


Abstract
Two characteristics of α-particles that enhance their potential for targeted radiotherapy are their high energy and approximately cellular range. Unfortunately, these properties also can have negative consequences, confounding the production of clinically relevant levels of radiopharmaceutical because of radiolytic effects. The purpose of this study was to evaluate the effect of radiation dose on the astatine species present before initiation of a labeling reaction and the potential role of these molecules in the efficiency of N-succinimidyl 3-211At-astatobenzoate (SAB) synthesis. The ranges of radiation dose evaluated were selected to reflect those that might be encountered in SAB synthesis for the preparation of clinical doses of 211At-labeled radiopharmaceuticals. Methods: The distribution of astatine species present in methanol, and the yields for the synthesis of SAB from N-succinimidyl 3-(tri-n-butylstannyl)benzoate as a function of radiation dose, were determined by high-performance liquid chromatography. Radiation doses in the range of 500–12,000 Gy were evaluated using different 211At time–activity combinations, and the effect of acetic acid, a normal component of astatodestannylation reactions, also was studied. Finally, the effect of the reducing agent sodium sulfite also was evaluated to characterize the nature of the species produced by radiolysis. Results: At radiation doses below 1,000 Gy, high-performance liquid chromatography analysis indicated that more than 90% of the 211At was present in methanol as a single species, At(1), whereas at higher doses, a second peak, At(2), emerged. At(1) decreased and At(2) increased in a radiation dose–dependent fashion, with At(2) becoming the predominant species at about 3,000 Gy. At(2) was identified as a reduced form of astatine, presumably astatide, which could not be efficiently oxidized to a species suitable for electrophilic astatination. In methanol/acetic acid, more than 95% of the astatine was present as At(2) even at doses below 1,400 Gy. Conclusion: The emergence of a reduced form of astatine, At(2), at higher radiation doses is consistent with the decline in SAB yields observed under these conditions. Alteration of the chemical form of the astatine by radiolysis could account for the declining yields noted in the preparation of clinical-level 211At-labeled radiopharmaceuticals and when the labeling chemistry is initiated hours after 211At production.
Targeted α-particle therapy is an appealing approach for therapeutic molecular medicine because the pathlength of α-particles approximates cellular dimensions. This offers the attractive possibility of selectively eliminating malignant cell populations by exploiting their expression, either native or induced, of specific molecular targets. The potential for minimizing collateral damage to surrounding tissues coupled with high cytotoxicity are additional characteristics of α-particles that have provided a further rationale for the development of targeted α-particle therapy. Translational efforts in this arena currently include the targeting of tumor vasculature, blood-borne tumors, and compartmentally spread cancers and the sterilization of residual tumor tissue after surgery (1–4).
Much of this work has focused on 211At, a radiohalogen with a 7.2-h half-life, because this radionuclide has many attractive features for targeted radiotherapy (5). These include the association of α-particle emission with 100% of 211At decays, no long-lived α-particle–emitting daughters, a half-life compatible with a variety of molecular carriers, and a potentially diverse chemistry. These features have led to the investigation of a wide variety of 211At-labeled radiotherapeutics, including peptides, proteins, thymidine, and metaiodobenzylguanidine analogs, as well as other small molecules (6–11). However, because of the scarcity of cyclotrons equipped with 25- to 30-MeV α-particle beams—a requirement for efficient 211At production—radionuclide availability is a major impediment to the clinical translation of these promising radiopharmaceuticals.
The fact that 211At-labeled therapeutics would by necessity generally be used at locations distant from a 211At production site presents a major challenge to the chemist because, under these circumstances, the astatine activity frequently arrives in a form that results in unacceptable labeling yields. The diminishing efficiency of electrophilic astatination reactions with the passage of time is well known, even when 211At is used at its site of production. These difficulties likely relate to the cumulative effect of 211At α-particles depositing large amounts of decay energy in a highly localized manner. The radiolytic consequences of α-particle decay also can interfere with the preparation of clinically relevant levels of 211At-labeled radiopharmaceuticals such as N-succinimidyl 3-211At-astatobenzoate (SAB), which is used for labeling monoclonal antibodies (mAbs) (12).
In previous publications, we have investigated the effects of increasing α-particle radiation dose on the tin precursor required for SAB synthesis (13) and the nature of the labeled products that were generated (14). The goal of the present study was to evaluate the effect of radiation dose on the astatine species present before initiation of the labeling reaction and the potential role of these molecules on labeling efficiency, using SAB as the model compound. Astatine is the heaviest of the halogens and has the lowest ionization energy of the group (920 kJ/mol). The element exists in multiple oxidation states—At−, At+, At+3 ( ), and At+5 (
), and At+5 ( )—and has an electronic configuration that makes it very reactive (15). For these reasons, we hypothesized that the chemical nature of 211At can be modified by interaction with highly reactive, radiolytically generated species coming from the solvent. Because previous work showed that methanol is a better solvent than benzene or chloroform for electrophilic astatination of organometallic derivatives at elevated radiation dose levels (13,14), all experiments described herein used methanol as the solvent.
)—and has an electronic configuration that makes it very reactive (15). For these reasons, we hypothesized that the chemical nature of 211At can be modified by interaction with highly reactive, radiolytically generated species coming from the solvent. Because previous work showed that methanol is a better solvent than benzene or chloroform for electrophilic astatination of organometallic derivatives at elevated radiation dose levels (13,14), all experiments described herein used methanol as the solvent.
MATERIALS AND METHODS
General
Methanol was obtained from Aldrich and was of anhydrous 99.8% grade (Sure/Seal bottles [Sigma-Aldrich] with <0.002% water and <0.0003% evaporation residue). All other solvents were of reagent grade or better and were used as purchased. The Na131I was obtained from Nordion Canada (NEZ035H; 65 GBq/mL; specific activity, 679 MBq/mg). The high-performance liquid chromatography (HPLC) analyses were performed on a System Gold HPLC system (Beckman) equipped with a diode array detector and a radioisotope detector. For reversed-phase chromatography, an Xterra (Waters) 4.6 × 250 mm (10-μm) column was used. The elution for reverse-phase chromatography was made with a gradient of solvent B (acetonitrile/acetic acid 0.1%) in solvent A (acetonitrile/water/acetic acid [5/95/0.1]) held at 28% of B for 15 min, followed by a 28%-to-100% linear gradient of B over 20 min, followed by 100% solvent B for the remainder of the HPLC run. The flow rate was 1 mL/min.
Electrophoresis was measured using a PowerPac Basic power supply (Biorad) run at 250 V and 50 mA for 15 min. Grade 1 glass fiber strips (1 × 20 cm; Whatman) were run in 0.05 M phosphate buffer, pH 7. The strips were analyzed immediately after the run using an imaging scanner (model 200; Bioscan).
SAB synthesis experiments were performed by astatodestannylation of N-succinimidyl 3-(tri-n-butylstannyl)benzoate (BuSTB), which was prepared as described previously (16,17). The purity of the tin precursor was confirmed before each set of experiments by thin-layer chromatography.
Production of 211At and Radiation Dose Calculations
The 211At was produced at the Duke University Medical Center cyclotron and an internal target system (18) by bombarding natural bismuth metal targets with 28.0-MeV α-particles using the 209Bi(α, 2n)211At reaction. The dry distillation system for isolation of the 211At from the bismuth cyclotron target was a variation of a system described previously (19,20). The current distillation device uses a polyetheretherketone (PEEK) capillary loop (1.6-mm [1/16-in] tubing, 1-mm inner diameter × 152-cm [5-ft] length) obtained from Upchurch Scientific instead of the glass vials that were used in the past (12) for trapping the 211At activity. One reason for this change was to delay the time at which the 211At first was exposed to the solvent. In addition, this change permitted a more accurate calculation of the radiation dose received by the solvent. During the distillation, the PEEK tubing was chilled using an ethanol dry-ice bath. A gas wash-bottle trap containing a reducing agent was placed downstream but before the vacuum pump, to retain any astatine that might escape from the PEEK tubing trap. Other aspects of the distillation procedure—the rest of the device, furnace, distillation temperature, quartz still, and argon flow—were similar to those used in the past (12). At the end of the distillation, the tubing was removed from the apparatus, and both ends were covered with all-purpose laboratory film (Parafilm; Alcan Inc.) and allowed to warm to room temperature. The astatine captured in the PEEK tubing was eluted just before each experiment into a small volume of methanol, normally in the range of 300–750 μL. The activity, time, and volume were recorded to calculate the radiation dose received by the solvent.
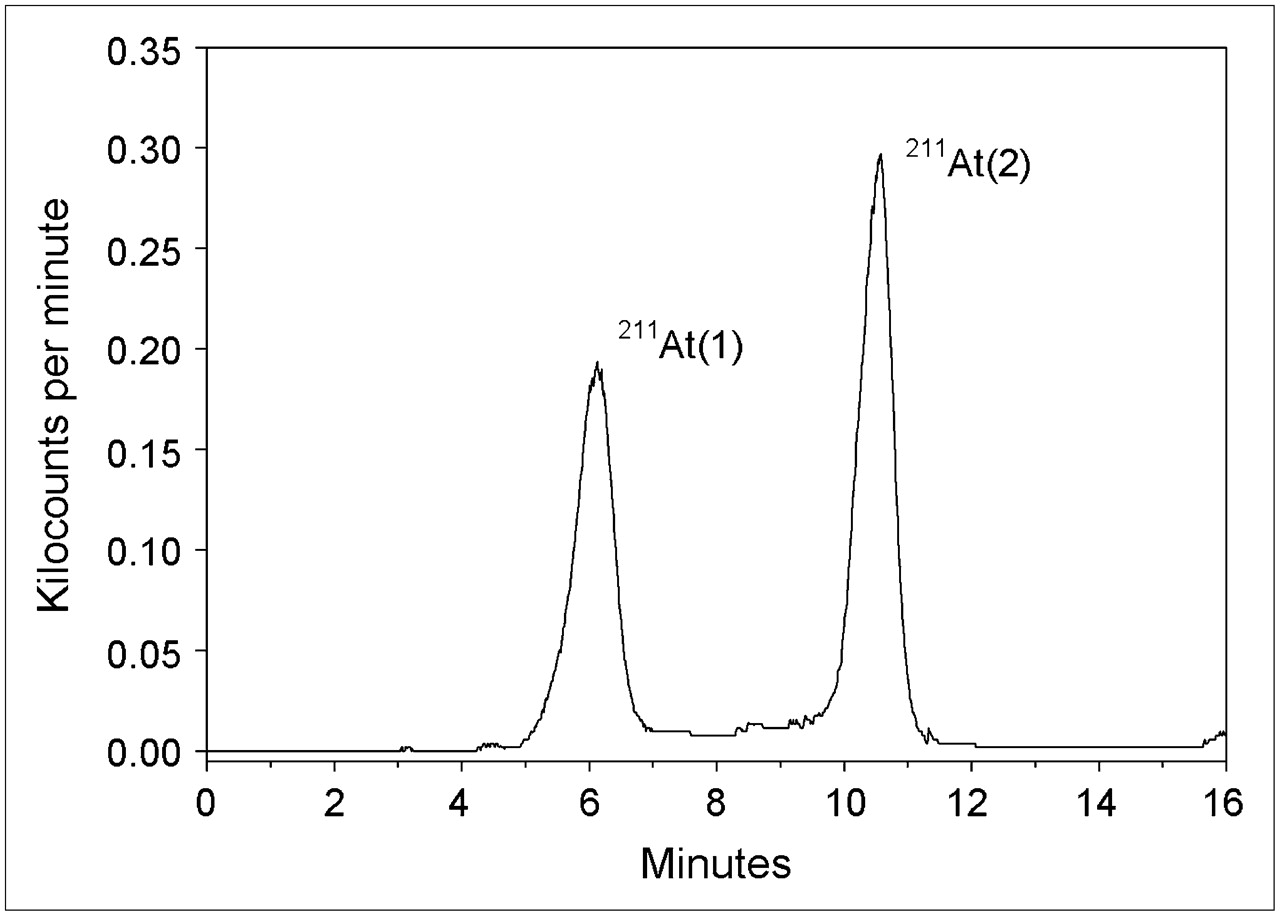
For the radiation dose calculation, all α-particle and α-recoil nuclei decay energy was assumed to be deposited in the solution because of the short range of these emissions (<100 μm) relative to the dimensions of the reaction mixtures. Uniform distribution of the reactants in the solvent also was assumed. The absorbed dose was calculated as (12):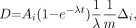 where D is expressed in grays, Ai is the initial activity in megabecquerels, λ is the decay constant for 211At (s−1), t is the exposure time in seconds, m is the mass of the solution (g), and Δi is the mean energy emitted per nuclear transition. Based on dose contributions from α-particles and α-recoil nuclei, a Δi of 1.09 × 10−3 Gy·g/MBq·s was calculated for 211At decay (21). A density of 0.791 g/mL was used to convert methanol volume to mass.
where D is expressed in grays, Ai is the initial activity in megabecquerels, λ is the decay constant for 211At (s−1), t is the exposure time in seconds, m is the mass of the solution (g), and Δi is the mean energy emitted per nuclear transition. Based on dose contributions from α-particles and α-recoil nuclei, a Δi of 1.09 × 10−3 Gy·g/MBq·s was calculated for 211At decay (21). A density of 0.791 g/mL was used to convert methanol volume to mass.
Radiolysis Experiments and SAB Synthesis
To study the influence of pH, we performed the experiments both with methanol alone and with methanol adjusted to an acidic pH (5.0–5.5) with acetic acid (0.67 mol/L), the conditions normally used for SAB synthesis (12). All the experiments were conducted under atmospheric conditions and at room temperature.
The radiation dose ranges evaluated herein were selected to reflect those that might be encountered in labeling SAB for the preparation of clinical doses of 211At-labeled radiopharmaceuticals. In a previous study, difficulties were encountered in reproducible SAB preparation and efficient mAb conjugation when 370 MBq of 211At-labeled mAb were required (12). Based on previously determined yields for SAB synthesis and mAb conjugation, and the times required for synthesis and purification, greater than 1,500 MBq of initial 211At activity would be needed to prepare this activity level of labeled mAb. At the volume used for SAB synthesis, a dose of 4,700 Gy would be delivered to the methanol reaction mixture (13). For this reason, experiments were performed under conditions resulting in the delivery of radiation doses both above and below this 4,700-Gy benchmark.
The activity levels of 211At used in each run ranged between about 40 and 160 MBq and were measured using a CRC-7 dose calibrator (Capintec). Samples for HPLC analysis were taken at various times that were selected on the basis of the approximate radiation dose levels of interest in each experiment. The radiation dose deposited in the reaction vessel ranged between about 500 and 12,000 Gy, with exposure periods ranging between minutes and about 24 h. The dose rates were kept below 1 Gy/s to facilitate comparison with previous experiments from our database. Samples were injected onto the HPLC column immediately after they were obtained. The areas of all astatinated species were integrated, and the results were expressed as the percentage of total activity eluted from the column and were plotted as a function of radiation dose (Gy).
When the synthesis of SAB was investigated, it was prepared by adding 50 μg of BuSTB in 50 μL of methanol, 100 μg of N-chlorosuccinimide (NCS) dissolved in 50 μL of methanol, and 20 μL of acetic acid to 0.4 mL of 211At/methanol in a reaction vial (Reacti-vial; Pierce), unless different conditions were specified. The reaction mixture was shaken for 20 min, and 50-μL aliquots were periodically removed for HPLC analysis. To study the effect of pH, parallel reactions were performed on the same day, with and without acetic acid at the same final volume. For the experiment directed at attempting to characterize the chemical form of 211At/methanol under high-level radiation dose conditions, a 30-μL aliquot of 211At/methanol was combined with 30 μL of sodium sulfite (2 μmol). Sodium sulfite is a reducing agent used routinely for the generation of astatide (15) and was allowed to react for 30 min. HPLC chromatograms were obtained for aliquots of the 211At in methanol, both before and after the addition of the sodium sulfite. Approximately equal levels of radioactivity were injected onto the HPLC column by adjusting the volume of the 2 aliquots that were analyzed.
RESULTS
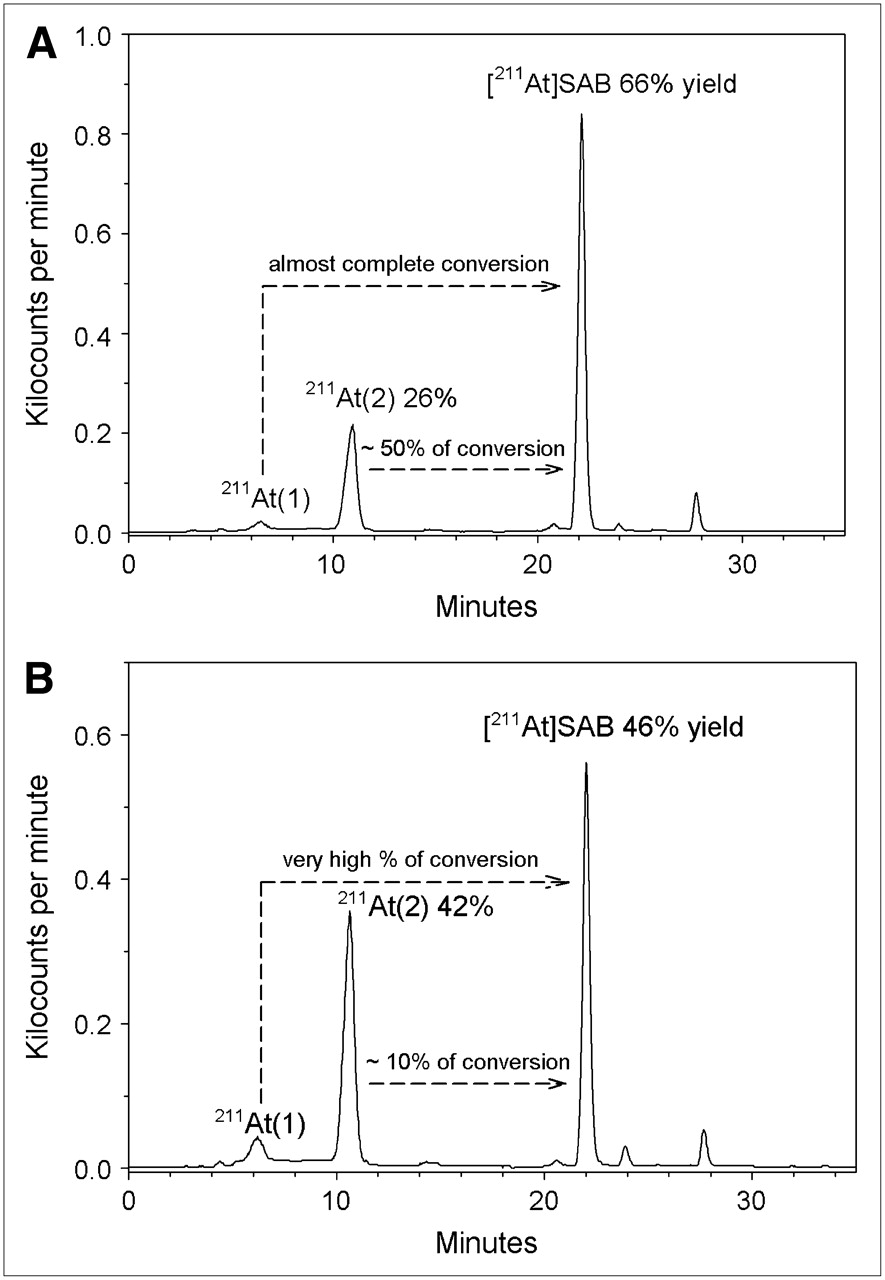
The effect of radiation dose on the chemical form of astatine was evaluated using HPLC. In a typical experiment, 211At was eluted into methanol (58 MBq/750 μL) and stored for 21.5 h at room temperature, resulting in a radiation dose of 3,630 Gy (Fig. 1). HPLC analysis indicated that essentially all 211At activity that eluted from the column (77%–89% of the total activity added to the column for all HPLC runs presented in this study) was found in 2 peaks eluting at 6.2 and 10.7 min, designated hereafter as At(1) and At(2), respectively. The first peak, observed at 6.2 min, corresponded to that observed in freshly prepared 211At/methanol solutions and those exposed to doses of less than 1,500 Gy.

Representative reverse-phase HPLC profile of radioactive species present in methanol solution after exposure to 211At. In this example, radiation dose received by solution was 3,630 Gy.
The effect of radiation dose on 211At/methanol solutions was further evaluated by taking aliquots at successive periods, yielding increasing calculated radiation doses. The results for an experiment performed with an initial activity concentration of 43.5 MBq/540 μL are shown in Figure 2. The distribution of eluted 211At activity between the 2 HPLC peaks clearly was dependent on the radiation dose received by the methanol, but not in a linear fashion. The contribution of At(2) increased gradually until about 2,200 Gy and then increased more rapidly, becoming the predominant species at radiation doses above about 3,200 Gy. Similar behavior, but with a slower increase of At(2) contribution, was observed in other experiments performed at higher dose rates, and experiments are currently in progress to explore potential dose rate effects in more detail.

Relative amounts of At(1) and At(2), as shown in Figure 1, in methanol (without acetic acid) as function of radiation dose absorbed by solvent.
Because acetic acid is a standard component in electrophilic astatination reactions, the 211At species generated in 211At/methanol solutions containing acetic acid (0.67 mol/L) was evaluated. At all doses investigated (1,000–20,000 Gy), at least 95% of eluted 211At activity was found in the At(2) form. Thus, at low radiation doses, astatine is present in different forms in methanol with or without acetic acid, whereas at higher radiation doses, At(2) predominates in both cases.
Based on the fact that methanol radiolysis is known to generate reducing species, particularly at an acidic pH (14), these observations suggested that At(2) is a reduced form of astatine, most likely astatide. To investigate this possibility further, 211At/methanol (160.6 MBq/460 μL) was treated with sodium sulfite. A small aliquot with a low level of 211At activity was used to minimize radiation dose deposition during treatment with the reducing agent. HPLC analysis was performed on an aliquot of 211At in methanol obtained before and 30 min after sodium sulfite addition. Treatment with sulfite decreased the fraction of eluted activity in the At(1) peak from 46% of eluted activity to less than 1%, whereas the contribution from the At(2) peak increased from 52% to 90%. Addition of the reducing agent resulted in almost complete conversion of At(1) to At(2) in 5 other paired experiments performed at different initial activity concentrations, providing further evidence that At(2) represents a reduced form of astatine. Furthermore, analysis of Na131I-iodide/methanol solutions under the same HPLC conditions showed that iodide eluted as a single peak at 10.2 min, a retention time quite similar to that observed for At(2).
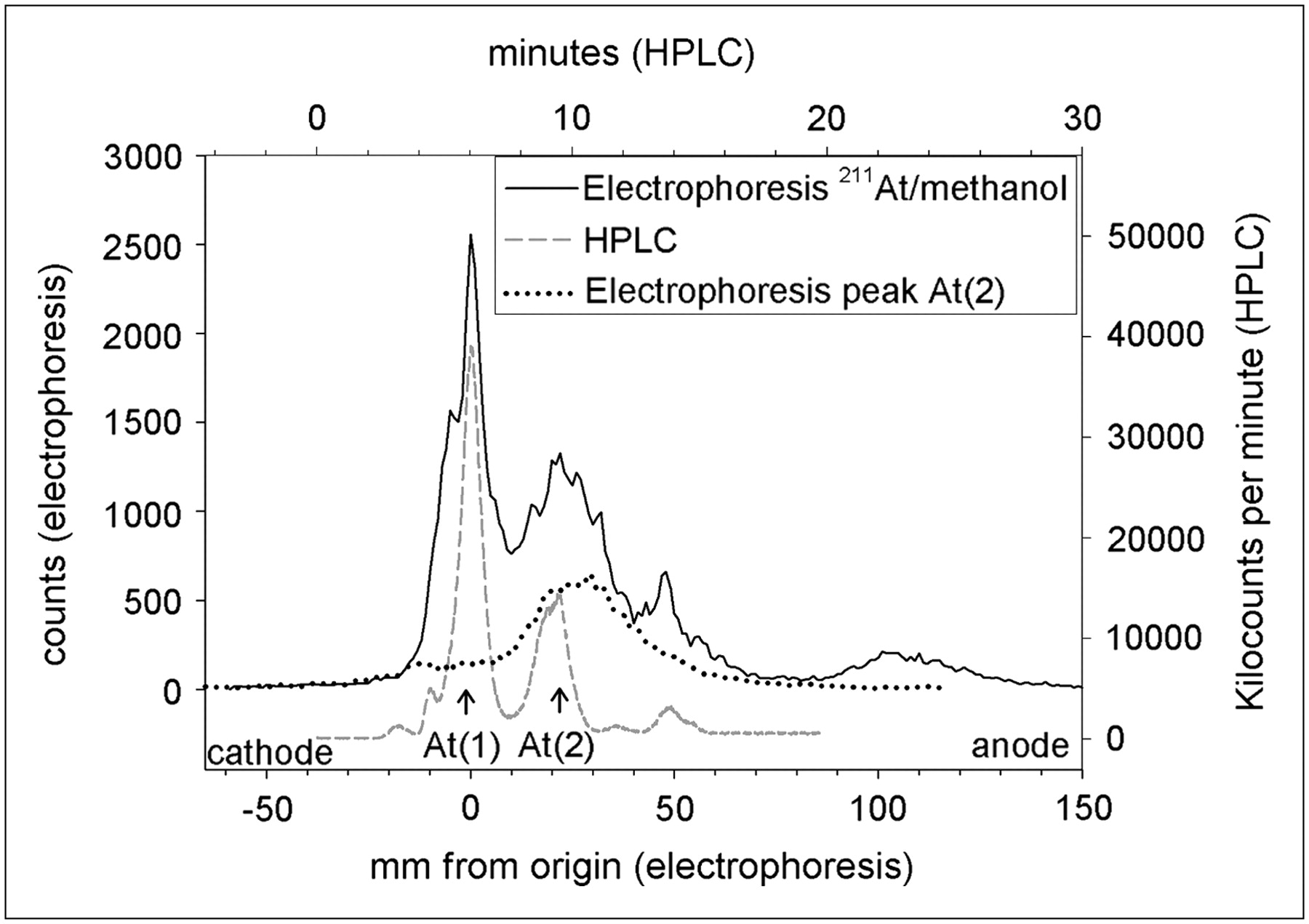
To better understand the chemical nature of the 2 species observed in the HPLC chromatograms, we analyzed 211At/methanol solutions by electrophoresis. Figure 3 shows a direct comparison of the HPLC and electrophoresis profiles obtained from the same methanol solution after exposure to 211At. Two peaks with similar relative magnitudes are observed with both techniques. In the electrophoresis profile, the larger peak is found at the origin, indicating that it is neutral, whereas the smaller peak migrates toward the anode, indicating that it is negatively charged. A sample was obtained from the At(2) peak, isolated by HPLC, and its electrophoretic behavior was evaluated. A single species migrating toward the anode was observed, confirming that At(2) is a negatively charged species.
Analysis of same 211At in methanol solution by reverse-phase HPLC (dashed line) and paper electrophoresis (solid line). Electrophoretic analysis of sample of At(2) peak observed on HPLC chromatogram also was performed (dotted line).
The peak corresponding to At(1) predominates in fresh 211At/methanol solutions at low radiation doses, conditions under which electrophilic astatination reactions give high reaction yields. The emergence of a reduced form of astatine, At(2), at higher radiation doses could account for the decline in SAB yields under these conditions. However, a potential compensatory strategy is also suggested: treatment of At(2) with an oxidant such as NCS to generate a reactive electrophilic astatine species.
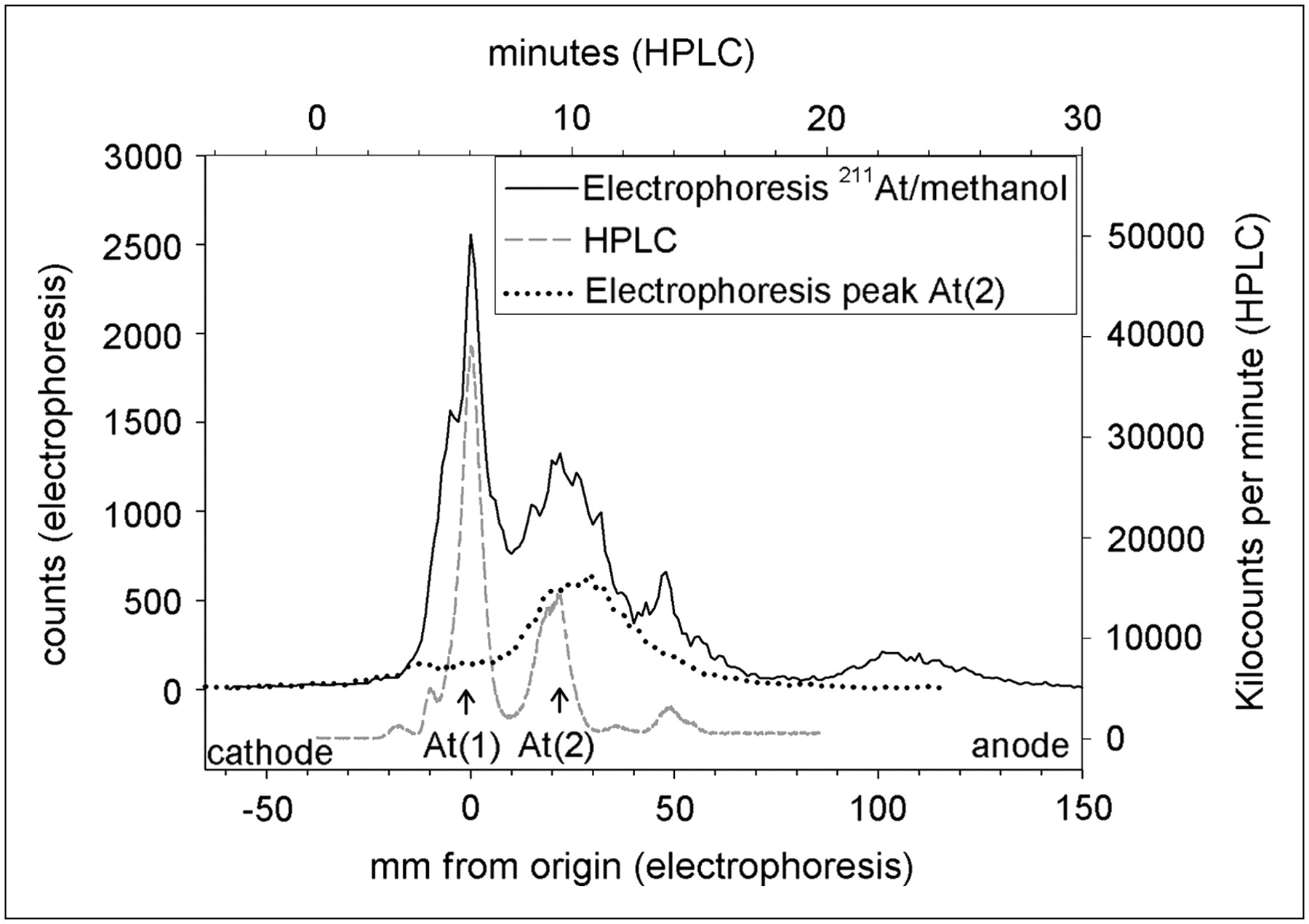
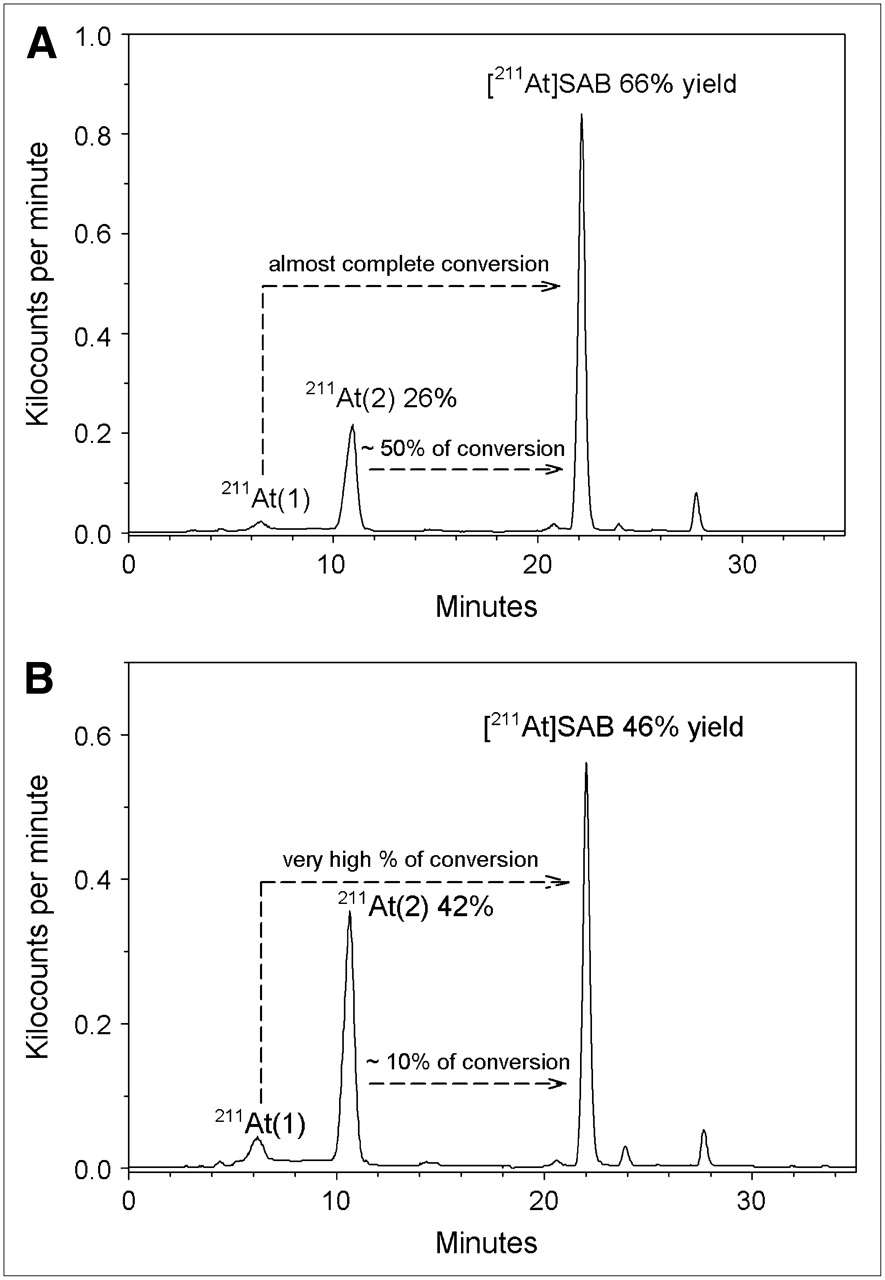
To investigate this possibility, we evaluated the synthesis of SAB from a 211At/methanol solution in which 46% and 52% of the activity were present as At(1) and At(2), respectively, determined by HPLC, at the initiation of SAB synthesis. The 211At and methanol (2 aliquots of 200 μL each) were added to 2 vials containing 80 μg of BuSTB and 200 μg of NCS in 100 μL of methanol; 1 reaction mixture also contained acetic acid (0.67 mol/L). The almost complete disappearance of the At(1) peak with concomitant SAB production suggests that that this astatine species is reactive toward electrophilic astatination (Fig. 4). In contrast, about half the At(2) generated by radiolysis before the SAB reaction was not oxidized by NCS to a species suitable for electrophilic astatination (Fig. 4A). Although a greater fraction of At(2) could be converted to SAB under acidic than under neutral reaction conditions (∼50% vs. ∼10%) by adding NCS to the reaction mixture, these results suggest that this strategy is not efficient for converting At(2) to the desired product.
Effect of presence (A) or absence (B) of acetic acid (0.67 mol/L) on synthesis of SAB from 211At/methanol solutions at radiation dose resulting in presence of At(1) and At(2). Arrows indicate percentage conversion of At(1) and At(2) to SAB, calculated by comparison to levels of At(1) and At(2) present at initiation of reaction and determined by HPLC.
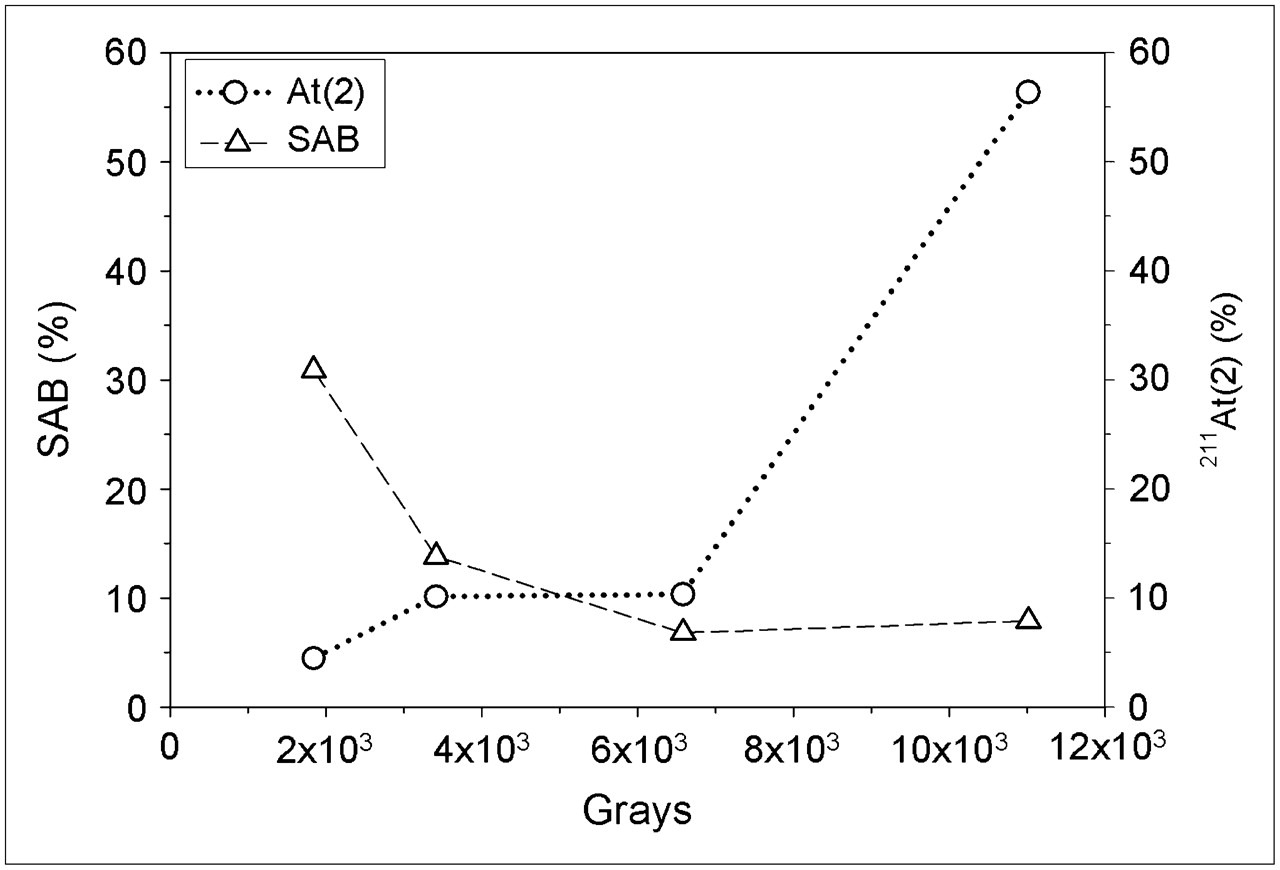
Next, 211At/methanol was reacted with BuSTB at 4 different radiation doses at a neutral pH without the addition of NCS to determine whether SAB yield could be related to At(2) fraction. These conditions were selected because both NCS and acetic acid could alter the chemical forms of astatine that are present. Previously (14), we observed that SAB could be generated in the absence of NCS; however, this radiolysis-induced SAB decreased with increasing radiation dose and was significantly higher without acetic acid. As shown in Fig. 5, an inverse relationship existed between SAB yield and the fraction of 211At/methanol present as At(2) at the initiation of the reaction. This finding further supports the hypothesis that, for SAB synthesis, At(2) is less reactive than At(1).

Production of SAB from reaction of BuSTB and 211At in methanol by radiolysis (i.e., no oxidant or acetic acid added), compared with percentage of 211At(2) present as function of radiation dose.
We next investigated the effect of acetic acid on SAB production in the presence of NCS in paired reactions using 211At/methanol solutions that had received a relatively low radiation dose (∼2,000 Gy) to minimize radiolysis-mediated production of the less reactive At(2) species. Under these conditions, SAB yields were essentially identical in the presence (73%) or absence (74%) of acetic acid. Thus, when the radiation dose received before initiation of the labeling reaction in methanol was low, running the reaction with NCS at a lower pH did not increase yields. On the other hand, the opposite behavior was observed at high radiation doses (Fig. 4), presumably reflecting more efficient conversion of At(2) to SAB in the presence of acetic acid. This greater efficiency is in contrast to the radiolysis-induced synthesis of SAB (i.e., in the absence of oxidant), which is less efficient in the presence of acetic acid (14).
DISCUSSION
Targeted α-particle radiotherapy is a promising approach to cancer treatment because, at least in principle, curative doses of radiation can be selectively delivered to malignant cells on the basis of their expression of oncogenically derived molecular markers. However, translation of concept to clinical practice has been slow because of many hurdles, including the need for more sophisticated dosimetric methodologies and normal-organ toxicity data, as well as limited availability of α-emitters with appropriate characteristics (2,3,22). Moreover, labeling strategies must be developed that are practical and reliable at the elevated dose levels required for patient treatment.
Such methodologies were critical to bringing 131I-tositumomab (Bexxar; GlaxoSmithKline) and 90Y-ibiritumonab tiuxetan (Zevalin; Biogen Idec, Inc.) to the marketplace; however, achieving similar success with an α-emitter such as 211At will be an even greater challenge. Compared with these β-emitters, 211At has a half-life shorter by an order of magnitude but deposits an energy per volume greater by 2 orders of magnitude than 90Y or 131I (23). Because of these differences, the potential impact of radiolytic effects on 211At labeling chemistry is considerably higher. Particularly for institutions distant from the site of 211At production, the radiation dose received before initiation of the labeling reaction will be considerable. Even when clinical-level 211At mAb labeling was performed with the radionuclide produced on site, radiation doses of up to approximately 4,000 Gy were delivered in the 5–40 min before SAB synthesis began (12).
The rapidly declining SAB yield with increasing radiation dose under conditions in which the tin precursor remained largely intact led us to hypothesize that this behavior was due to radiation-induced changes in the chemical form of astatine (14). The results of the current study are consistent with this hypothesis.
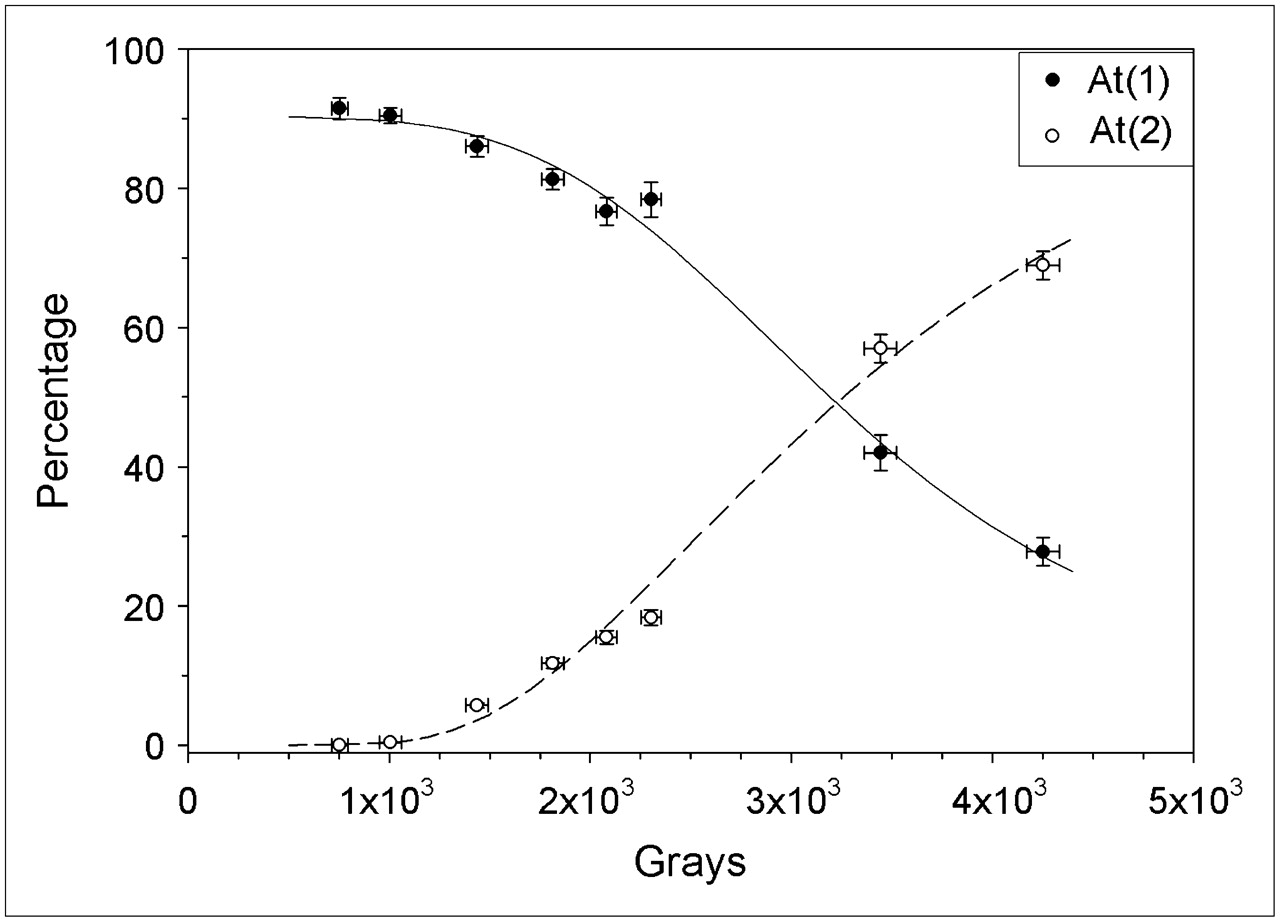
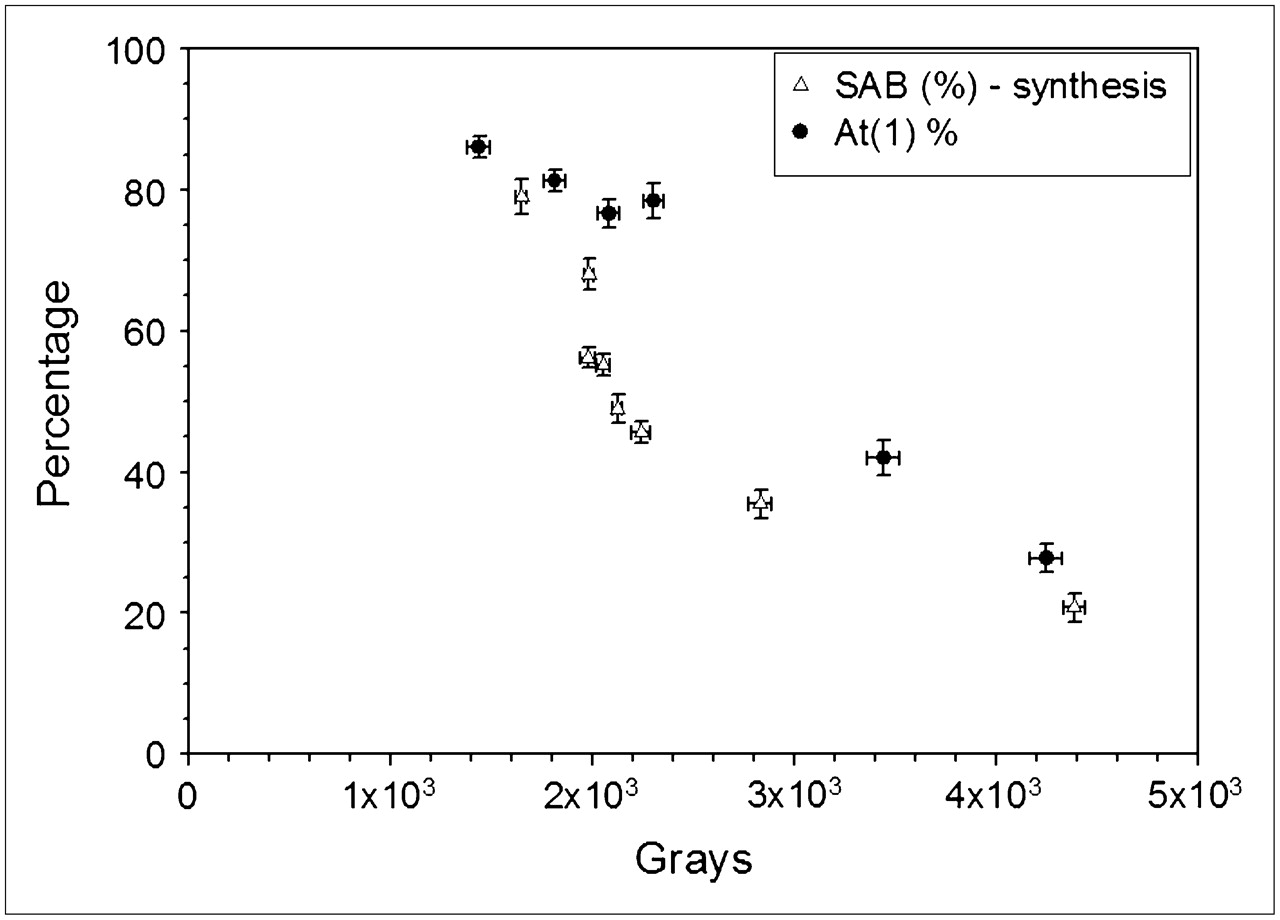
In Figure 6, the fraction of activity that would be present as At(1) at the beginning of a hypothetical reaction (Fig. 2) is compared with SAB yields determined previously (14) as a function of the radiation dose received by the 211At/methanol solution before initiation of the destannylation reaction. Although this comparison must be done with caution, it is worth noting the at least qualitatively similar decline in At(1) fraction and SAB yield as a function of radiation dose. This finding suggests that At(2) is in a chemical form from which At+, the species generally considered to be required for electrophilic astatodestannylaton, cannot be efficiently generated.

Unfortunately, definitive chemical identification of At(1) and At(2) could not be done because the lack of a stable astatine isotope limits the use of standard analytic techniques. Because iodine is the element most similar to astatine, its chemistry might shed some light on the behavior of astatine. Of potential relevance to the current work is a theoretic study of complex formation of iodine with methanol (24). These complexes are formed through acid–base types of interactions in which iodine functions as the acid or electron acceptor. Methanol contains relatively mobile protons, making acid–base types of interactions and the formation of complexes of this kind possible. If astatine also interacted with methanol in a similar fashion, the peak At(1) could be a complex of this kind. However, we cannot be certain at this time even whether this peak corresponds to a single astatine species.
In contrast to low-linear-energy transfer radiation, such as β-emitters, the dominant mechanism for α-particle–mediated radiolysis is spur overlap, a consequence of their densely ionizing nature (25,26). As the number of α-decays per unit time (i.e., dose rate) increases, the probability of intertrack overlap increases until, at very high dose rates, a nearly homogeneous environment is created (27). This decreases the yield of radicals and favors the formation of molecular products within the spur (26). An important consequence of methanol radiolysis in that regard is the generation of hydrogen and formaldehyde, which are reducing species (28). These molecules can then interact with astatine atoms that have not decayed, potentially altering their oxidation state and chemical form. Because of its carrier-free nature, when 211At is isolated from the cyclotron target in the absence of oxidant, the number of moles of reducing species produced from methanol radiolysis could exceed those of astatine by several orders of magnitude, even at relatively low radiation doses. This could lead to the generation of reduced forms of astatine, which are not suitable for electrophilic astatination. Our results demonstrating an increasing fraction of At(2) with increasing radiation dose are consistent with this hypothesis.
We hypothesize that At(2) is astatide for several reasons. First, At(1) is nearly completely converted to At(2) by treatment with the reducing agent sodium sulfite. Second, the retention time of At(2) was nearly identical to that of sodium 131I-iodide. And third, At(2) was demonstrated to be an anionic species by electrophoresis, whereas At(1) was shown to be a neutral species. It is worth noting that the yields of SAB generally are higher when astatine is present in an organic solvent, where it is presumed to be in the zero valence state (29–31). Thus, it appears that radiolysis of methanol could have 2 deleterious effects on electrophilic astatination reactions—consumption of the oxidant added to the reaction to create the electrophilic astatine species and reduction of the 211At, making generation of electrophilic astatine more difficult.
The reactions occurring during methanol radiolysis provide a rationale for the dose- and pH-dependent increase in At(2) production. In methanol, radiation renders the spur more acidic than the bulk because of the formation of CH3OH2+, and acidic pH increases the production of reducing species in methanol (28). Thus, as the radiation dose increases, the spur becomes more acidic, which, in turn, increases generation of reducing species. We speculate that the sequence “more acidic spur → more reducing species” results in increased At(2) production, which will continue increasing incrementally as the spur overlap increases. Consistent with this speculation is the observation of increased production of At(2) at lower radiation doses in the presence, compared with the absence, of acetic acid.
Alteration of the pH at a microscopic level could also account for the observation that the addition of acetic acid did not increase SAB yield when a fresh 211At/methanol solution was used. An acidic pH is well known to be required for efficient electrophilic halogenation (30). The radiation doses received during these SAB syntheses were only about 20% of those received during a clinical therapy–level SAB synthesis. We speculate that even this radiation dose might have been sufficient to create a microscopic-level acidic pH environment, obviating the addition of acetic acid to the SAB synthesis reaction mixture. Recently, the addition of acetic acid was reported to have no effect on the synthesis of analogous 211At-labeled N-succinimidyl benzoates from their corresponding tin precursors at similar activity levels (32).
CONCLUSION
211At is one of the most promising α-particle emitters for targeted radiotherapy. However, the radiolysis effects induced by these high-linear-energy particles can interfere with labeling chemistry either when performed at clinically relevant activity levels or when the reaction is initiated hours after 211At production. Previous studies using the well-known protein conjugation agent SAB revealed 2 problems: a competitive reaction of radiolysis-generated species with the tin precursor (13) and a reaction of the 211At with the solvent itself at high radiation doses (14). Our results presented here demonstrate for, what is to our knowledge, the first time another significant impediment to successful astatine chemistry: alteration of the chemical forms of 211At with increasing radiation doses from forms suitable for electrophilic labeling reactions to forms that are not. We show that in methanol (the optimal solvent identified to date for therapy-level astatodestannylation), astatine is present at low radiation doses in a form from which At+ can readily be generated, perhaps stabilized in a complex with methanol. With increasing deposition of radiation dose to the solvent, conversion to a reduced species, probably astatide, occurs, accounting for the decline in labeling yields at elevated radiation doses. More solvents need to be investigated to gain a better understanding of radiolytic effects on reactions of this type. In conclusion, the results of this study underscore the potential importance of radiolysis-mediated effects on the chemistry of α-particle–emitting radiopharmaceuticals and the need to evaluate labeling chemistry at the high radiation doses required for clinical use.
Acknowledgments
We thank Marc Hens and Kevin Alston for excellent technical assistance. This work was supported in part by grants CA42324 and NS20023 from the National Institutes of Health, grant DE-FG02-05ER63963 from the U.S. Department of Energy, and a grant from the Pediatric Brain Tumor Foundation.
Footnotes
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication November 29, 2006.
- Accepted for publication March 23, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Effective Treatment of Human Breast Carcinoma Xenografts with Single-Dose 211At-Labeled Anti-HER2 Single-Domain Antibody Fragment
- An Improved 211At-Labeled Agent for PSMA-Targeted {alpha}-Therapy
- Direct Procedure for the Production of 211At-Labeled Antibodies with an {varepsilon}-Lysyl-3-(Trimethylstannyl)Benzamide Immunoconjugate