


Notwithstanding the major improvements in diagnosis and therapy, coronary heart disease remains the leading cause of death in the United States. About every 26 s, an American will undergo a coronary event, and about every minute someone will die from one (American Heart Association computation based on latest available mortality data) (1). The event causing death is most often a thrombus in the coronary artery, set off by rupture of an atheroma located in the vessel wall (2). Atheromas likely to rupture (vulnerable plaques) have See page 563
many characteristics similar to an abscess. Vulnerable plaques have a large hypoxic, metabolically active core, containing lipid, oxidized lipid, and numerous inflammatory cells (primarily foam cells or macrophages). Vulnerable lesions have a thin cap (<65 μm) separating the region of intense inflammation from blood in the vessel lumen (3). The lipid-laden inflammatory cells in the necrotic core secrete proteolytic enzymes (such as matrix metalloproteinases), which weaken the thin fibrous cap. Disruption of the fibrous cap allows the thrombogenic inflammatory material in the lesion to contact the blood, leading to formation of the critical thrombus (Fig. 1) (4).

Molecular factors involved in plaque evolution. Numerous activated macrophages and other inflammatory cells accumulate in atheromas, accounting for increased 18F-FDG metabolism in these lesions. Inflammatory cells secrete proteolytic enzymes, such as matrix metalloproteinases, weakening thin fibrous cap. Rupture of fibrous cap follows, leading to thrombus formation. ICAM = intercellular adhesion molecule; MMP = matrix metalloproteinase; SMC = smooth muscle cell; VCAM = vascular cell adhesion molecule. (Reprinted with permission of (4). See original article for detailed discussion.)
Atheromas concentrate 18F-FDG because macrophages in the lesion require glucose to perform their phagocytic activities. Glucose is delivered to the lesion by diffusion from the plasma, through the extracellular fluid. Concentration of 18F-FDG in areas of inflammation appears to require a chronic lesion. Studies by Som et al. in 1980 evaluated 18F-FDG uptake in a sterile turpentine abscess but found little uptake of 18F-FDG in the lesion (5). In retrospect, the lack of uptake was likely due to imaging of the lesion performed at an early phase. This problem was not clarified until 1995, when Yamada et al. performed serial autoradiographic studies in animals with sterile abscesses (6). These investigators found minimal uptake in the first 24 h but progressively increasing uptake in inflammatory cells of the abscess wall up to 4 d. In 1997, the concept of using 18F-FDG PET to detect vascular inflammation due to atheromas was suggested by Vallabhajosula and Fuster (7). In 2002, the initial study by Rudd et al. in 8 patients with carotid atherosclerosis demonstrated the potential relationship of focal 18F-FDG uptake to symptomatic cerebrovascular disease (8). Tawakol et al. confirmed a correlation between macrophage density and 18F-FDG uptake in inflamed human carotid endarterectomy specimens from symptomatic patients (9). Vascular uptake of 18F-FDG in large vessels is reproducible over a 2-wk interval (10), but the pattern of uptake changes in approximately half the patients over longer intervals of time (11). These changes in uptake likely reflect the waxing and waning course of atheroma and must be considered when the use of 18F-FDG vascular imaging is suggested to define the efficacy of therapy. Despite this fluctuating uptake, serial 18F-FDG imaging has already been used to demonstrate a substantial decrease in carotid inflammation after therapy with simvastatin (12), and a recent study suggested that behavioral modification may reverse 18F-FDG uptake in large vessels in correlation with elevation of high-density lipoprotein and atherogenic risk reduction (13).
Although imaging atheromas in large vessels is useful, metabolic vascular imaging will likely have its greatest value in the coronaries. On the basis of the model of therapeutic response monitoring in oncology, the hope is that vulnerable plaques will be not only detected early but also monitored serially for response to therapy. Dunphy et al. have already observed increased 18F-FDG uptake in the coronary arteries in association with cardiac risk factors and coronary calcium (14). However, their observation was hindered by high myocardial background activity in half of the patients. One of the major challenges to imaging coronary atheromas by 18F-FDG PET is, therefore, bound to be the variable levels of myocardial background activity often encountered.
In this issue of The Journal of Nuclear Medicine, Wykrzykowska et al. have taken the next step toward the validation of noninvasive metabolic imaging of vulnerable plaque in the human coronary tree by focusing on the suppression of myocardial background (15).
The cardiomyocyte is an omnivore, readily consuming free fatty acids (FFAs), glucose, lactate, pyruvate, and to a lesser extent, ketones (Fig. 2) (16–18). FFAs are the major substrate used by the myocardium under normal aerobic conditions, particularly in the fasted state. Glucose becomes the preferred substrate for oxidative metabolism in fatty acid–depleted states and when the concentrations of glucose and insulin are high, as in the immediate postprandial state. Although a 6-h fast increases lipolysis and FFA supply, fasting alone does not reliably suppress myocardial 18F-FDG uptake (19–21). Indeed, heterogeneity of 18F-FDG accumulation in the myocardium in the fasted state was recognized in the early days of clinical 18F-FDG imaging for myocardial viability (22,23). Shreve et al. noted that myocardial 18F-FDG uptake remains variable even after fasting upward of 18 h (24).
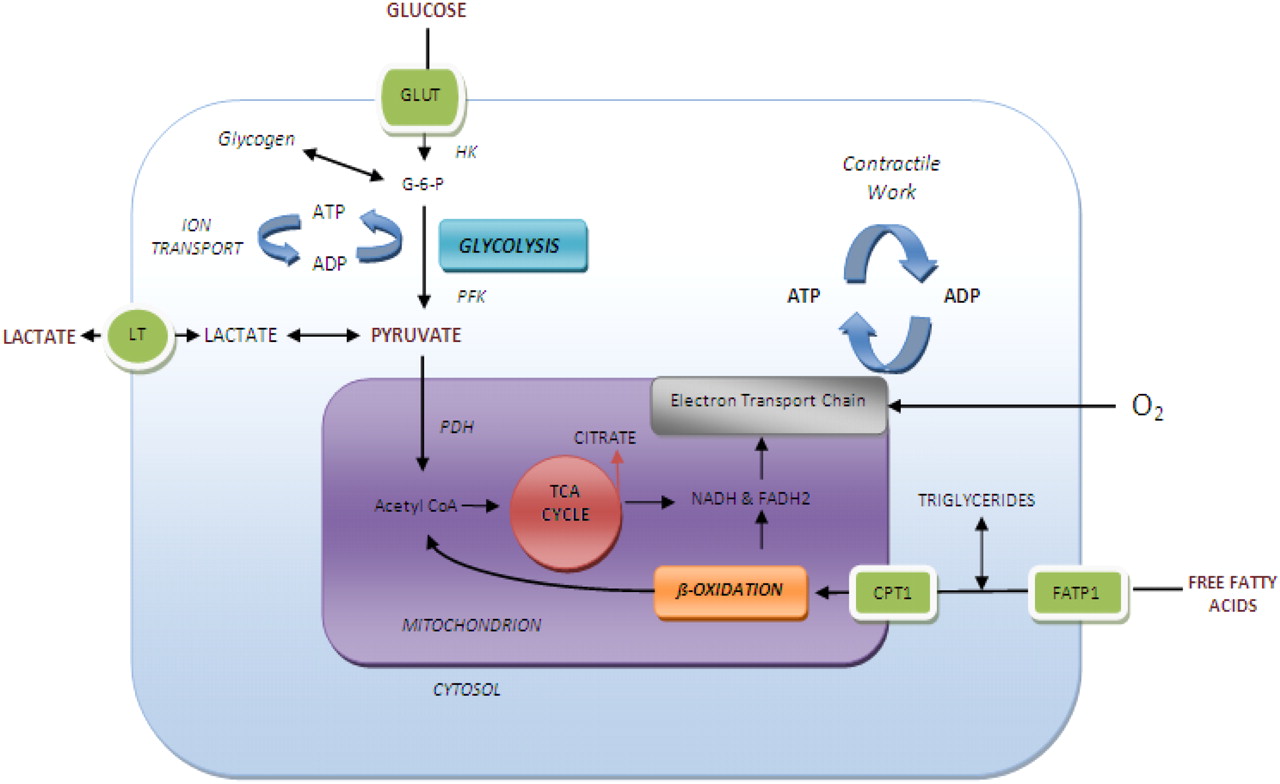
Schematic depiction of myocardial substrate metabolism. ADP = adenosine diphosphate; ATP = adenosine triphosphate; CoA = coenzyme A; CPT1 = carnitine palmitoyl transferase; FADH2 = flavin adenine dinucleotide-reduced; FATP1 = fatty acid-transport protein 1; G-6-P = glucose 6-phosphate; GLUT = glucose transporters; HK = hexokinase; LT = lactate transporter; NADH = nicotinamide adenine dinucleotide-reduced; PDHa = active dephosphorylated pyruvate dehydrogenase; PFK = phosphofructokinase; TCA = tricarboxylic acid. (Adapted with permission of (18).)
What is the solution, then? On the basis of their earlier studies (25), the investigators at the Beth Israel Deaconess Medical Center in Boston recommended that the patient ingest a high-fat, low-carbohydrate, protein-preferred meal the night before the scan and a vegetable oil drink the morning of the scan. This preparation exploits the glucose–fatty acid cycle, the inverse metabolic relationship between glucose and fatty acids, which was first described by Randle in the 1960s (26). Increased FFA availability promotes FFA oxidation and inhibits glucose use (Fig. 3) (27). Conversely, glucose availability promotes glucose oxidation and inhibits FFA use (28). The inhibitory effect of elevated FFA concentrations on glucose use by skeletal muscle has been demonstrated repeatedly in humans. As early as 1978, Phelps et al. showed that myocardial 18F-FDG uptake under aerobic conditions can be reduced in humans by preparation with a high-fat meal (29). In theory, this metabolic switch should be further enhanced by lipid loading or drugs that acutely increase FFA availability—the converse of glucose loading or acipimox administration—before 18F-FDG myocardial viability studies, when homogeneous 18F-FDG uptake is desired (30). Yet, given the ubiquity of glucose consumption at the cellular level, even in the face of upregulated FFA oxidation, this approach is not as easy as it may initially seem (31). The success of any intervention is further confounded by the interplay of patient-specific comorbidities and hormonal milieu. For instance, hypertension and high adrenergic states are known to promote glucose over FFA use, perhaps because of increased myocardial workload (despite the concomitant rise in plasma fatty acid levels). Paradoxically, β-blockers are thought to decrease FFA oxidation in the setting of heart failure, independent of substrate availability, favoring glucose oxidation and consequently 18F-FDG use (32). As intracellular calcium concentration is a stimulus for glucose transport and oxidation, calcium channel blockers may be helpful in lieu of β-blockers for heart rate reduction before coronary artery 18F-FDG inflammation studies (33). Other drugs that can potentially interfere with suppression of myocardial 18F-FDG uptake include the antianginal drug trimetazidine (an FFA oxidation inhibitor), which increases myocardial glucose use (32). Similarly, rosiglitazone, a peroxisome proliferator–activated receptor-α activator, enhances insulin-stimulated myocardial glucose uptake in patients with type 2 diabetes, most probably because of its suppression of serum FFAs (34). On the other hand, patients taking levothyroxine exhibited reduced myocardial 18F-FDG uptake in 1 study (35), although this is somewhat controversial, as chronic thyroid hormone administration increases myocardial glucose transport and glycolysis (33,36). Despite the multiplicity of confounding factors, it is conceivable that they could be overcome in the acute setting by a standardized dietary or pharmacologic preparation (16).

Glucose-fatty acid cycle. Suppression of glucose metabolism by increased FFA is highlighted by red arrows. Oxidation of FFAs generates acetyl-CoA (coenzyme-A; inhibitor of pyruvate dehydrogenase) and citrate (inhibitor of phosphofructokinase), leading to accumulation of glucose-6-phosphate (G-6-P; inhibitor of hexokinase), and consequently, inhibition of intracellular glucose transport and use. CPT1 = carnitine palmitoyl transferase 1; FATP1 = fatty acid-transport protein 1; GLUT4 = glucose transporter 4; HK = hexokinase; IR = insulin receptor; PDH = pyruvate dehydrogenase; PFK = phosphofructokinase. (Adapted with permission of (27).)
A critical question is whether diet or pharmacologic intervention will indirectly affect lesion uptake. This is not a factor in oncologic imaging, because of the uncoupling of the glucose–fatty acid cycle in malignancy. In fact, 1 study demonstrated that tumor uptake of 18F-FDG is not enhanced by suppressing serum FFA with acipimox (37). Does the same hold true for inflammation? An earlier study by the Beth Israel group suggested that uptake at nonvascular inflammatory sites is unaffected by the high-fat, low-carbohydrate diet preparation (25). Although vulnerable plaques have increased angiogenesis in the vasa vasorum, the supply of oxygen is insufficient for the high level of metabolism, rendering the local environment hypoxic (38). This result seems to promote a glycolytic state over oxidative metabolism, even in the presence of an altered nutrient environment.
Once the hurdle of myocardial background has been cleared, the next challenge is the fundamental disparity between the coronary lesion size and the limited spatial resolution of PET. Autopsy studies reported by Virmani et al. detailed the spectrum of coronary atheromas (39). Thin fibrous cap atheromas—the precursors of acute plaque ruptures—have a necrotic core with a mean length of 8 mm and an area of 1.7 mm2 comprising about 25% of the total atheroma area. This size translates to a volume of less than 0.1 mL for most inflamed lesions. A recent investigation using intracoronary ultrasound and multislice CT reported a mean volume of 0.09 mL for significant plaques (40). Taken together, the data suggest that vulnerable lesions reside at the edge of the spatial resolution of PET (4–6 mm at the center of the field of view; volume resolution, ∼0.125 mL). However, this hurdle may be overcome by the intense signal emanating from vulnerable plaques due to the high density of activated macrophages. As the target-to-background ratio increases, the size required for detection decreases (41). Thus, the molecular signal from the coronary tree must be optimized by limiting degrading or blurring factors, including photon attenuation by intervening body tissue, coronary tree motion (respiratory and cardiac), and partial-volume error. In the era of hybrid PET/CT systems, CT attenuation maps have improved the efficiency of attenuation correction. With motion, the lower the heart rate, the better. The coronary tree moves least during diastole; therefore, prolonging diastole with heart rate reduction may be useful (assuming no significant augmentation of myocardial 18F-FDG uptake). Electrocardiogram gating has long been used for blood-pool and myocardial perfusion SPECT and is available for PET/CT. Although the effective counting rate will suffer, itself a cause of image degradation, list-mode acquisition may reduce this problem (42). The next hurdle is respiratory motion. Although breath-hold acquisition is the mainstay of clinical CT, PET requires at least 3 min per field of view for data acquisition, rendering breath-hold impractical. A recent comprehensive review by Nehmeh and Erdi details multiple methods of respiratory correction now under development (43). Partial-volume error can introduce significant variability to the quantification of lesion uptake, depending on the lesion geometry, spatial resolution of the camera, and method of reconstruction. As with respiratory gating, several approaches are under development to mitigate the effects of partial-volume error, of particular importance for therapeutic response monitoring (44).
Prospective studies are needed to determine the prognostic significance of lesion intensity, number of discrete lesions per vascular territory, and total number of territories involved. As mentioned, the intensity of uptake correlates with the number of macrophages in a lesion, presumably an indicator of lesion vulnerability. Summing the total number of lesions should provide evidence for risk, akin to sum scores in SPECT and PET perfusion imaging. Finally, prospective investigation of patients without concurrent malignancy and correlation with a more proximate invasive or noninvasive (CT) coronary angiography should provide more robust information. We eagerly await the authors' pending prospective study.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication October 7, 2008.
- Accepted for publication October 15, 2008.





{kind=link}
{kind=link}
{kind=link}