTc-mebrofenin excretion, becoming abnormal immediately after the onset of inflammation and remaining abnormal during persistent inflammation under these conditions, indicated that the identification of specific cellular mechanisms responsible for transporting 99mTc-mebrofenin into bile will be significant for various applications. Here, we report studies that led us to the ABC subfamily C member 2 (Abcc2)—which mediates the secretion of amphiphilic glutathione, glucuronide, and sulfate conjugates into bile (5,10)—as the major relevant bile canalicular transporter of 99mTc-mebrofenin. We performed these studies in well-defined rat models, syngeneic animals deficient in dipeptidyl peptidase IV (DPPIV) enzyme activity to serve as recipients for localizing healthy DPPIV-positive transplanted cells (9), animals with CCl4-induced liver injury (8), and animals mutant in the Abcc2 gene (11), which is also known as multidrug resistance protein-2.
MATERIALS AND METHODS
Animals and Surgical Procedures
A total of 40 DPPIV-negative (DPPIV−) Fischer 344 (F344) rats were studied. These animals were 6–8 wk old, weighed 120–180 g, and were provided by the Special Animal Core of Marion Bessin Liver Research Center. Healthy F344 rats were from the National Cancer Institute. HsdAmc:TR-Abcc2 mutant rats in Wistar background and their healthy counterparts, HsdRccHan: WIST rats, were from Harlan. Animal Care and Use Committees for Albert Einstein College of Medicine and Long Island Jewish Medical Center approved the experimental protocols.
For surgery, rats were anesthetized with ketamine and xylazine (Fort Dodge Animal Health). Hepatocytes were isolated from F344 rats by standard 2-step collagenase perfusion of the liver. Cell viability was examined using trypan blue dye exclusion. In DPPIV− rats, 2 × 107 hepatocytes were injected over 9–12 s via the splenic pulp (9,12). To induce chemical liver injury, CCl4 was administered intramuscularly to DPPIV− rats. Animals were sacrificed at 6 h, 1 d, or 3 d or at 1, 2, 3, and 4 wk after cell transplantation or CCl4 treatment (n = 3 each). Carbon was administered 60 min before sacrifice to demonstrate Kupffer cell activation, as described previously (12). A 10 or 15 mg/kg dose of cyclosporin A was given by gavage 2–3 h before 99mTc-mebrofenin studies to some HsdAmc:TR-Abcc2 and F344 rats (n = 6 each) (13).
Drugs and Chemicals
CCl4, cyclosporin A, mineral oil, and chemicals were purchased commercially (Sigma Chemical Co.). CCl4 was suspended in mineral oil (1:1, v/v), and cyclosporin A was solubilized in ethanol. A kit was used for preparing 99mTc-mebrofenin (Choletec; Bracco Diagnostics).
99mTc-Mebrofenin Imaging
Commercially available mebrofenin was mixed with 99mTc-sodium pertechnetate (185–222 MBq) in 3 mL of normal saline. This solution was diluted in normal saline and then mixed with 99mTc-mebrofenin (7.4 MBq), and 1 mL was injected into the splenic pulp. Beginning immediately after the injection of 99mTc-mebrofenin, 10-s dorsal images were acquired for 60 min with a γ-camera (Argus; ADAC Laboratories) (8). Data were analyzed on a commercially available nuclear medicine workstation by drawing regions of interest over the liver and generating time–activity curves. The time to maximal accumulation of 99mTc-mebrofenin, Tpeak, and percentage of Tpeak 99mTc-mebrofenin activity remaining at 60 min after injection were determined.
Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated with TRIzol (Invitrogen Corp.) and treated with RNase-free DNase (Qiagen Inc.), and cDNA was prepared from 2–4 μg of RNA with an Omniscript RT Kit (Qiagen). TNF-α, macrophage inflammatory protein-2, monocyte chemotactic protein-1, and interferon-inducible protein-10 were analyzed by semiquantitative RT-PCR, as described previously (14). Real-time quantitative PCR (qPCR) was performed with commercially available primers for rat TNF-α and β-actin (MGC124630, reference sequence NM012675; and PRR06570B, reference sequence NM031144, respectively; SA Biosciences). After DNase treatment, 1 μg of total RNA was converted to cDNA with a RT2 PCR Array First Strand Kit (SA Biosciences). Real-time PCR was performed with a RT2 Real-Time SYBR Green PCR Master Mix (SA Biosciences), according to the manufacturer's instructions, in an ABI Prism 7000 Sequence Detection System (Applied Biosystems). PCR products were analyzed in 2% agarose gels with ethidium bromide to verify single products. Fold changes in gene expression were calculated on the basis of PCR cycle threshold (Ct) values between experimental and control samples after normalizing data in each sample with β-actin. Real-time qPCR for biliary transporters used a Brilliant SYBR GreenQPCR kit from Qiagen. Briefly, 5 μg of total RNAs were reverse-transcribed with Superscript II (GIBCO-BRL). Primers were designed to generate 150–base pair amplicons (Table 1). PCR was performed in a PRISM 7900HT system (Applied Biosystems Inc.), with 1 cycle for 15 min at 95°C and 40 cycles at 94°C for 15 s, 55°C for 30 s, and 72°C for 30 s. Product sizes were verified in 2% agarose gels. Data were analyzed by software (SDS, version 2.1; Applied Biosystems Inc.) to obtain Ct values. Ct values for 18S RNA served as a denominator for normalization. Gene expression was expressed as fold change above controls.
qPCR Primers for Bile Canalicular Transporters
Tissue Studies
Liver samples were cooled to −80°C in methylbutane or fixed in 10% buffered formalin. Paraffin-embedded tissue sections were stained with hematoxylin and eosin according to standard methods. To visualize transplanted cells, 5-μm-thick cryosections were prepared and stained histochemically for DPPIV, as described previously (12). Incorporation of carbon in Kupffer cells was also analyzed as described previously (12). The number of Kupffer cells with carbon was counted in 50 consecutive high-power fields per tissue (n = 3–4 rats, each under ×200 magnification with centering on portal areas).
Western Blot Analysis
Tissue samples weighing 100 mg were homogenized in 1 mL of 1 mM sodium bicarbonate with protease inhibitors; the clear supernatant was incubated with 100 mM sodium bicarbonate with protease inhibitors for 15 min, followed by centrifugation under 100,000g for 1 h at 4°C. The pellet was resuspended in water and disrupted by ultrasonication. Bradford reagent was used for measuring protein content, and equal amounts of protein were resolved in 10% sodium dodecyl-polyacrylamide gels to prepare transblots with standard methods. Transblots were probed with antibodies (dilution, 1:1,000) in phosphate-buffered saline against Abcb11 (PC-064, Kamiya Biomedical Co.), Abcc2 (Gene Tex Inc.), and solute carrier (SLC) organic anion transporter family member 1B1 (Slco1b1) (from Allan Wolkoff, Albert Einstein College of Medicine). Antibody binding was demonstrated with 1:5,000 dilutions in phosphate-buffered saline of peroxidase-conjugated antirabbit IgG for Abcb11 and Slco1b1 (NA934W; GE Healthcare U.K. Ltd.) or antimouse IgG for Abcc2 (A3673; Sigma), followed by detection with enzymatic chemiluminescence (NEL104; Perkin Elmer LAS Inc.). Blots were reprobed after regeneration with Restore Western Blot Stripping Buffer (Pierce Biotechnology Inc.).
Blood Tests
Serum was stored at −20°C for total bilirubin, alanine aminotransferase, and alkaline phosphatase measurements with an automated clinical system.
Statistical Analysis
Data are shown as mean ± SEM. Significances were analyzed by SigmaStat 3.0 software (Jandel Scientific) with the t test, Mann–Whitney rank sum test, or ANOVA with the Holm–Sidak method, as appropriate. A P value of less than 0.05 was considered significant.
RESULTS
Evidence for Inflammation After Cell Transplantation and CCl4 Treatment
In DPPIV− rats, transplanted cells were identified in the liver, and hepatic morphology was normal throughout the studies. In animals treated with CCl4, liver necrosis was observed after 1 d, whereas liver morphology was normal at subsequent times. Kupffer cells were activated with carbon incorporation after CCl4 treatment or cell transplantation (compared with controls), increasing after 1 d by 6 ± 1-fold and 3 ± 0.3-fold, respectively, and remaining elevated at lower levels for 2 wk (Fig. 1). This change was more pronounced in CCl4-treated rats. Also, hepatic expression of TNF-α, macrophage inflammatory protein-2, monocyte chemotactic protein-1, and interferon-inducible protein-10 mRNAs increased within 6 h after CCl4 treatment or cell transplantation, as indicated by nonquantitative RT-PCR (not shown). Increased expression of TNF-α messenger RNA as a major mediator of the Kupffer cell response was verified by qPCR. For instance, after cell transplantation (compared with untreated controls) TNF-α expression increased significantly by 22 ± 3-fold, 56 ± 1-fold, 10 ± 1-fold, and 4 ± 0.5-fold at 6 h, 1 d, 3 d, and 2 wk, respectively (P < 0.05, ANOVA with Holm–Sidak test).

Changes in Kupffer cell activation. Shown is analysis of kinetics by which Kupffer cells were activated after CCl4 treatment or cell transplantation. Data indicated that Kupffer cells were activated more after CCl4 treatment than after cell transplantation. Asterisks indicate significant differences between normal controls and activated cells, although activated cells were present for long time after both CCl4 treatment and cell transplantation. Data were from multiple animals per time point (n = 3 each). Ctr = controls.
These changes in Kupffer cell activation and cytokine expression were accompanied by the inhibition of 99mTc-mebrofenin excretion from the liver, such that more than 90% of Tpeak 99mTc-mebrofenin activity was retained 1 d after either CCl4 treatment or cell transplantation, compared with retention of less than 30% of 99mTc-mebrofenin activity in healthy control rats (P < 0.001, t test). Previously, we reported that hepatic excretion of 99mTc-mebrofenin was impaired for 2 wk after the administration of cells or CCl4 to animals only once (8,9).
Expression of Bile Canalicular Transporters Changed After Onset of Inflammation
We considered that hepatic inflammation would alter expression of bile canalicular transporters because downregulation of hepatic transporters had previously been demonstrated in lipopolysaccharide-induced inflammation models of cholestasis in rats and mice (5). If there were a causal relationship between these 2 processes, this change should have occurred early and persisted during the period of abnormal 99mTc-mebrofenin excretion. qPCR assays showed significant changes in expression of Abcb4, Abcb11, and Abcc2 after CCl4 treatment or cell transplantation (Fig. 2). Compared with healthy controls, 6 h after CCl4 treatment expression decreased for Abcb4 (38% ± 0.6%), Abcb11 (17% ± 0.4%), and Abcc2 (18% ± 1%). Decreased gene expression was also observed at 6 h after cell transplantation (Abcb4 [13% ± 4%], Abcb11 [16% ± 3%], and Abcc2 [14% ± 10%]) (P < 0.05, ANOVA with Holm–Sidak test). After 4 wk, expression of Abcb4 and Abcb11 returned to normal or near-normal; Abcc2 expression was still decreased to 44% ± 1% in CCl4-treated animals and 66% ± 15% in cell-transplanted animals (P < 0.05, ANOVA with Holm–Sidak test). No consistent change was observed in Abcg2 gene expression (not shown).
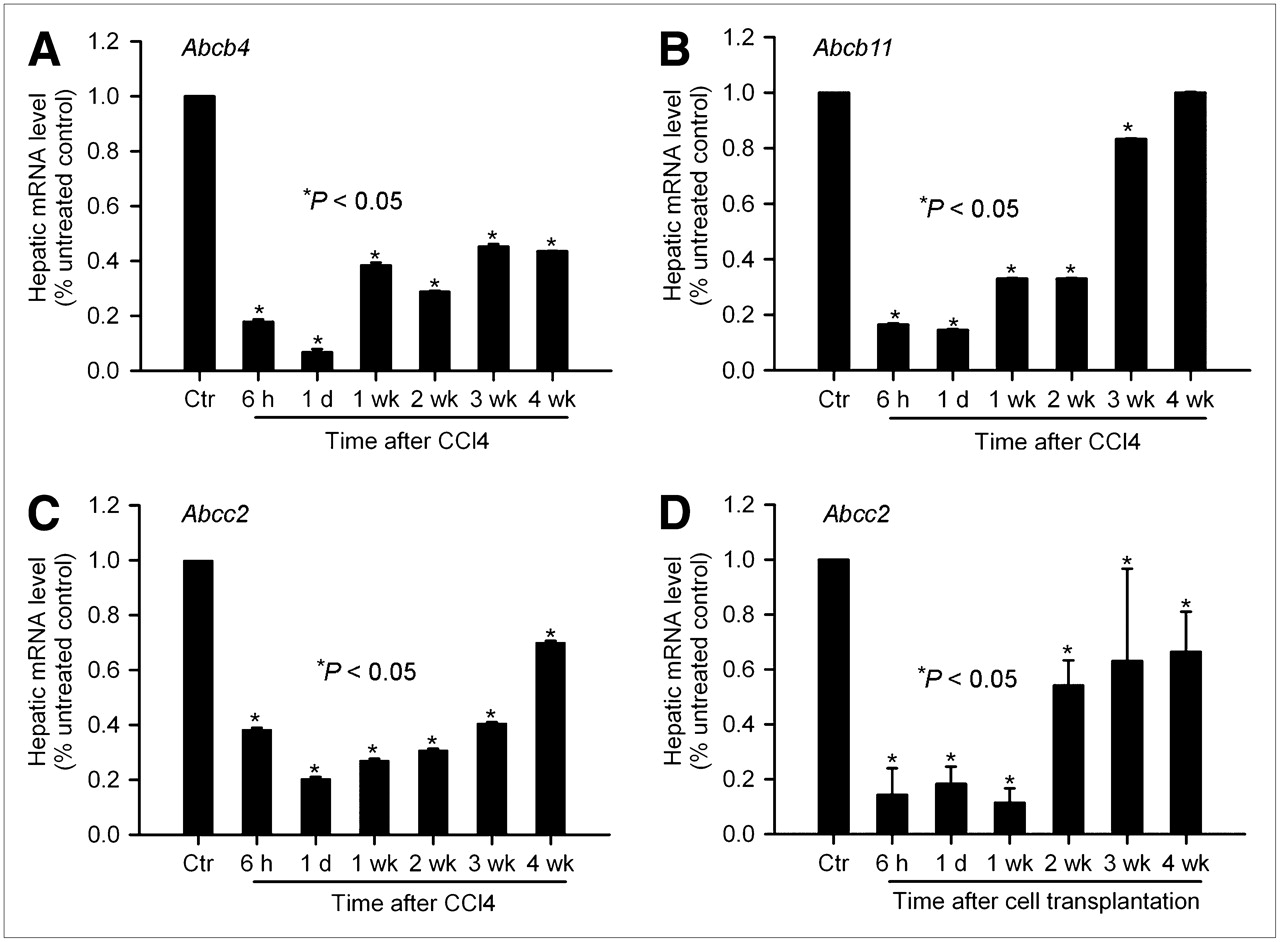
Perturbations in expression of canalicular transporters by hepatic inflammation. qPCR analysis of expression of Abcb4 (A), Abcb11 (B), and Abcc2 (C) genes from control rats and rats treated with CCl4 (A–C) or cell transplantation (D). After CCl4 treatment and cell transplantation, gene expression was promptly and persistently downregulated. Expression of Abcc2 gene was perturbed most profoundly, particularly in animals treated with CCl4. Subsequently, Abcb11 expression showed earliest recovery, and Abcc2 was expressed at subnormal levels through 4-wk period. Ctr = controls.
We verified the findings at the protein level by Western blots and found that Abcb11 protein level decreased shortly after either cell transplantation or CCl4 treatment, although the level returned to normal after 3 wk (Fig. 3). The Abcc2 protein level also decreased promptly after cell transplantation or CCl4 treatment. However, Abcc2 levels had not returned to normal even 4 wk after CCl4 treatment. By contrast, expression of Slco1b1 in the same tissue samples was unaffected by cell transplantation or CCl4 treatment. These findings were reproduced in samples from additional animals, and it was verified that inflammatory manipulations most prominently affected Abcc2 expression.

Changes in expression of canalicular proteins. Western blots are shown from same liver samples with stripping and reprobing of transblots. Expression of Abcb11 declined initially 1 d and 1 wk after CCl4 treatment (lanes 2 and 3) and returned to normal subsequently. Abcc2 expression also declined after 1 d but did not recover for the entire 4-wk duration of study. Expression of Slco1b1 was unaffected. Ctr = controls.
Role of Abcc2 in Hepatic Excretion of 99mTc-Mebrofenin
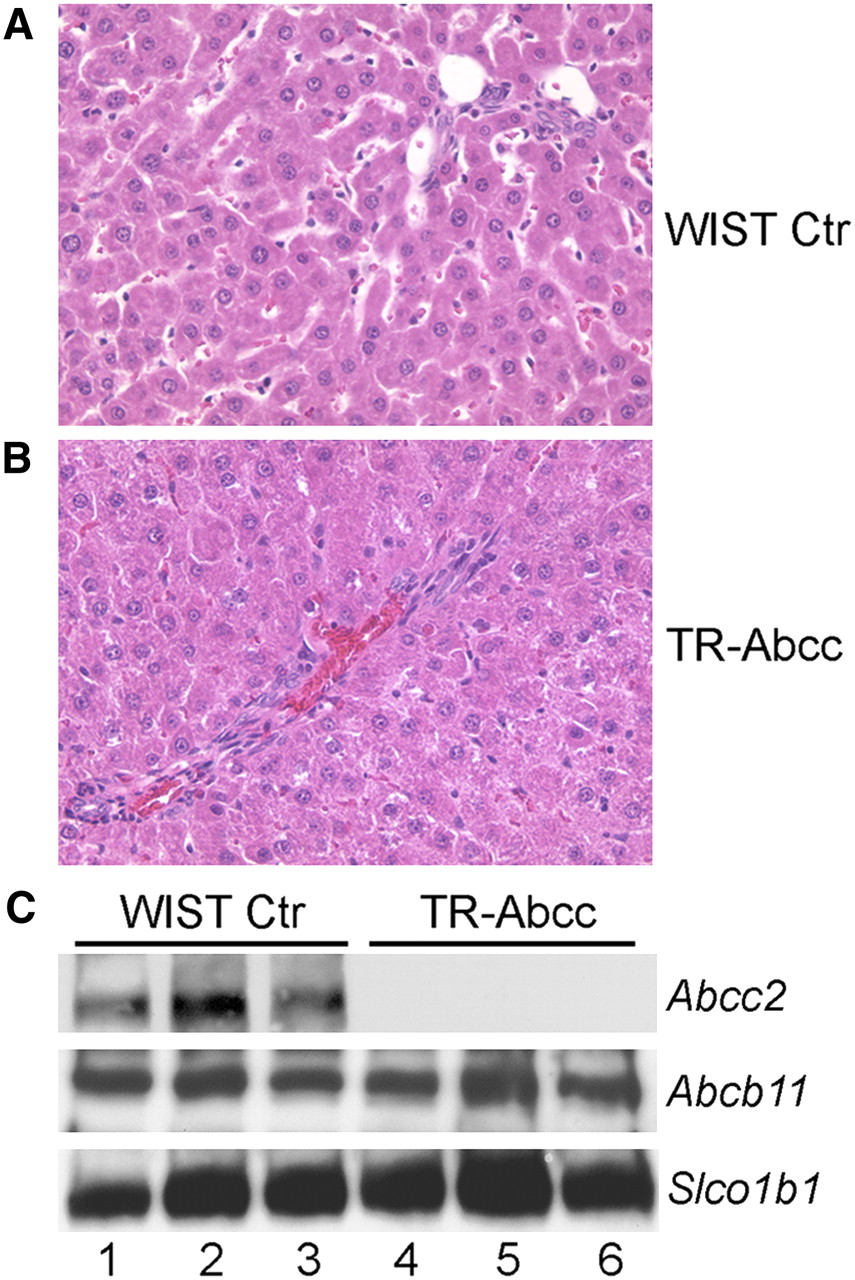
We reasoned that if Abcc2 were responsible for excreting 99mTc-mebrofenin, studies in the setting of an Abcc2-null phenotype would be helpful. Therefore, because the Abcc2 gene is naturally inactivated in HsdAmc:TR-Abcc2 mutant rats (10,11), we studied the capacity of the rats for excreting 99mTc-mebrofenin. First, to establish that the phenotype of these animals was correct, we measured total serum bilirubin, which should have been elevated. Serum bilirubin was 2.02 ± 0.28 mg/dL in HsdAmc:TR-Abcc2 mutant rats, compared with 0.23 ± 0.05 mg/dL in healthy control HsdRccHan:WIST rats (n = 6 each) (P = 0.002, Mann–Whitney rank sum test). Serum alanine aminotransferase and alkaline phosphatase levels were normal in both animal groups. Also, liver histology was normal in HsdAmc:TR-Abcc2 mutant rats and HsdRccHan:WIST rats (Figs. 4A and 4B). Western blots showed absence of Abcc2 protein in HsdAmc:TR-Abcc2 mutant rats. Abcb11 and Slco1b1 proteins were expressed at normal levels in these animals. These findings verified the Abcc2-null state in HsdAmc:TR-Abcc2 rats and indicated that the animals were appropriate for our studies.
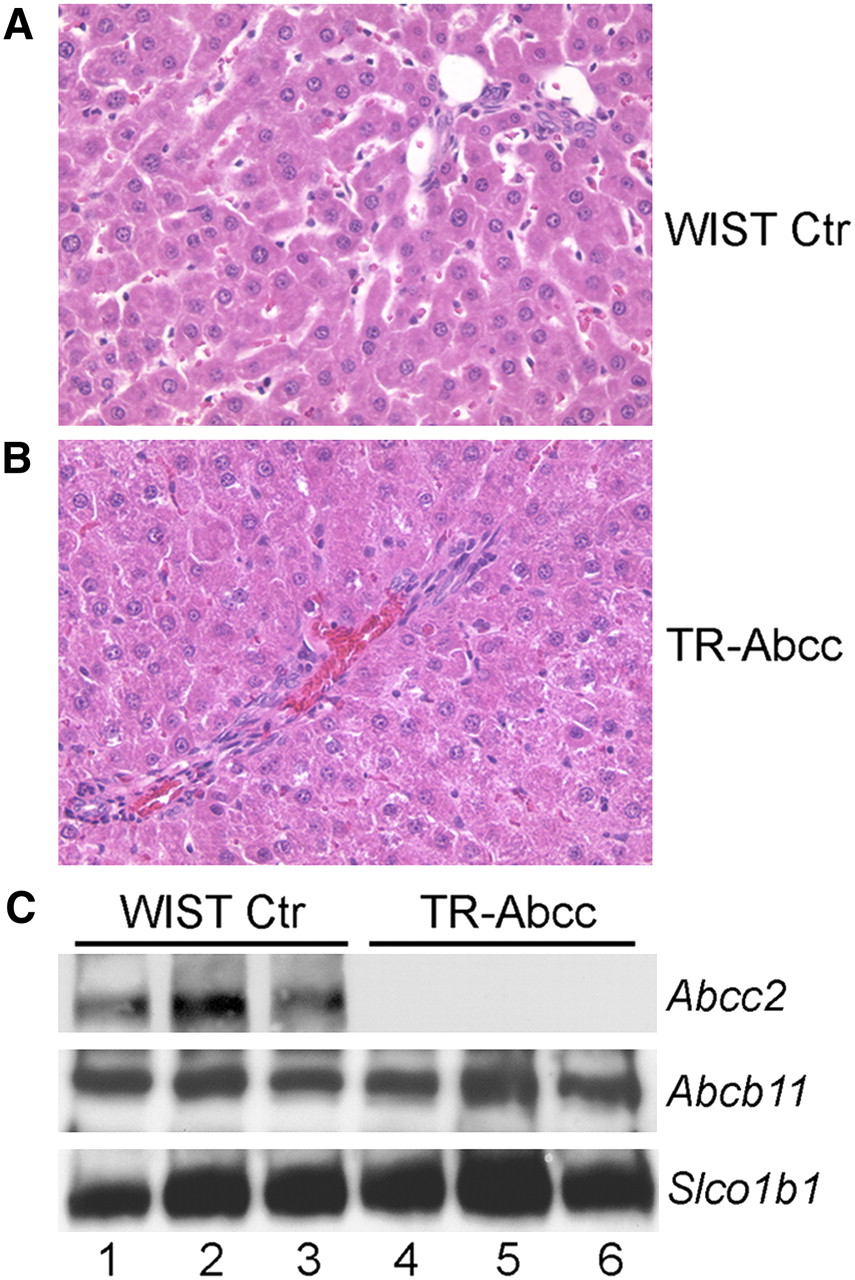
Phenotype of HsdRccHan:WIST rats (A) and HsdAmc:TR-Abcc2 mutant rats (B). Liver morphology was normal in these animals, with absence of inflammatory changes, biliary proliferation, or cholestasis. (C) Western blots of same samples for Abcc2, which was present in HsdRccHan:WIST rats (lanes 1–3) and absent in HsdAmc:TR-Abcc2 mutant rats (lanes 4–6) and for Abcb11 and Slco1b1, which were expressed normally. Each lane represents liver from different animal. Ctr = controls. (Original magnification in A and B, ×200; hematoxylin and eosin stain.)
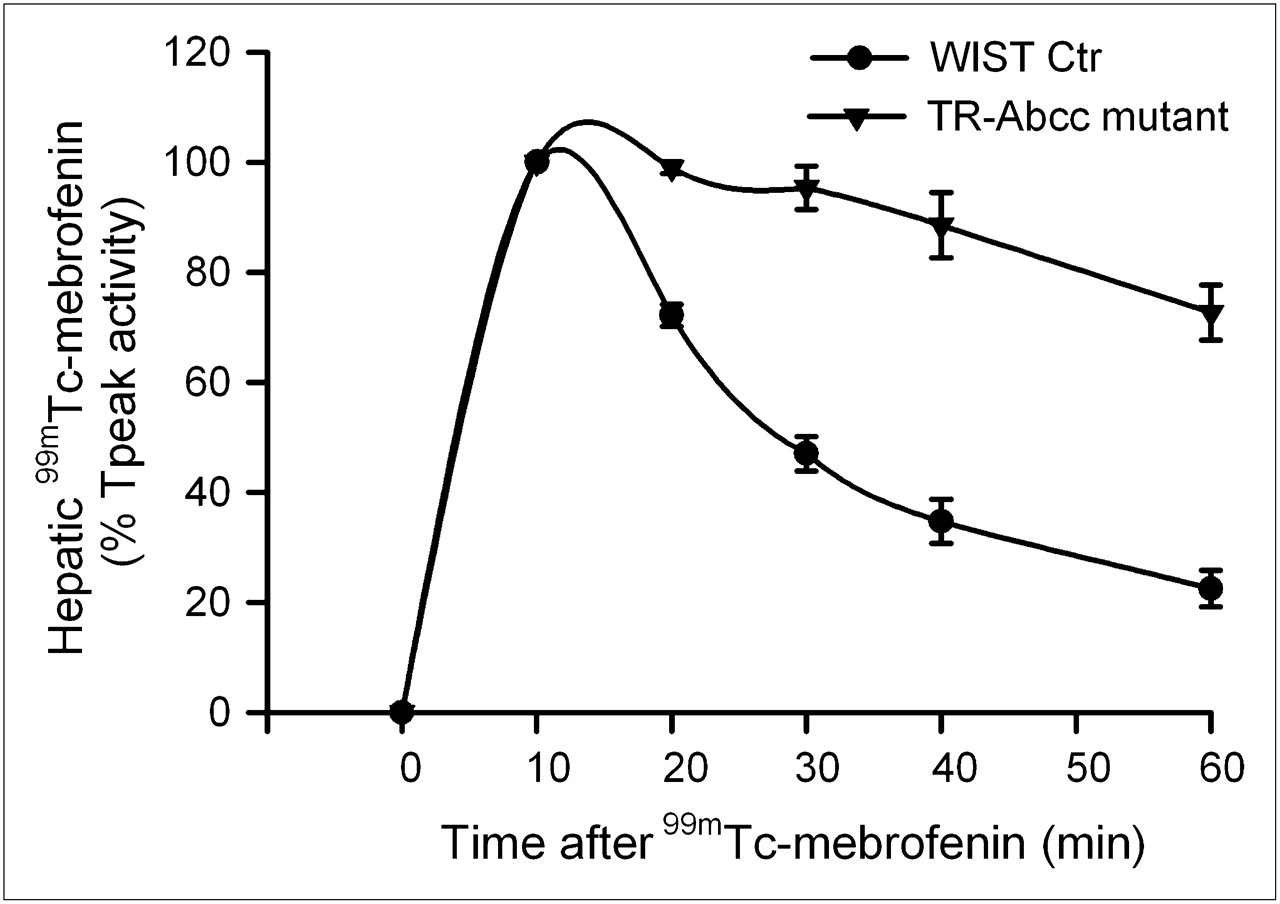
When HsdAmc:TR-Abcc2 mutant rats and their healthy counterpart rats were given 99mTc-mebrofenin (n = 6 each), no differences were observed in Tpeak values, indicating that the initial hepatic accumulation of 99mTc-mebrofenin was unaltered (Fig. 5). However, hepatic excretion of 99mTc-mebrofenin was significantly different. Much of the 99mTc-mebrofenin activity at 60 min was retained in HsdAmc:TR-Abcc2 mutant rats (73% ± 5%), compared with healthy HsdRccHan:WIST rats (22% ± 3%) (P < 0.001, t test).
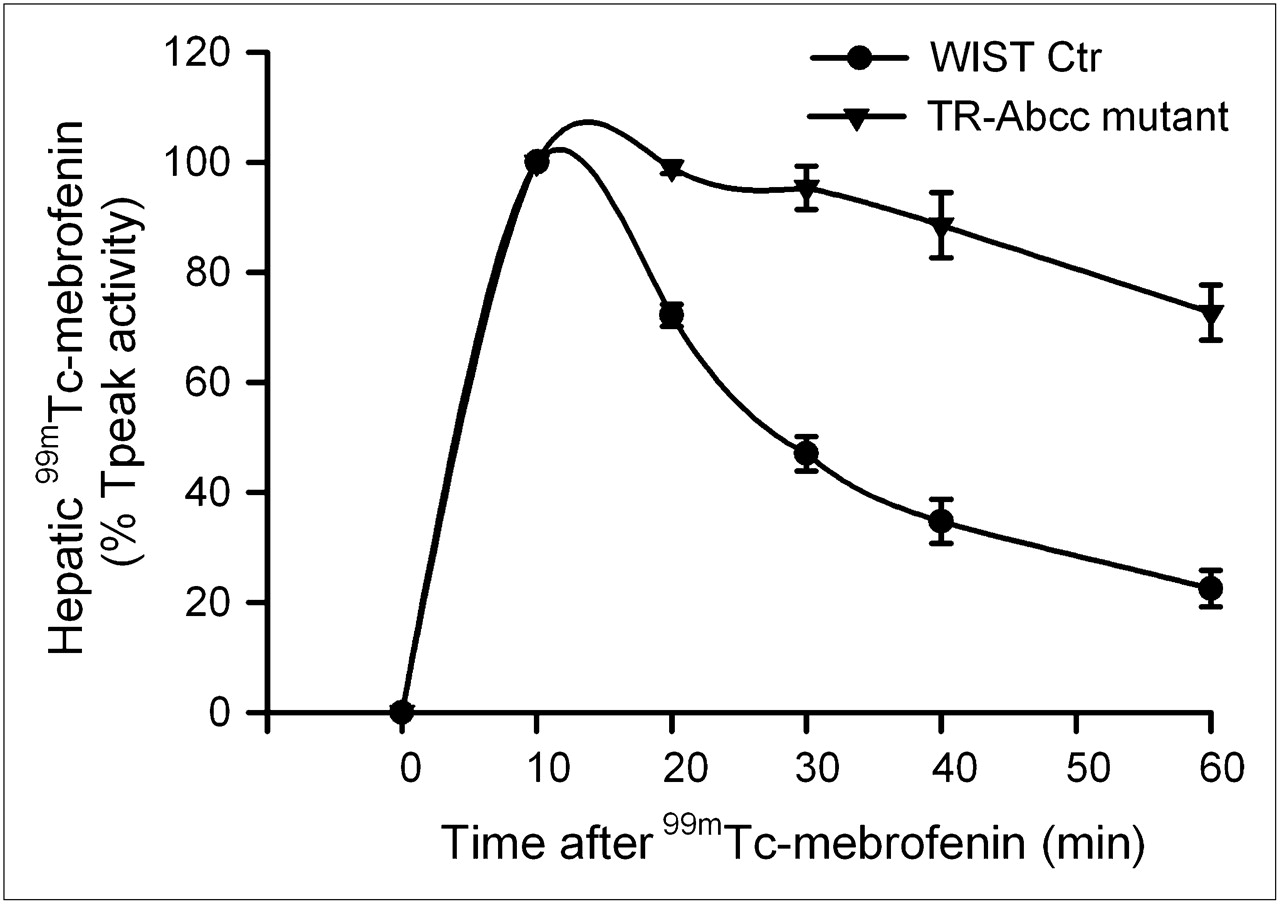
Scintigraphy showing impaired hepatic excretion of 99mTc-mebrofenin. Time–activity curves from healthy control HsdRccHan:WIST rats and HsdAmc:TR-Abcc2 mutant rats (n = 6 each). Initial accumulation of 99mTc-mebrofenin was similar in both groups. However, 99mTc-mebrofenin was promptly excreted in control rats, whereas its excretion was markedly impaired in HsdAmc:TR-Abcc2 mutant rats, resulting in retention of 22% ± 3% vs. 73% ± 5% of the Tpeak activity at 60 min (P < 0.001, t test). Ctr = controls.
Because HsdAmc:TR-Abcc2 mutant rats did excrete a small fraction of 99mTc-mebrofenin activity, we considered whether alternative pathways could have contributed to this result. In view of the simultaneous decline in Abcc2 and Abcb11 expression after inflammatory perturbations, we examined whether drugs known to alter bile canalicular transporter activity could be helpful. Because cyclosporin A has been associated with the regulation of Abcb11-dependent and Abcc2-dependent canalicular transport of bile salts and drugs (13,15), we determined its effects on 99mTc-mebrofenin excretion. When a 10 or 15 mg/kg dose of cyclosporin A was administered to healthy rats, accumulation of 99mTc-mebrofenin was not altered. However, more 99mTc-mebrofenin was retained in the liver, 70% ± 6% and 77% ± 4%, respectively, of Tpeak values at 60 min, which was similar to the abnormality in HsdAmc:TR-Abcc2 mutant rats (Fig. 6A). By contrast, in HsdAmc:TR-Abcc2 mutant rats, excretion of 99mTc-mebrofenin was impaired from the outset, with a retention of 77% ± 3% of Tpeak values at 60 min, as before, and did not significantly change after the administration of cyclosporin A. Again, the accumulation of 99mTc-mebrofenin in animals treated with cyclosporin A was unchanged, indicating that excretion of 99mTc-mebrofenin was dependent most on Abcc2 and that cyclosporin A most likely interfered with only this pathway of hepatic 99mTc-mebrofenin excretion.
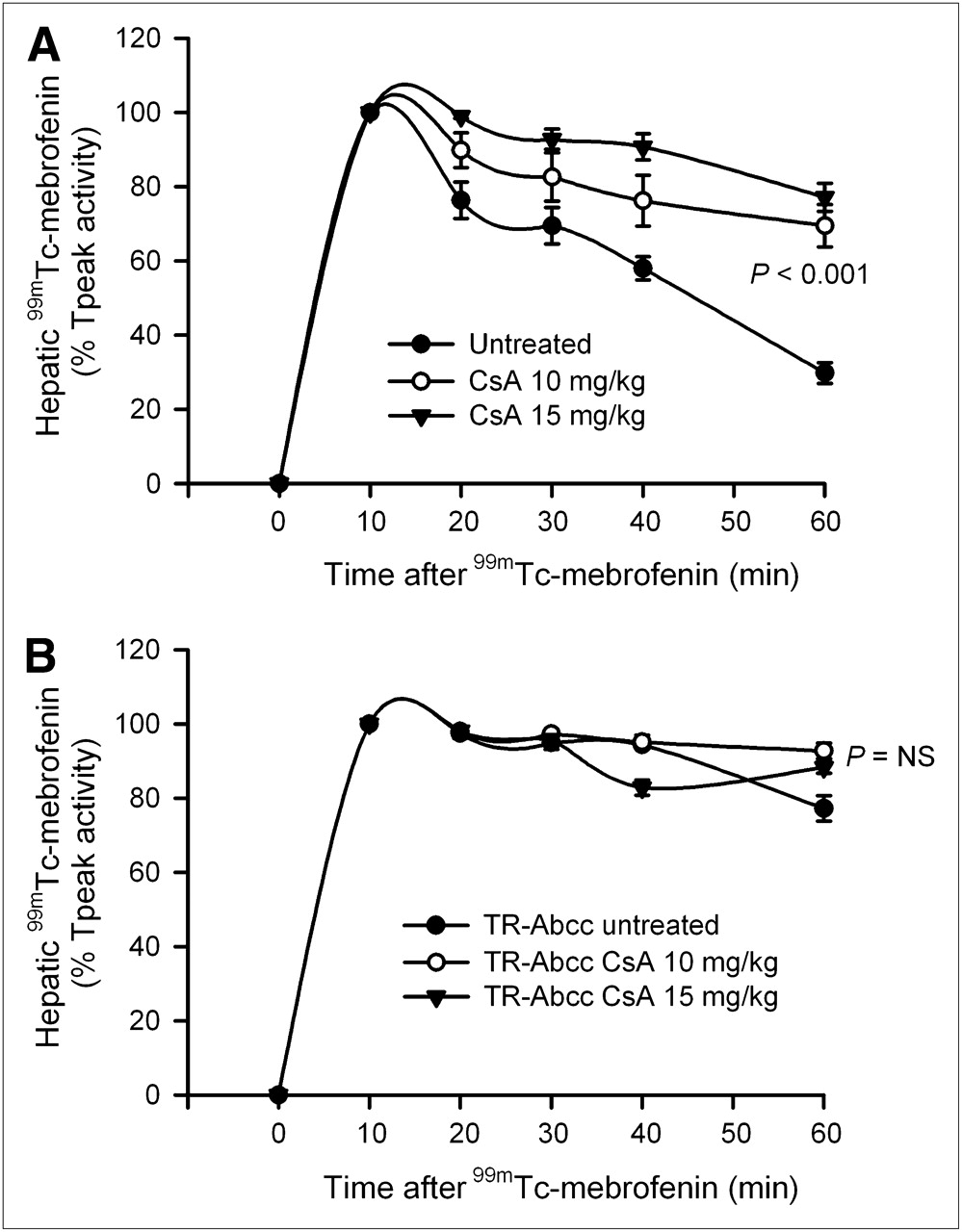
Regulation of hepatic 99mTc-mebrofenin excretion by cyclosporin A. (A) Time–activity curves in healthy F344 rats with and without prior administration of cyclosporin A (n = 6 each). In these animals, initial accumulation of 99mTc-mebrofenin was unchanged. However, 99mTc-mebrofenin excretion was impaired with retention of 70% ± 6% and 77% ± 4% of the Tpeak activity at 60 min in recipients of 10 and 15 mg/kg doses of cyclosporin A (compared with only 30% ± 3% in control animals), respectively (P < 0.001, ANOVA). (B) Changes in 99mTc-mebrofenin excretion in HsdAmc:TR-Abcc2 mutant rats with or without cyclosporin A (n = 6 each). In these animals, 99mTc-mebrofenin excretion was already impaired and cyclosporin A did not cause further significant impairment. CsA = cyclosporin A; NS = not significant, ANOVA.
DISCUSSION
These studies demonstrated that in experimental models of hepatic inflammation, excretion of 99mTc-mebrofenin was impaired in the long term. Because the expression of bile canalicular transporters was also affected by hepatic inflammation, correlations could be made between 99mTc-mebrofenin excretion and downregulated expression of specific transporters to incriminate Abcc2 as the likely 99mTc-mebrofenin transporter. This correlation was directly established in HsdAmc:TR-Abcc2 mutant rats, which constituted an excellent model of Abcc2 deficiency (10,11). Further studies in animals treated with cyclosporin A verified that Abcc2 was responsible for most 99mTc-mebrofenin excretion. These findings indicated that as a ligand for Abcc2, 99mTc-mebrofenin was an excellent reporter for demonstrating the integrity of this cellular pathway, which will be significant for interpreting abnormalities in 99mTc-mebrofenin imaging and for developing new applications with 99mTc-mebrofenin or related analogs.
The mechanism by which 99mTc-mebrofenin is incorporated in hepatocytes is not fully defined, although the uptake process is thought to be sodium-independent, without requiring the Slco1a1 gene, which should be expressed on the sinusoidal domain of hepatocytes (3). However, alternative sinusoidal transporters (i.e., Slco1b1 and Slco1b3) did allow 99mTc-mebrofenin uptake to otherwise nonpermissive Xenopus oocytes. We found that expression of Slco1b1 was unaffected by hepatic inflammation in rats, as was in agreement with normal initial hepatic accumulation of 99mTc-mebrofenin, indicated by Tpeak values under conditions studied here and as discussed in detail previously (8). Similarly, fractional retention of peak hepatic 99mTc-mebrofenin over 60 min was established as an effective parameter for assessing 99mTc-mebrofenin excretion, and this excretion was rapidly and persistently inhibited by hepatic inflammation due to cytokines, such as interleukin-6 and TNF-α (8), which are major mediators of phagocyte and macrophage responses. Here, we found that multiple cytokines were activated by inflammatory perturbations, including significant and prolonged activation of TNF-α. In agreement with our findings, inflammatory cytokines downregulated the expression of bile canalicular transporters in the short term, including Abcc2, Abcb11, and other genes (16–18), although not Slco1b1 (19). Here, we found that downregulation of canalicular transporters can continue in the long term during ongoing activation of inflammatory cells and local release of relevant cytokines. The role of nuclear receptor–based signaling in regulating bile canalicular transporter expression under various experimental conditions and disease states has also been established (5). Therefore, the assessment of hepatobiliary transport with noninvasive 99mTc-mebrofenin imaging will be effective for further biologic and clinical studies of Abcc2 regulation.
The transport abnormalities in HsdAmc:TR-Abcc2 mutant rats have been characterized previously (11). These rats constitute an authentic model of Dubin–Johnson syndrome in people, in whom the Abcc2 gene is mutated (10), except that the liver in Dubin–Johnson syndrome is characterized by an accumulation of a dark pigment and lipofuscin, which was not observed in the liver of HsdAmc:TR-Abcc2 mutant rats. Our finding of 99mTc-mebrofenin retention in HsdAmc:TR-Abcc2 mutant rats indicated clearly that Abcc2 was required for transporting much of the 99mTc-mebrofenin into bile. This correlated with long-standing observations in Dubin–Johnson syndrome of abnormal handling of 99mTc-mebrofenin analogs with hepatic accumulation and inability to clear radiotracers over time (20). These findings established that 99mTc-mebrofenin imaging will provide a specific diagnostic test for Dubin–Johnson syndrome and for other disorders characterized by the loss of Abcc2 activity. Moreover, our studies with cyclosporin A demonstrated that pharmacologic interactions involving Abcc2 can be studied in intact animals by 99mTc-mebrofenin assays. Excretion of 99mTc-mebrofenin is not affected by elevated levels of serum bilirubin (2), which constitutes another substrate of Abcc2 (21), suggesting that Abcc2 likely has greater affinity for 99mTc-mebrofenin than for bilirubin. On the other hand, increased retention of 99mTc-mebrofenin after treatment of animals with cyclosporin A indicated that this drug could more preferentially bind Abcc2. The excretion of some 99mTc-mebrofenin activity in HsdAmc:TR-Abcc2 mutant rats suggested that pathways other than Abcc2 might play smaller additional roles. In Dubin–Johnson syndrome, compensatory upregulation of the Abcc3 gene at the lateral membrane of hepatocytes could result in reflux of conjugated bilirubin into the blood (11), although the Abcc3 gene was not upregulated in Abcc2 knockout mice (21), raising doubts about that alternative for 99mTc-mebrofenin removal. However, increased expression of Abcc3 and some other transporters capable of excreting substrates into other locations, for example, the gastrointestinal tract (11), is not excluded. Also, the possibility that additional bile canalicular transporters (e.g., Abcb4) could excrete small amounts of 99mTc-mebrofenin is not excluded. Abcb4 has a similar minor role in excreting sestamibi (largely excreted by the Abcb1 protein (22)); however, liver injury and inflammation in mutant mice lacking Abcb4 might produce confounding data in demonstrating this possible minor role of Abcb4 in 99mTc-mebrofenin excretion.
Taken together, our findings indicate that the identification of perturbations in molecular pathways represented by the Abcc2 gene through assays of 99mTc-mebrofenin will provide further ways to approach a variety of physiologic, pathobiologic, and pharmacologic mechanisms.
CONCLUSION
Studies in animal models demonstrated that 99mTc-mebrofenin was largely excreted by Abcc2-mediated hepatobiliary transport. This mechanism should guide analysis of pathophysiologic perturbations in the hepatic handling of 99mTc-mebrofenin and diagnosis of impairments in Abcc2 gene function.
Acknowledgments
We thank Chaoying Zhang for contributing technical assistance. This work was supported in part by National Institutes of Health grants R01 DK46952, P30 DK41296, P30 CA13330, M01 RR12248, and R37 HD20362.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication January 21, 2009.
- Accepted for publication March 16, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}