


Abstract
Herpes simplex virus type 1 thymidine kinase (HSV1-TK) is a widely used reporter for in vivo noninvasive monitoring of therapeutic gene expression, immune cell trafficking, and protein–protein interactions in various animal systems. However, the stability of HSV1-TK limits its application in studies that require rapid turnover of the reporter. The purpose of this study was to create a destabilized HSV1-TK as a transcription reporter that allows for dynamic studies of short-time-scale gene expression events. Methods: A destabilized HSV1-TK was created by targeting inactivating mutations in the nuclear localization signal of HSV1-TK and fusing the degradation domain of mouse ornithine decarboxylase to the C-terminal end. The protein or enzyme stability was determined by Western blot analysis and HSV1-TK enzyme activity assay, respectively. The proteasome inhibition assay was used to test whether the rapid turnover of the destabilized HSV1-TK was processed in a 26S proteasome–dependent manner. The suitability of destabilized HSV1-TK as a transcription reporter was tested by linking it to a tetracycline-turnoff–expressing system. The dynamic transcriptional events mediating a series of doxycycline inductions were monitored by destabilized HSV1-TK or by native HSV1-TK and were determined by an in vitro HSV1-TK enzyme activity assay and in vivo small-animal PET imaging. Results: The destabilized HSV1-TK, unlike wild-type HSV1-TK, was unstable in the presence of cycloheximide and had a short half-life of protein and enzyme activity. The rapid turnover of the destabilized HSV1-TK was processed in a 26S proteasome–dependent manner. Furthermore, the destabilized HSV1-TK had low cytotoxicity when it was highly expressed in living cells. The results of dynamic gene expression studies in vitro and in vivo showed that the destabilized HSV1-TK is an optimal reporter for monitoring short-time-scale dynamic transcriptional events mediating a series of doxycycline inductions, whereas the wild-type HSV1-TK is not optimal to achieve this purpose. Conclusion: The use of destabilized HSV1-TK as a transcription reporter together with a molecular probe, which has a short physical and biologic half-life, allows more direct monitoring of transcription induction and easier monitoring of its coincidence with other biochemical changes.
- herpes simplex virus type 1 thymidine kinase (HSV1-TK)
- mouse ornithine decarboxylase (MODC)
- doxycycline (Dox)
- nuclear localization signal (NLS)
- PET imaging
Wild-type herpes simplex virus type 1 thymidine kinase (HSV1-TK) and a mutant HSV1-TK gene, HSV1-sr39tk, are the most commonly applied reporter genes in current molecular imaging studies using radiolabeled probes and PET (1). This reporter system has been widely used in noninvasive monitoring of therapeutic gene expression in vivo and in immune cell trafficking and protein–protein interactions in various living animals (2–4). However, our previous study showed that HSV1-TK is highly stable in mammalian cells (5). This stability limits its application for monitoring short-time-scale dynamic processes, such as kinetic gene expression controlled by inducible promoters or by a less stable protein with a more rapid turnover. Thus, the highly stable reporter protein of HSV1-TK poses a significant disadvantage for dynamic studies of short-time-scale gene expression events because of poor temporal resolution (5). Furthermore, the HSV1-TK enzyme has been shown to have cytotoxicity on its own when used as a selection marker or therapeutic gene in transgenic mice or in gene therapy strategies because of the nuclear tropism of its protein and the presence of a putative cryptic testis-specific promoter (6–8).
In mammalian cells, cellular proteins differ widely in their stabilities. Rapid turnover in protein is often caused by signals that induce protein degradation. In some cases, one of the best characterized of these proteolytic signals has been identified as a region enriched in proline (P), glutamate (E), serine (S), and threonine (T) residues (PEST) and flanked by positively charged residues such as lysine (K), arginine (R), or histidine (H) (9–11). In other cases, the signal is a modification of phosphorylation (12) or a protein–protein interaction (13). Before degradation, most short-lived proteins need ubiquitin modification, which is catalyzed by several ubiquitin-modifying enzymes. Only these ubiquitinated proteins are recognized and degraded by a 26S proteasome (14). In the bioinformatics analysis of the HSV1-TK amino sequence, there are no potential proteolytic cleavage sites and the protein half-life of HSV1-TK was estimated to be about 30 h on the basis of the N-end rule. This may explain in part why HSV1-TK is stable in mammalian cells (5).
Mouse ornithine decarboxylase (MODC) is the key enzyme in the biosynthesis of polyamines (15–17). This protein is known to be one of the shortest-lived proteins in mammalian cells, with a half-life of approximately 30 min. In contrast, ornithine decarboxylase (ODC) of Trypanosoma brucei is quite stable when expressed in mammalian cells (18). Comparison of the primary sequence of these 2 proteins shows over 60% homology (19). However, MODC has extra sequences at its C terminus that are not needed for ODC activity. This C terminus contains a PEST sequence, and its deletion from MODC prevents its rapid degradation (11). Also, the protein fused with the C-terminal extension of ODC becomes unstable. Therefore, this region functions as the “degradation domain” of MODC (20). Furthermore, it has been shown that the rapid turnover of ODC is brought about by the 26S proteasome but that ODC is highly unusual in that ubiquitination is not required for this degradation (21–23). Instead, a noncovalent association with a protein termed antizyme directs ODC to the proteasome. Antizyme increases the degradation of ODC by enhancing its interaction with the proteasome but does not increase the rate of proteasomal processing. It was also shown that proteasomes actually start degradation of ODC at the COOH terminus (24). Therefore, rapid degradation of mammalian ODC requires a degradation domain that contains a PEST motif located at the COOH terminus of the protein.
To increase the temporal resolution of the HSV1-TK reporter system in molecular–genetic imaging, it is necessary to develop a HSV1-TK with low protein stability and low cytotoxicity. We hypothesized that HSV1-TK targeted the inactivating mutations in the nuclear localization signal (NLS) located in the N terminus and fused with the degradation domain of MODC at the C-terminal end, thus overcoming the limitation of high protein stability and cell toxicity in living mammalian cells. This modified HSV1-TK may degrade through the 26S proteasome because of the attachment of the PEST degradation signal of MODC to its carboxyl-COOH-terminal region. In this study, we successfully generated a destabilized and low-cytotoxicity HSV1-TK as a transcription reporter that allowed for direct monitoring of transcription induction and its coincidence detection with other biochemical changes in living animals in molecular–genetic imaging studies.
MATERIALS AND METHODS
Plasmid Construct
The HSV1-TK gene was amplified from the plasmid pORF-HSV1-TK (InvivoGen) using polymerase chain reaction (PCR). The primers used, (5′-GAAGATCTATGGCCTCGTACCCCGGCCA-3′) and (5′-CCCAAGCTTGTTAGCCTCCCCCATCTCCCGGG-3′), contain the restriction enzyme sites of BglI and HindIII. The PCR product was subcloned into the pd2EGFP-N1 vector (Clontech), from which was deleted enhanced green fluorescence protein (EGFP) using BglI and HindIII restriction enzymes. In this constructed vector (pCMV-HSV1-TK:dMODC), the stop codon of HSV1-TK was deleted to make a continuous open reading frame with the C terminus of MODC, and the HSV1-TK fused with the degradation domain of MODC was controlled by human cytomegalovirus (CMV) immediate early promoter. In addition, the HSV1-TK gene was amplified with another reverse primer (5′-CCTCTACCCCCTCCGATTGACT-3′) to contain stop codon and was inserted in the pd2EGFP-N1 vector, from which was deleted EGFP and MODC using BglI and HpaI restriction enzymes. This constructed vector, termed pCMV-HSV1-TK, acted as the control vector. Furthermore, the HSV1-TK was digested with BglII and XmaI from the pCMV-HSV1-TK vector. This fragment was inserted into the pEGFP-N1 and pd2EGFP vectors, which were digested with BglII and XmaI. These constructed vectors were termed pCMV-HSV1-TK:GFP and pCMV-HSV1-TK:GFP:dMODC, respectively. Inactivation of the NLS signal of HSV1-TK was based on a protocol described elsewhere (25). Briefly, the codons for Arg25-Arg26 amino acids in HSV1-TK were replaced by the BamHI restriction site, 5′-GGATCC-3′ (to encode Gly25-Ser26), by PCR. The resulting vectors were termed pCMV-mNLSHSV1-TK, pCMV-mNLSHSV1-TK:dMODC, pCMV-mNLSHSV1-TK:GFP, and pCMV-mNLSHSV1-TK:GFP:dMODC, respectively.
For real-time monitoring of the protein degradation of mNLSHSV1-TK:dMODC and HSV1-TK, the mNLSHSV1-TK:dMODC or HSV1-TK cDNA sequence was subcloned into the pEGFP-N1 vector (Clontech) to create a construct that would express mNLSdHSV1-TK or HSV1-TK protein tagged with GFP at its carboxyl terminus. The HSV1-TK was digested with BglII and XmaI from the pCMV-HSV1-TK vector. This fragment was inserted into the pEGFP-N1 vector, which was digested with BglII and XmaI. This constructed vector was termed pCMV-HSV1-TK:GFP. In addition, the mNLSHSV1-TK:dMODC:GFP was digested with BglII and EcoRI from pCMV-mNLSHSV1-TK:dMODC and inserted into pEGFP-N1, which was digested with BglII and EcoRI. This constructed vector was termed pCMV-mNLSHSV1-TK:dMODC:GFP. In the tetracycline-turnoff–expressing mNLSHSV1-TK:dMODC and mNLSHSV1-TK, the mNLSHSV1-TK:dMODC and mNLS HSV1-TK genes were amplified using PCR from the plasmid pCMV-mNLSHSV1-TK:dMODC and pCMV-mNLSHSV1-TK, respectively. The primers used (5′-CGGGATCCTACAGCTGTTAAAGAAGGAGG-3′, 5′-ACGCGTCGCCTTTTGCTAGCGAATTCTCA-3′, 5′-CGGGATCCTACAGCTGTTAAAGAAGGAGG-3′, and 5′-ACGCGTCGCCTTCCAAAGAATTCTCA-3′) contain the restriction enzyme sites of BamHI and SalI for insertion into the pRevTRE vector (Clontech). The mNLSHSV1-TK:dMODC and mNLSHSV1-TK genes were then inserted into the response retroviral vector pRevTRE under the control of the minimal immediate early promoter of cytomegalovirus (PminCMV). These vectors were termed pRevTRE-mNLSHSV1-TK:dMODC and pRevTRE-mNLSHSV1-TK, respectively.
Determination of Protein Localization and Sensitivity of mNLSHSV1-TK in Cells
We investigated whether the HSV1-TK–targeted inactivating mutations in the NLS block nuclear import of its protein and increase total cellular enzymatic activity. The constructed DNAs, pCMV-mNLSHSV1-TK:GFP and pCMV-HSV1-TK:GFP, were introduced into the NG4TL4 cells using jetPEI (Polyplus-Transfection). After 48 h, the subcellular localization of the HSV1-TK:GFP or mNLSHSV1-TK:GFP protein in transduced tumor cells was visualized by fluorescence microscopy (Nikon) using a 488-nm excitation beam and 510-nm emission filters. Furthermore, the medium was changed before the experiment, and 3H-ganciclovir (3,700 Bq/0.5 mL/well) was added to each well and incubated at 37°C for 2 h. At the end of each incubation period, the wells were washed 3 times in phosphate-buffered saline (PBS). The cells were harvested using a cell scraper, and the radioactivity in the cell pellet was measured using a Beckman LS 6500 scintillation counter.
Determination of Protein and Enzyme Stability of mNLSHSV1-TK:dMODC in Living Cells
The pCMV-mNLSHSV1-TK:dMODC and pCMV-mNLSHSV1-TK plasmids were transfected into the NG4TL4 cells using jetPEI. After 24 h, transfected cells were treated with cycloheximide (50 μg/mL; Sigma) dissolved in minimal essential medium at 0, 3, and 6 h. At each time point, cells were trypsinized, counted by hemocytometry, and concentrated by centrifugation. The supernatant was removed and then dissolved in 40 μL of PBS and stored at −80°C for Western blot analysis and HSV1-TK enzyme activity assay.
Western Blot Analysis
Cells were lysed in a homogenization buffer containing pepstatin (1.45 mM), leupeptin (2.1 mM), dithiothreitol, triethanolamine (50 mM), and ethylenediamine tetraacetic acid/ethylene glycol tetraacetic acid (0.1 mM). Total protein (5–20 mg) was loaded in Laemmli buffer onto a 7.5% polyacrylamide stacking gel and run at 40 V and then 100 V through a 7.5% separating gel using a Mini Cell (Bio-Rad). The proteins were transferred to nitrocellulose membranes (Bio-Rad no. 162-0146) for 1 h using a Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad). The membrane was then blocked in 13 PBS/0.05% polysorbate 20/5% nonfat dry milk overnight at 4°C. After 2 washes in 13 PBS/0.005% polysorbate 20, the anti-HSV1-TK monoclonal antibody (kindly provided by Dr. William Summers, Yale University) was added for 1.5 h. After another 3 washes, secondary antibody was added for 1.5 h. Using an enhanced chemiluminescence kit (Amersham Life Science no. RPN2106), the membrane was developed on Kodak film in the dark room.
HSV1-TK Enzyme Activity Assay
The HSV1-TK enzyme activity assay was based on a protocol described elsewhere (5). Briefly, the stored cells were lysed in a lysis buffer and then the cell extracts were incubated for 60 min with 8-3H-ganciclovir to determine the formation of phosphorylated 8-3H-ganciclovir. The extracts were then separated from phosphorylated 8-3H-ganciclovir using a DE-81 filter (Whatman). These DE-81 filter papers were digested by cellulase and counted in a scintillation counter (Beckman LS 6500).
Proteasome Inhibition Assay
Two vectors, pCMV-HSV1-TK:GFP and pCMV-mNLSHSV1-TK:dMODC:GFP, were transfected into NG4TL4 cells growing in microscope slide culture chambers using jetPEI. After 24 h, cells were treated with cycloheximide (50 μg/mL), with or without 40 μM MG-132. After an additional 6 h of incubation, cells were washed with PBS and fixed in 5% formaldehyde. Cells were examined and photographed under these conditions using fluorescence microscopy. Cells were also trypsinized and counted by hemocytometry, and concentrated by centrifugation. The supernatant was removed and then dissolved in 40 μL of PBS and stored at −80°C for the HSV1-TK enzyme activity assay.
In Vitro Cell Proliferation Assay
Wild-type NG4TL4, NG4TL4-HSV1-TK, NG4TL4-mNLSHSV1-TK, and NG4TL4-mNLSHSV1-TK:dMODC cells were seeded into a 96-well plate (Falcon; Becton Dickinson Labware) at 1 × 106 cells per well in minimal essential medium containing 10% fetal calf serum. The number of cells was quantified using trypan blue staining. Morphologic changes were also observed by microscopy.
Tetracycline-Turnoff–Expressing mNLSHSV1-TK:dMODC and mNLSHSV1-TK Cell Lines
The procedures for establishing a tetracycline-turnoff–expressing cell line are detailed in our previous report (5). Briefly, the NG4TL4 cells infected with pRevtet-off virus were separately subjected to the infectious supernatant containing pRevTRE-mNLSHSV1-TK:dMODC virus, pRevTRE-mNLSHSV1-TK, and pRevTRE-HSV1-TK virus, and then the cells were selected with 600 μg of hygromycin per milliliter to establish a tetracycline-turnoff-mNLSHSV1-TK:dMODC–expressing cell line (NG4TL4/tet-off-mNLSHSV1-TK:dMODC), a tetracycline-turnoff-mNLSHSV1-TK–expressing cell line (NG4TL4/tet-off-mNLSHSV1-TK), and a tetracycline-turnoff HSV1-TK–expressing cell line (NG4TL4/tet-off-HSV1-TK), respectively.
Comparison of Doxycycline-Mediated Induction of mNLSHSV1-TK:dMODC and HSV1-TK
One × 106 NG4TL4/tet-off-mNLSHSV1-TK:dMODC cells, NG4TL4/tet-off-mNLSHSV1-TK cells, and NG4TL4/tet-off-HSV1-TK cells were seeded separately in 10-cm Petri dishes and incubated for 12 h. Cells were then treated with a series of concentrations of doxycycline (0, 0.01, 0.1, 1, and 10 ng/mL) dissolved in minimal essential medium at 0, 6, 12, 24, 48, and 72 h. At each time point, cells were trypsinized and concentrated by centrifugation. The supernatant was removed, dissolved in 40 μL of PBS, and stored at −80°C for in vitro assay of HSV1-TK enzyme activity and for quantitative PCR.
Quantitative PCR
Total RNA from the cells was obtained using the RNeasy Mini Kit (Qiagen Inc.) according to the manufacturer's protocol and reverse-transcribed with Omniscript RT (Qiagen) using random hexamers (Applied Biosystems). Quantitative PCR was performed in an Opticon 2 Monitor (MJ Research) and SYBR Green I dye (Applied Biosystems). The specific primers for β-actin were 5′-TCA AGA TCA TTG CTC CTC CTG AGC-3′ (forward) and 5′-TAC TCC TGC TTG CTG ATC CAC ATC-3′ (reverse), and the specific primers for HSV1-TKHSV-TK were 5′-ACA AAA AGC CAC GGA AGT CC-3′ (forward) and 5′-AGT TGC GTG GTG GTG GTT T-3′ (reverse). The amplification conditions were 1 cycle of 2 min at 50°C and 1 cycle of 10 min at 95°C, followed by 35 cycles of 15 s at 95°C, 30 s at 60°C, and 30 s at 72°C.
Dynamic Gene Expression Study
One × 106 NG4TL4/tet-off-mNLSHSV1-TK:dMODC cells, NG4TL4/tet-off-mNLSHSV1-TK cells, and NG4TL4/tet-off-HSV1-TK cells were seeded separately in 10-cm Petri dishes and incubated for 12 h to attach to the dishes. These cells were then monitored for HSV1-TK activity at a series of observation times (0, 3, 6, 12, 24, 48, and 72 h). During these times, the cells were treated with 10 ng of doxycycline per milliliter at 0 and 24 h to turn off the expression of the reporter genes, and then the medium was changed to remove doxycycline at 12 and 48 h. With this approach, the dynamic transcriptional events could be conducted by adding and removing the doxycycline. The mNLSHSV1-TK:dMODC, mNLSHSV1-TK, and HSV1-TK activity at each time point was determined using the same methods as described above.
Imaging of mNLSHSV1-TK:dMODC, mNLSHSV1-TK, and HSV1-TK Reporter Gene Expression Mediated by Tetracycline-Turnoff–Expressing System In Vivo
One × 105 of the NG4TL4/tet-off-mNLSHSV1-TK:dMODC cells, NG4TL4/tet-off-mNLSHSV1-TK cells, NG4TL4/tet-off-HSV1-TK cells, and NG4TL4 control cells were implanted, with each cell line grown as a separate tumor at 3 different subcutaneous sites in Friend virus B-type (FVB/N) female mice. After 2 wk, when tumor sizes varied between 0.5 and 1.0 cm, animals were injected with 9.25 × 106 Bq of 18F-9-(4-fluoro-3-hydroxy-methyl-butyl)guanine (FHBG) and imaged on a small-animal PET scanner (microPET; Concorde Microsystems). After the PET scanning, the mice were given 3 mL of doxycycline (500 g/mL) intraperitoneally, and doxycycline (500 g/mL) was added to the cage water supply for 24 h. The mice were scanned again with 18F-FHBG at 24 h after administration of the doxycycline. The animal experiments were approved by the Laboratory Animal Care Panel of the National Yang-Ming University.
Statistical Analysis
All values are shown as mean ± SD. Statistical analyses were performed using an unpaired Student t test to evaluate the significance of differences in values between 2 treated groups. A 2-tailed value of P less than 0.05 was considered significant.
RESULTS
mNLSHSV1-TK Protein Localization and Sensitivity in Cells
Fluorescent microscopic analysis of transfected NG4TL4 cells demonstrated distinct differences in subcellular localization of native and NLS mutant HSV1-TK:GFP fusion proteins. The wild-type HSV1-TK:GFP localized predominantly to the cell nucleus (Fig. 1A). The mNLSHSV1-TK:GFP exhibited pancellular distribution with no visible differences between the nucleus and cytoplasm (Fig. 1B). On the other hand, the result from the 3H-ganciclovir cellular uptake assay showed that the mNLSHSV1-TK:GFP–transfected cells had a greater accumulation of 3H-ganciclovir than did the HSV1-TK:GFP–transfected cells (P < 0.001). Furthermore, compared with mNLSHSV1-TK:GFP–transfected cells (P < 0.0001) and HSV1-TK:GFP–transfected cells (P < 0.0001), the control NG4TL4 cells had no significant accumulation of 3H-ganciclovir (Fig. 1C). The transduction efficiency with the reporter constructs was about 56%, as determined by fluorescence-activated cell sorting.
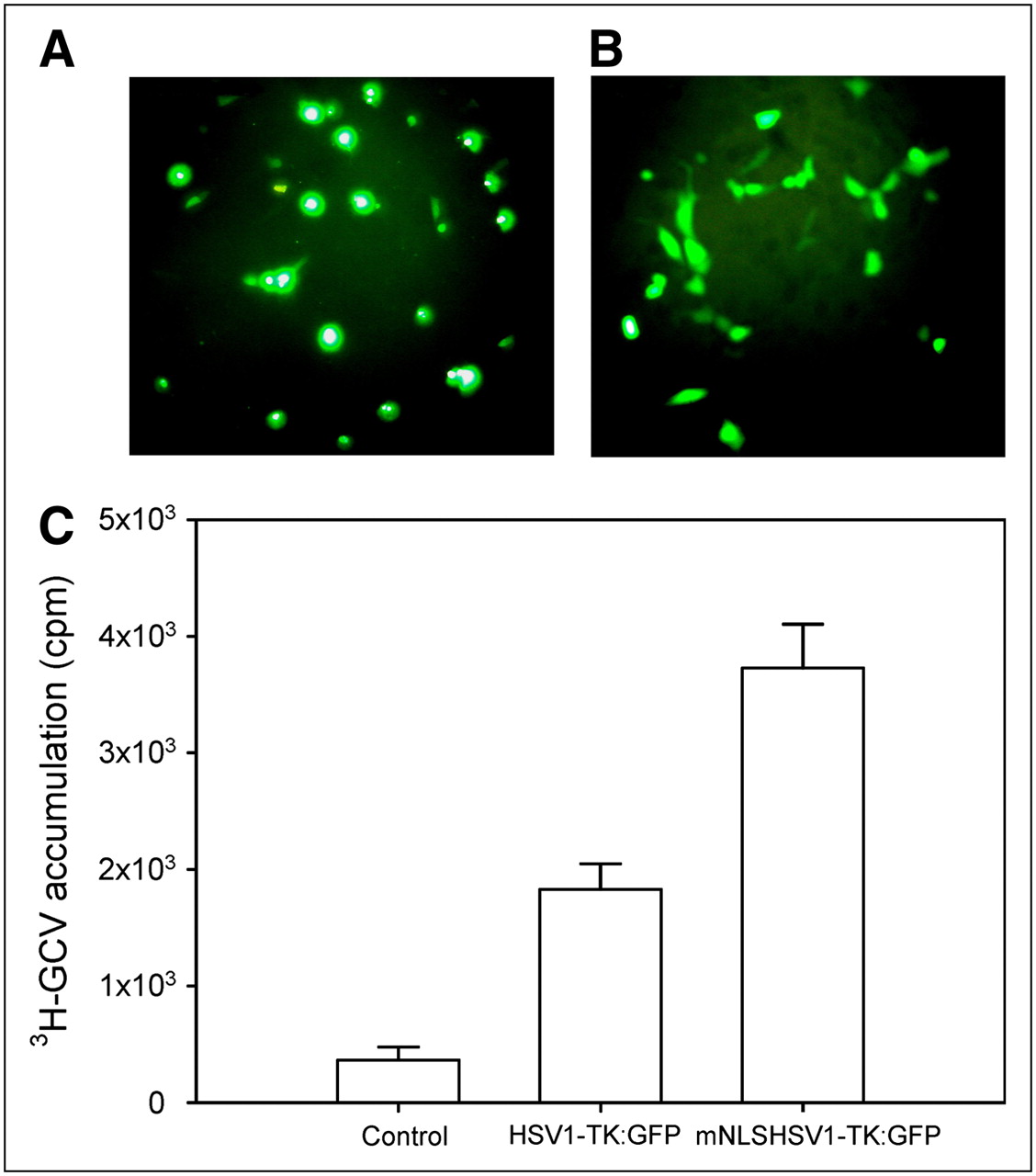
(A) Fluorescent photomicrographs of NG4TL4 cells expressing native HSV1-TK:GFP. (B) Fluorescent photomicrographs of NG4TL4 cells expressing mNLSHSV1-TK:GFP with mutations in N-terminal NLS. (C) 8-3H-ganciclovir accumulation in NG4TL4 control cells, HSV1-TK:GFP–transfected NG4TL4 cells, and mNLSHSV1-TK:GFP–transfected NG4TL4 cells. Bars represent mean ± SD of 3 independent experiments.
mNLSHSV1-TK:dMODC Protein and Enzyme Stability in Cells
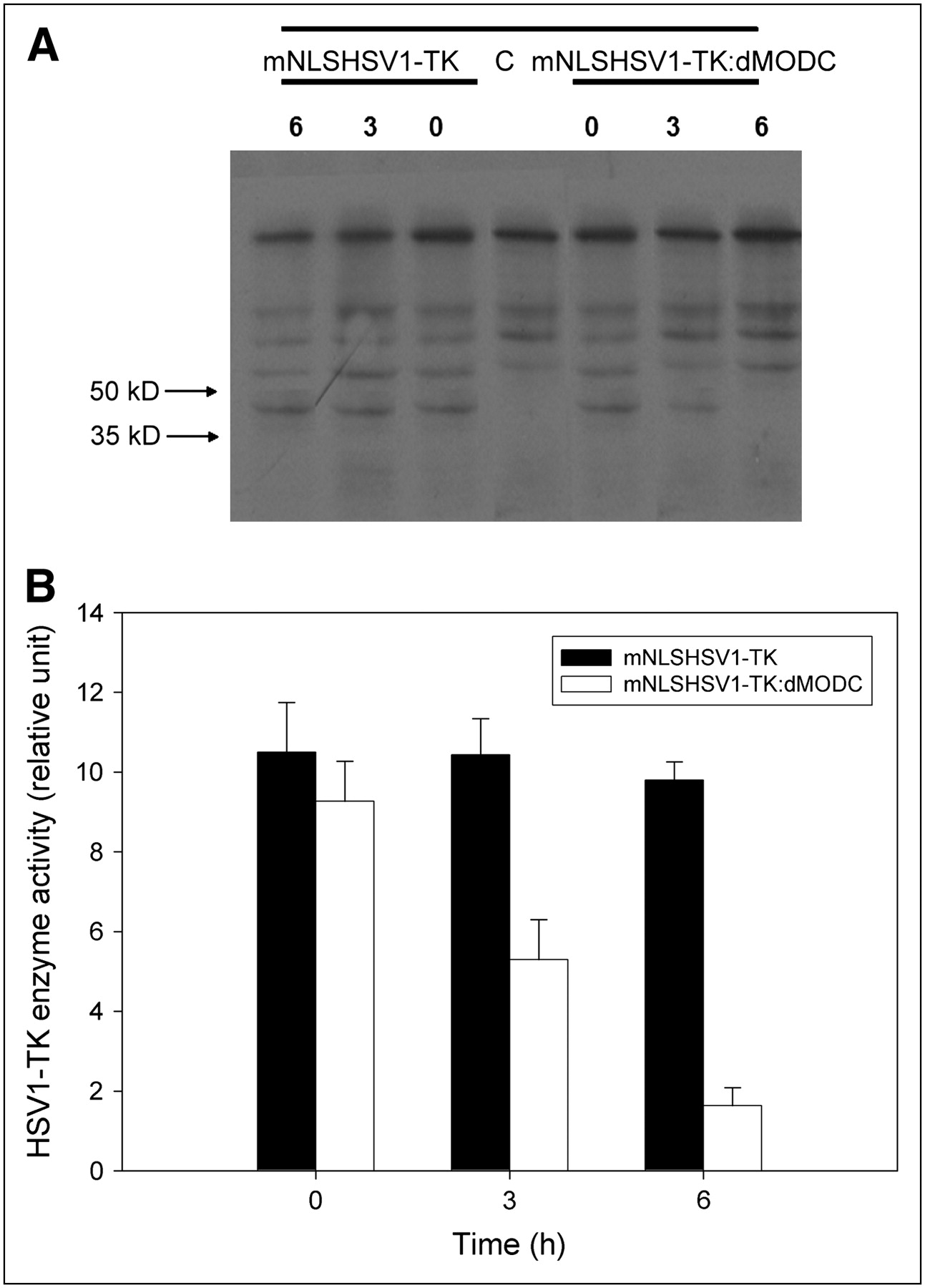
A specific band of TK proteins in the Western blot analysis was located at 35–55 kDa (Fig. 2A). No detectable change in the level of TK was found between mNLSHSV1-TK–transfected cells and mNLSHSV1-TK:dMODC–transfected cells at 0 h of cycloheximide treatment. However, the mNLSHSV1-TK:dMODC fusion protein was unstable, as determined by a measured decline after cycloheximide treatment. About half the mNLSHSV1-TK:dMODC fusion protein was degraded within 3 h of cycloheximide treatment, and no protein level was determined at 6 h of cycloheximide treatment. In contrast to mNLSHSV1-TK:dMODC, mNLSHSV1-TK was still stable during the observation period. Therefore, we concluded that the half-life of the destabilized HSV1-TK was approximately 3 h. Furthermore, similar results were found in the enzyme activity of mNLSHSV1-TK and mNLSdHSV1-TK:dMODC in NG4TL4 cells with cycloheximide treatment using the HSV1-TK enzyme activity assay (Fig. 2B).
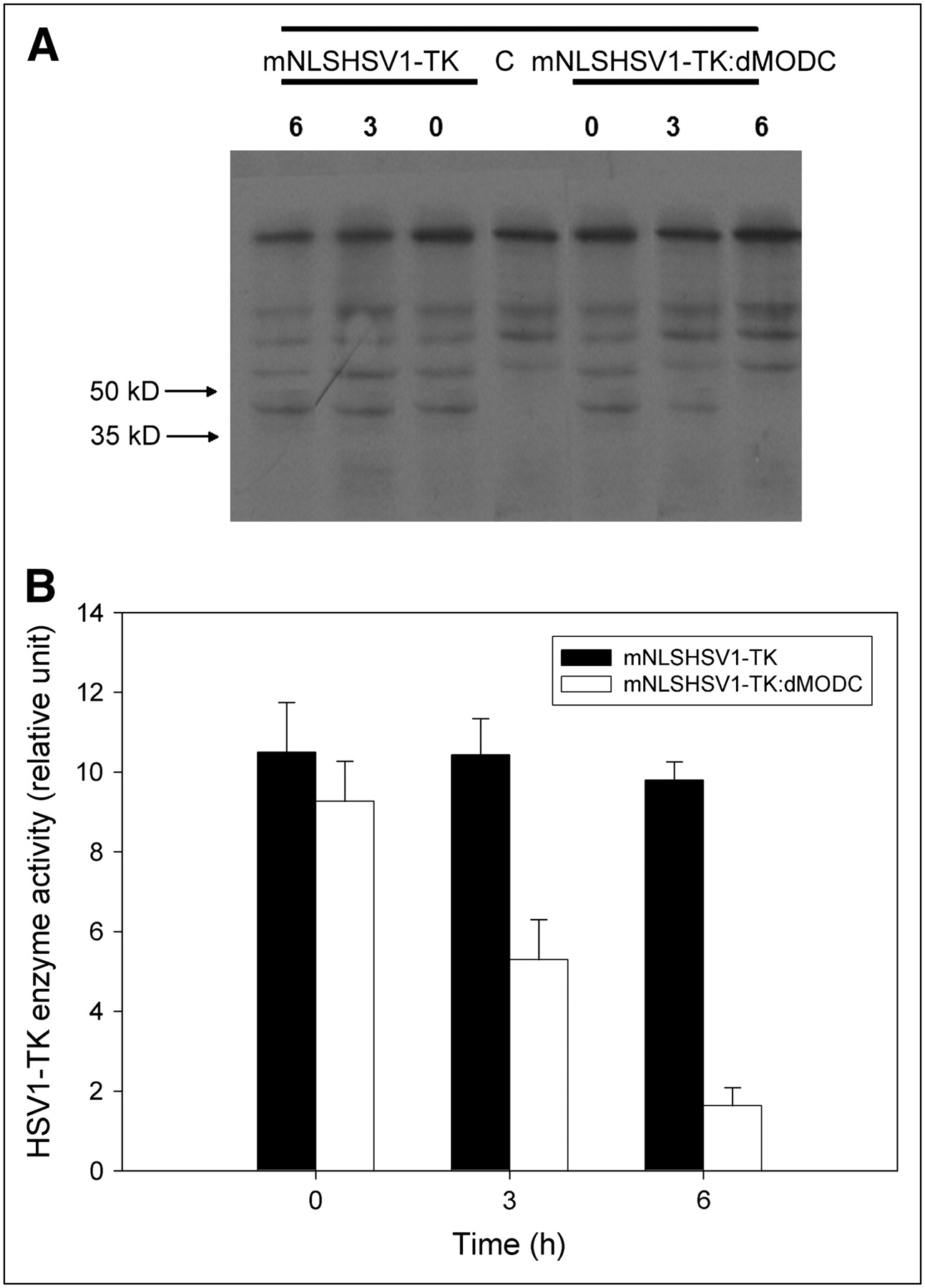
(A) Western blot analysis of protein stabilities of mNLSHSV1-TK and mNLSHSV1-TK:dMODC in living NG4TL4 cells, with or without treatment with 0.1 mg of cycloheximide per milliliter. Cell lysates were subjected to sodium dodecyl sulfate (SDS) gel electrophoresis and transferred onto membrane. mNLSHSV1-TK and mNLSHSV1-TK:dMODC were detected with monoclonal antibody against TK. (B) Stability of mNLSHSV1-TK and mNLSHSV1-TK:dMODC in living NG4TL4 cells, with or without treatment with 0.1 mg of cycloheximide per milliliter. HSV1-TK activity of lysed cells was determined by in vitro HSV1-TK enzyme activity assay. Bars represent mean ± SD of 3 independent experiments.
mNLSHSV1-TK:dMODC Protein Degradation in Living Cells
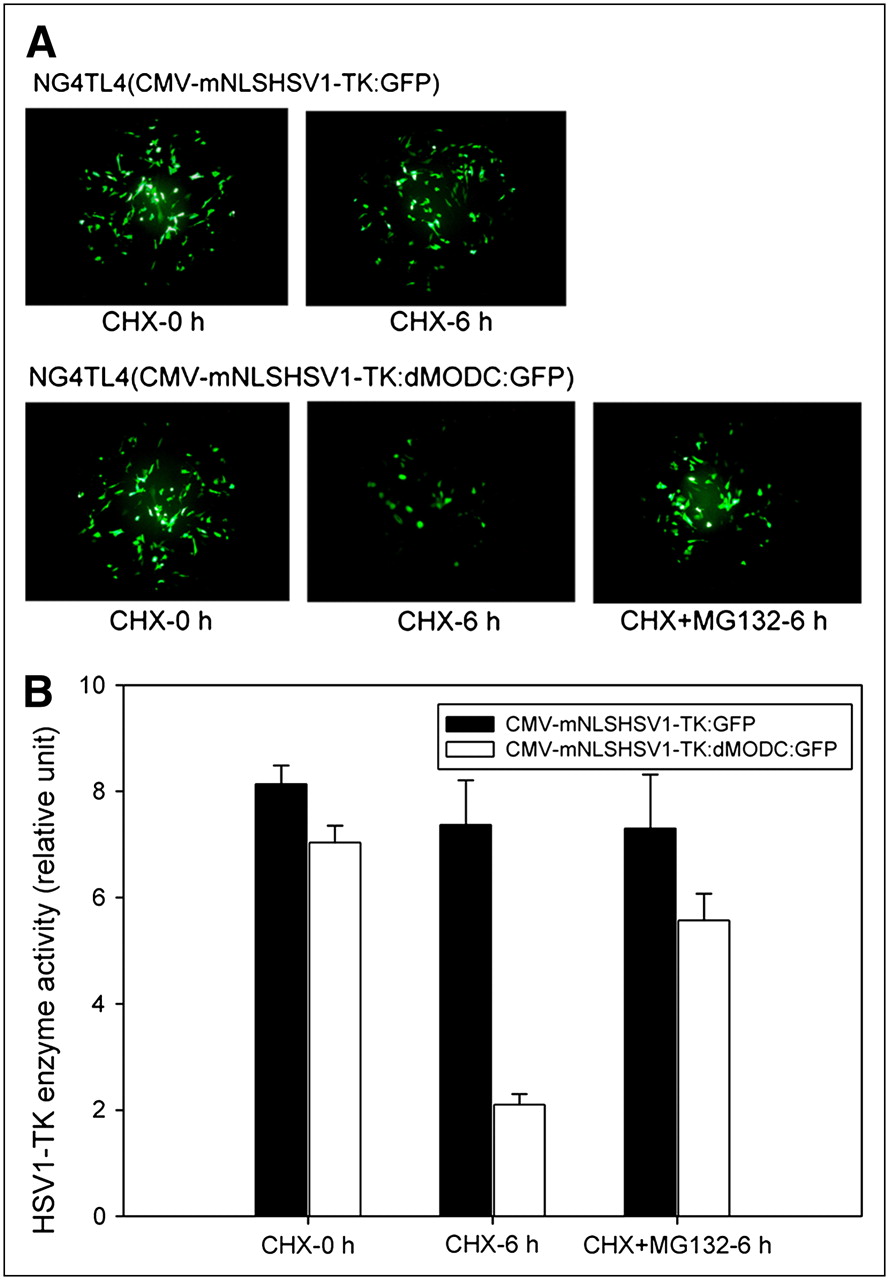
The results from the proteasome inhibition assay showed that no significant decay of fluorescence intensity was found in NG4TL4 cells transfected with pCMV-mNLSHSV1-TK:GFP, with or without cycloheximide (Fig. 3A). In contrast, NG4TL4 cells transfected with pCMV-mNLSHSV1-TK:dMODC:GFP had a significant decay of fluorescence intensity after cycloheximide treatment, and this decay could be inhibited using the proteasome inhibitor MG-132. Furthermore, the HSV1-TK enzyme activity assay demonstrated that no significant decay of TK enzyme activity was obtained in NG4TL4 cells transfected with pCMV-HSV1-TK:GFP, with or without cycloheximide (P = 0.52) (Fig. 3B). However, NG4TL4 cells transfected with pCMV-mNLSHSV1-TK:dMODC:GFP had a significant decay of TK enzyme activity after cycloheximide treatment (P < 0.0001), and this effect could be inhibited by the addition of MG-132 (P < 0.001) as well.

(A) Fluorescence stabilities of HSV1-TK:GFP and mNLSHSV1-TK:dMODC:GFP in presence of cycloheximide, and with or without MG-132 (26S proteasome inhibitor), were examined with fluorescence microscope. (B) TK enzyme stabilities of HSV1-TK:GFP and mNLSHSV1-TK:dMODC:GFP in presence of cycloheximide, and with or without MG-132 (26S proteasome inhibitor), were measured by HSV1-TK enzyme activity assay. NG4TL4 cells were transfected with vectors expressing these 2 proteins. After 24 h, transfected cells were treated with 100 mg of cycloheximide per milliliter for 6 h, with or without 40 μM MG-132. Fluorescence stabilities and TK enzyme stability of these fusion proteins were examined with fluorescence microscope and HSV1-TK enzyme activity assay, respectively.
Toxicity of HSV1-TK and mNLSHSV1-TK:dMODC in Transduced Cells
There was no significant difference in growth rate between NG4TL4 control cells and mNLSHSV1-TK:dMODC–transfected NG4TL4 cells, as determined by a comparison of their cell growth curves (Fig. 4). However, growth inhibition was observed in HSV1-TK– and mNLSHSV1-TK–transfected NG4TL4 cells, compared with NG4TL4 control cells and mNLSHSV1-TK:dMODC–transfected NG4TL4 cells.
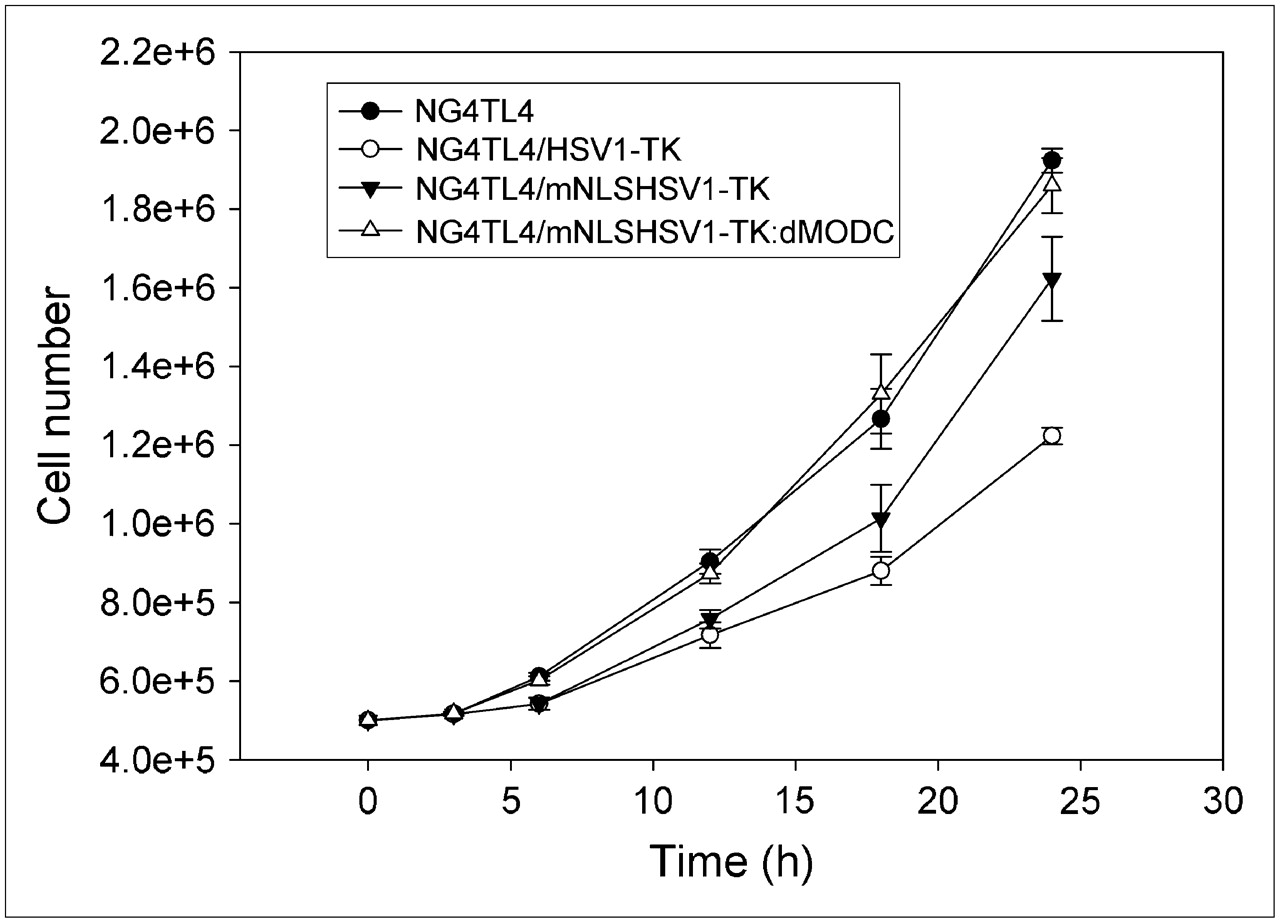
In vitro cell growth of NG4TL4 control cells, HSV1-TK–transfected NG4TL4 cells, mNLSHSV1-TK–transfected NG4TL4 cells, and mNLSHSV1-TK:dMODC–transfected NG4TL4 cells. Cell growth was analyzed using trypan blue staining assay. Data points represent mean ± SD of 3 independent experiments.
Tetracycline-Turnoff–Expressing mNLSHSV1-TK:dMODC and HSV1-TK
The results from quantitative PCR showed that all messenger RNA levels in all HSV1-TK, mNLSHSV1-TK, and mNLSHSV1-TK:dMODC showed a dose-dependent tendency at 6 h after treatment with doxycycline (Fig. 5A). The enzyme activity of mNLSHSV1-TK:dMODC also showed a dose-dependent tendency at 6 h after treatment with doxycycline (Fig. 5B). However, this effect was not observed in the enzyme activity of HSV1-TK or mNLSHSV1-TK after treating with doxycycline in the tetracycline-turnoff–expressing cell lines. The results suggested that mNLSHSV1-TK:dMODC as a reporter is more sensitive and quicker than native HSV1-TK or mNLSHSV1-TK for demonstration of the transcription intensities triggered by upstream transcription events.

(A) Doxycycline-mediated inhibition of HSV1-TK messenger RNA for NG4TL4/tet-off-HSV1-TK cells, NG4TL4/tet-off-mNLSHSV1-TK cells, and NG4TL4/tet-off-mNLSHSV1-TK:dMODC cells. (B) Doxycycline-mediated inhibition of HSV1-TK enzyme activity for NG4TL4/tet-off-HSV1-TK cells, NG4TL4/tet-off-mNLSHSV1-TK cells, and NG4TL4/tet-off-mNLSHSV1-TK:dMODC cells. Stably transformed cell lines were treated with 0, 0.01, 0.1, 1, and 10 ng of doxycycline per milliliter. After doxycycline treatment, changes in messenger RNA levels and enzyme activity of HSV1-TK were assayed at various time points using real-time PCR and in vitro assay for HSV1-TK enzyme activity, respectively. Bars represent mean ± SD of 3 independent experiments.
Doxycycline-Mediated Dynamic Gene Expression of HSV1-TK and mNLSHSV1-TK:dMODC
In our manually regulated gene expression model, the short-time-scale gene expression events can be mimicked through a series of doxycycline inductions. The mNLSHSV1-TK:dMODC as a reporter was able to demonstrate exactly the kinetic gene expression triggered by a series of doxycycline inductions (Fig. 6). However, there were no significant changes in enzyme activity for HSV1-TK or mNLSHSV1-TK.

Dynamic gene expression of HSV1-TK, mNLSHSV1-TK, or mNLSHSV1-TK:dMODC mediated by series of doxycycline inductions. HSV1-TK, mNLSHSV1-TK, and mNLSHSV1-TK:dMODC gene expression of NG4TL4/tet-off-HSV1-TK cells, NG4TL4/tet-off-mNLSHSV1-TK, and NG4TL4/tet-off-mNLSHSV1-TK:dMODC cell lines mediated by series of doxycycline inductions were monitored using in vitro assay for HSV1-TK enzyme activity. Open and closed arrows indicate changes in medium with or without doxycycline, respectively. Data points represent mean ± SD of 3 independent experiments.
mNLSHSV1-TK:dMODC and HSV1-TK Reporter Gene Expression Mediated by Tetracycline-Turnoff–Expressing System In Vivo
PET studies were performed on mice bearing multiple subcutaneous tumors grown from transduced NG4TL4 cell populations expressing native HSV1-TK protein or different mutant HSV1-TK proteins, such as mNLSHSV1-TK and mNLSHSV1-TK:dMODC. These tumors are identified as outlined in Figure 7. The nontransduced NG4TL4 tumor (wild-type) served as a negative control. Highly specific localization of 18F-FHBG–derived radioactivity in tumors expressing native (TK) and mutant HSV1-TK proteins (mNLSTK and dTK) was seen before doxycycline treatment (Fig. 7). The levels of 18F-FHBG radioactivity in nontransduced NG4TL4 tumors (wild-type) were low and similar to body background. Furthermore, higher levels of 18F-FHBG accumulation were observed in tumors expressing mNLSHSV1-TK (mNLSTK) (1.745 ± 0.083 %ID/g) than in tumors expressing native HSV1-TK (TK) (0.867 ± 0.053 %ID/g) or mNLS-HSV1-TK:dMODC (dTK) (0.745 ± 0.043 %ID/g) before doxycycline treatment (Table 1). After 24 h of doxycycline induction, the accumulation of 18F-FHBG in the tumors expressing mNLSHSV1-TK:dMODC (0.063 ± 0.024 %ID/g) had been almost fully suppressed. However, a remnant accumulation of 18F-FHBG was found in tumors expressing native HSV1-TK (TK) (0.683 ± 0.118 %ID/g) and tumors expressing mNLS-HSV1-TK:dMODC (dTK) (1.234 ± 0.112 %ID/g), indicating that the previous enzyme of HSV1-TK or mNLSHSV1-TK cannot be turned over in such a short observation time when its transcription has been suppressed by doxycycline.

Sequential PET studies of tumors growing in FVB/N female mice. NG4TL4/tet-off-mNLSHSV1-TK:dMODC cells (dTK), G4TL4/tet-off-mNLSHSV1-TK cells (mNLSTK), NG4TL4/tet-off-HSV1-TK cells (TK), or NG4TL4 control cells (wild-type [WT]) were injected subcutaneously into 3 separate sites in single mice. When tumors reached at least 5 mm, mouse was imaged with 18F-FHBG on sequential days. Doxycycline (500 μg/mL) was administered intraperitoneally and also added to cage water supply for 1 d. Mouse was scanned with 18F-FHBG again 24 h after exposure to doxycycline.
Quantitative Data from Sequential PET Studies of Tumors Growing in FVB/N Female Mice
DISCUSSION
In this research, we first targeted inactivating mutations in the NLS located in the N terminus of HSV1-TK, which suppresses the nuclear import of its protein. Our results indicate the HSV1-TK–targeted inactivating mutations in the NLS suppress nuclear import of its protein and further increase the total cellular enzymatic activity. These results also correspond to the published results of other research groups (25,26). In mammalian cells, most intracellular proteins are degraded in the cytoplasm (27,28). Therefore, we fused the mNLSHSV1-TK with the PEST domain from MODC to improve its high protein stability in cells. Our results demonstrate that mNLSHSV1-TK:dMODC decreases its protein and enzyme stability in living cells. However, the protein and enzyme stability of mNLSHSV1-TK was still stable even if it was located in the cytoplasm during the observation period. In the past, the PEST domain in protein degradation has been supported by mutational transfer of the PEST domain from MODC to metabolically stable proteins (11). An attachment of the PEST degradation signal of MODC (amino acids 422–461) to the carboxyl-COOH-terminal region of the EGFP reporter gene decreased the half-life of the protein 6-fold in mammalian cells (29). In this study, our results prove that mNLSHSV1-TK became attached to the PEST domain of MODC at the C terminus, mNLSHSV1-TK:dMODC, and decreased its protein and enzyme stability in living cells. Its protein or enzyme half-life was estimated to be about 2–3 h. The availability of this short-lived HSV1-TK is essential for use in a more responsive reporter system.
To test the hypothesis that rapid turnover of mNLSHSV1-TK:dMODC is also brought about by the 26S proteasome, one can apply the well-established fact that protease inhibitors directed toward the catalytic function of the proteasome can be used to monitor proteasome function (30). The inhibitor MG-132 has frequently been used to turn off the proteasome and allow targeted proteins to accumulate in the cell (31). In this study, we used a similar approach together with optical imaging and an HSV1-TK enzyme activity assay to test this hypothesis. Our results demonstrated that NG4TL4 cells transfected with pCMV-mNLSHSV1-TK:dMODC:GFP had significant decay of TK enzyme activity and fluorescence intensity after cycloheximide treatment, and this effect could be also inhibited by the addition of MG-132. These results indicate that the protein degradation of destabilized HSV1-TK, mNLSHSV1-TK:dMODC, is processed in a manner dependent of the 26S proteasome.
The wild-type HSV1-TK has shown that it has cell toxicity on its own in gene therapy or in transgenic mice because of the nuclear tropism and high stability of its protein (6–8). Our destabilized HSV1-TK (mNLSHSV1-TK:dMODC) has improved the nuclear tropism and high protein stability of wild-type HSV1-TK. Therefore, the destabilized HSV1-TK may be low in cytotoxicity when it is highly expressed in living cells because of the prevention of a high accumulation of HSV1-TK protein in cell nuclei. To test this hypothesis, we determined and compared the cell growth of NG4TL4 control cells, HSV1-TK–transfected NG4TL4 cells, mNLSHSV1-TK–transfected NG4TL4 cells, and mNLSHSV1-TK:dMODC–transfected NG4TL4 cells using the trypan blue staining assay. Our results showed that the destabilized HSV1-TK has low cytotoxicity when it is highly expressed in living cells. On the other hand, our results from the doxycycline-mediated dynamic gene expression studies in vitro and in vivo indicated that destabilized HSV1-TK has better temporal resolution than does native HSV1-TK. Destabilized HSV1-TK poses significant advantages for dynamic studies of short-time-scale gene expression events and is suitable for detecting dynamic changes in the rate of gene expression that might evolve within a few hours, such as in circadian rhythm studies or signal transduction research.
In vivo bioluminescence reporter imaging is suitable for monitoring transcription because of its relatively fast induction and the considerably short biologic half-life of both luciferin and luciferase (32,33). However, this advantage over the longer-lived EGFP has recently been mitigated by the development of short-lived, rapidly degradable variants of EGFP (29). These short-lived variants have been used for higher-temporal-resolution imaging of EGFP chimeric proteins within cells. Recently, this destabilized EGFP was also applied to the real-time monitoring of the signal transduction activity of hypoxia-inducible factor 1 in tumor xenografts (34). Although the luciferase- and destabilized EGFP–based optical imaging systems have high temporal resolution to monitor short-time-scale gene expression events or other biochemical processes in vivo, they still are not applicable for fully 3-dimensional tomography to monitor or quantify gene expression intensity for transgenes or endogenous genes. In addition, the penetration of emitting light photons limits these impinging modalities in large-animal imaging or clinical use (35). In contrast to optical imaging systems, PET has higher sensitivity and better signal penetration. Furthermore, gene expression intensity can be quantified in 3-dimensional mode. Most important, the ability to perform translational research from a cell culture setting to preclinical animal models to clinical applications is a unique and powerful feature of PET reporter-gene imaging (36). Therefore, we suggest that our destabilized HSV1-TK as a reporter together with a molecular probe, such as 11C-2′-fluoro-2′-deoxyarabinofuranosyl-5-ethyluracil, which has a short biologic and physical half-life, can increase temporal resolution in PET reporter systems and is suitable for monitoring short-time-scale dynamic processes, such as kinetic gene expression controlled by inducible promoters or by a less stable protein with a more rapid turnover.
CONCLUSION
The HSV1-TK–targeted inactivating mutations in the NLS located in the N terminus and fused with the degradation domain of MODC at the C-terminal end improve its high protein stability and cell toxicity in living mammalian cells. The rapid turnover of this destabilized HSV1-TK is processed in a 26S proteasome–dependent manner. Its use as a transcription reporter allows direct monitoring of transcription induction and easier monitoring of its coincidence detection with other biochemical changes in living animals using small-animal PET imaging.
Acknowledgments
This study was supported by grants NSC95-3112-B-010-004, NSC94-3112-B010-011, NSC93-2623-C-010-003, and NSC91-3112-P-075-001-Y from the National Science Council, Taipei, Taiwan. We thank the Molecular-Genetic Imaging Core/NRPGM, Taiwan, for technical support. We thank Professor Jolyon H. Hendry and Tsuey-Ling Jan for editorial assistance.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication December 20, 2006.
- Accepted for publication September 29, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}