


Abstract
This study aimed to determine the magnitude and distribution of tissue angiotensin-converting enzyme (ACE), mast-cell chymase, and angiotensin II, type 1, plasma membrane receptor (AT1R), in relation to collagen replacement in infarcted and noninfarcted left ventricular myocardial segments. A new radiotracer, 18F-fluorobenzoyl-lisinopril (FBL), was synthesized without compromising its affinity for tissue ACE. Methods: Five- to 10-μm contiguous short-axis slices of explanted hearts from 3 patients with ischemic cardiomyopathy were incubated in vitro with FBL, with and without 10−6 M lisinopril. Tissue radioactivity was recorded as a function of position in photostimulating luminescence units (PSL). Immunohistochemistry studies were performed with mouse monoclonal antibody against ACE, anti–mast cell chymase, and polyclonal antibody against the human AT1R. Results: There was specific binding of FBL to ACE; mean FBL binding was 6.6 ± 5.2 PSL/mm2, compared with 3.4 ± 2.5 PSL/mm2 in segments incubated in solution containing cold, 10−6 M lisinopril (P < 0.0001). Mean FBL binding was 6.3 ± 4.5 PSL/mm2 in infarcted, 7.6 ± 4.7 PSL/mm2 in periinfarcted, and 5.0 ± 1.0 PSL/mm2 in remote, noninfarcted (P < 0.02 vs. periinfarcted) segments. The autoradiographic observations concerning FBL binding were confirmed by ACE and AT1R immunoreactivity. Distribution of mast cell chymase differed from ACE, as a higher number of mast cells was present in the remote, noninfarcted myocardium than in the periinfarcted myocardium (5.1 ± 3.2 vs. 3.2 ± 2.2 mast cells per field, P < 0.001). The number of mast cells in ischemic hearts exceeded that in normal hearts (4.2 ± 2.7 vs. 1.5 ± 1.2 mast cells per field, ×200, P < 0.001). Conclusion: FBL binds specifically to ACE. The binding is nonuniform in infarcted, periinfarcted, and remote, noninfarcted segments, and there is apparently increased ACE activity in the juxtaposed areas of replacement fibrosis. On the other hand, the distribution of mast cell chymase appears nonuniform and disparate from ACE.
The renin–angiotensin system is an adept regulator of human physiology and is frequently activated in patients with ischemic left ventricular (LV) dysfunction and heart failure. Accumulating evidence from clinical and experimental studies indicates that the renin–angiotensin system and its primary effector peptide, angiotensin II, are linked to the pathophysiology of interstitial fibrosis, cardiac remodeling, and heart failure (1). In animal models, increased expression of angiotensin-converting enzyme (ACE) has been associated with cardiac fibrosis (2). In patients with heart failure, inhibition of the renin–angiotensin system with ACE has proven to favorably affect LV remodeling and patient outcome (3,4).
Recent data have demonstrated that renin–angiotensin and kallikrein–kinin systems can be synthesized locally in the heart (5). This emerging concept implies that, in myocytes, LV remodeling, and vascular tone, the local tissue effects of angiotensin may have more of a role than do the circulating plasma effects. The discovery of the tissue ACE system and its ability to locally produce effector hormones (autocrine effects) has encouraged us to study the potential of ACE imaging in monitoring LV remodeling (6,7). The capacity to image tissue ACE will allow the study of this important disease first in vitro using explanted human hearts and ultimately in vivo in heart failure patients.
To date, there are two 18F-radiolabeled ACE inhibitors: 18F-captopril (8) and 18F-fluorobenzoyl-lisinopril (FBL) (9). Although captopril has a higher affinity for plasma ACE, lisinopril is thought to have a higher affinity for tissue ACE. It has been known since the early development of ACE inhibitors that the normalized oral doses of most inhibitors have equivalent effects on serum ACE but differential effects on tissue, as distinguished by both their magnitude and their duration (10). This effect may be explained by the structure of 1 of the 2 catalytic domains of tissue ACE in complex with an inhibitor (11). The fact that 1 of the 2 active sites on the tissue ACE is a more lipophilic pocket would explain the differential binding in tissue. ACE found in plasma is produced by proteolytic cleavage of the membrane-bound ACE, and therefore both binding sites are equally available (12).
In this study, we determined whether FBL can bind specifically to tissue ACE in explanted human hearts in vitro. In addition, we examined histologically the magnitude and distribution of tissue ACE, chymase, and angiotensin II, to establish their relationship to matrix and replacement collagen in the infarcted and noninfarcted segments of LV myocardium. The clinical significance of this study is in the potential to monitor tissue ACE as a function of progressive heart failure by external imaging. If the finding of increased ACE is reversible—that is, with ACE inhibitors—noninvasive imaging of radiolabeled ACE would allow monitoring of both the progression of disease and the effect of medical and interventional therapies in such patients before collagen replacement ensues.
MATERIALS AND METHODS
Patient Population
The 3 subjects in this study represented a subset of 13 men with ischemic cardiomyopathy awaiting cardiac transplantation who were enrolled in a study determining the relationship of thallium uptake to myocardial fibrosis (13). All patients had severe (New York Heart Association functional class III or IV) heart failure or angina and were listed as outpatients at the time of enrollment. All had angiographically documented multivessel coronary artery disease. Patients with recent acute myocardial infarction or unstable angina were excluded from the study.
Tissue Handling and Picrosirius Red Staining for Collagen
The apical surface of the midventricular transverse section of the LV fixed in 10% phosphate-buffered formaldehyde was used for collagen staining and quantification. After the assessment of gross myocardial fibrosis, LV sections were divided into 8 equal sectors representing the ventricular septum and the anterior, lateral, and inferior walls (2 regions each). Each region was embedded in paraffin and then cut at a 5- to 10-μm thickness for staining with hematoxylin and eosin and with picrosirius red (Pfaltz and Bauer), a collagen-specific stain. Stained sections were used for providing a general roadmap for the expression of ACE, angiotensin II, type 1 plasma membrane receptor (AT1R), and chymase in relation to replacement and matrix collagen.
Radiochemistry
We have developed a 3-step method for the radiosynthesis of N-succinimidyl-4-18F-fluorobenzoate, beginning with our synthesis of 18F-fluorobenzoic acid (14). The coupling of this intermediate to lisinopril was attempted in several solvents and under different conditions of pH and temperature. These coupling reactions lacked reproducibility and were usually accompanied by significant amounts of hydrolysis of N-succinimidyl-4-18F-fluorobenzoate to 18F-fluorobenzoic acid. Reproducibility was obtained only by purification of the active ester by high-performance liquid chromatography before coupling. The product of the coupling reaction with lisinopril was also purified by high-performance liquid chromatography. The radiochemical yield averaged 15% (n = 3), uncorrected, after a synthesis time of 130 min (9).
Autoradiographic Techniques for Presence of Tissue ACE
After removal of the paraffin, the slide-mounted sections were incubated in no-carrier-added 18F-FBL, with and without a micromolar concentration of FBL, to determine nonspecific binding. After 1 h of incubation, the slide-mounted slices were removed from the solution, air dried, and placed on a phosphor-imaging plate with a pixel size of 25 μm (BAS-SR2025; Fuji). After exposure overnight (∼18 h), the plates were scanned using a Bio-imaging Analysis System 5000 (Fuji).
Immunohistochemical Staining
Following standard preparation techniques, human monoclonal anti-ACE antibody (Biotrack Inc.) and rabbit anti-AI and anti-AII antibodies (Bachem Laboratories) were used. Briefly, 0.4% pepsin in 0.01 HCl at 38°C during 7 min was used as an antigen retrieval technique. The primary antibodies were added at 1:100 dilutions and incubated overnight. As the blocking solution, 2% bovine serum albumin plus 10% normal horse serum in phosphate-buffered saline was used. A secondary antibody, avidin-biotin-peroxidase complex, and diaminobenzidine were used to develop the color (Vector Laboratories). ACE and AT1R expressions were assessed semiquantitatively on a 4-point scale (0 = none, 1+ = mild, 2+ = moderate, and 3+ = heavy in the infarcted, periinfarcted, and remote areas of the LV myocardium). This was done by systematically scanning each microscopic slide at a magnification of ×7.8 and visually grading each field for the degree of expression of ACE or AT1R. A mean value was then derived from the sum of all scores obtained divided by the number of microscopic fields examined.
Peroxidase Method to Examine Histochemically the Distribution of AT1R Receptors
A polyclonal antibody against the human AT1R (Santa Cruz Biotechnology, Inc.) was used (1:100 dilution) in conjunction with the Vectastain Elite ABC kit. For antigen retrieval, 5- to 10-μm-thick sections were immersed in a 0.4% pepsin solution in 0.1 N HCl for 10 min at 37°C, followed by blocking with 2% bovine serum albumin and 10% normal goat serum. The sections were then incubated overnight with AT1R antibody. Bronchial and vascular smooth muscle cells from lung tissue of patients without pulmonary disease served as positive controls.
Mast Cell Chymase
Toluidine blue staining and monoclonal antibody against chymase were used to identify myocardial mast cells histologically and by immunohistochemistry. For the mast cell chymase staining, a heat-induced epitope retrieval technique with a citrate buffer in a microwave at 90°C during 8 min was used. The primary antibody used was a mouse monoclonal anti–mast cell chymase (Neomarkers; LabVision) at a 1:200 dilution and overnight incubation. To block nonspecific staining, NEN blocking agent (FP 1020; Perkins Elmer) in Tris-HCl buffer was used. The number of mast cells per high power field (×200) was counted in periinfarcted and remote areas of LV myocardium.
Quantitative Analysis of Volume Fraction of Collagen
Collagen volume fractions were quantified in tissue sections using a semiautomated computerized image analysis system (Quantimet 520; Cambridge Laboratories, Inc.), similarly to the technique described previously (13). Collagen volume fraction measured by this method has been shown to correlate closely with the hydroxyproline content of the myocardial tissue (15). Analysis was performed with a standard light microscope and a mounted video camera system connected to a host computer and a digitizer. First, by systematically scanning a microscopic slide (at a magnification of ×7.8), we obtained a composite digitized image. The volume fraction of interstitial and replacement collagen was then calculated as the percentage area of tissue section occupied by each. Intramural vessels, perivascular collagen, endocardium, and trabeculae were excluded from this particular analysis. Perivascular collagen was defined as that collagen in the adventitia of intramural coronary arteries. A segment with replacement fibrosis involving less than 50% of the inner LV wall was considered a nontransmural infarct, and that involving more than 50% of the inner LV wall, a transmural infarct. Myocardial segments adjacent to a nontransmural infarct or patchy, scattered areas of replacement fibrosis were designated as periinfarcted segments. Segments without transmural, nontransmural, or patchy replacement fibrosis were designated as remote, noninfarcted segments. In the latter segments, the presence or absence of interstitial fibrosis (matrix expansion) and microscopic (<1 cm2 in surface area) or patchy (>1 cm2 in surface area) replacement fibrosis was determined by light microscopy.
Quantitative Analysis of Autoradiographic Slides
The quantitative data, expressed as differential uptake ratio per square meter (DUR/m2), were determined by correlating counts per minute (cpm) to photostimulating luminescence units (PSL) determined by the Fuji software for analyzing the phosphor-imaging data. For each study, adjacent sagittal heart slices at 6 different locations throughout the heart were used to determine the linear relationship of PSL to cpm (cpm = slope × PSL). One slice was exposed to the phosphor-imaging plate overnight, and the adjacent slice was counted on a 1480 Wallac Wizard γ-counter. Using the PSL units associated with the phosphor-imaging slice and the cpm from the adjacent slice, the slope of the line was determined for each study (the slope varied from 0.169 to 0.192 between previous studies) (16). Using this relationship, segments of interest were converted from PSL/m2 to cpm/m2, which were used to calculate DUR/m2 by the following equation:
Statistical Analysis
Data are expressed as mean ± SD or frequency (%). Student t tests were used to demonstrate significant results using the InStat3 statistical program (GraphPad Software, Inc.). ANOVA was applied for multigroup comparisons using the same program. Although the samples for the group comparisons of infarcted, periinfarcted, and remote, noninfarcted areas were acquired from different areas of the heart, myocardial segments were not completely independent.
RESULTS
Autoradiographic Evidence for Presence of Tissue ACE and Its Relationship to Collagen
There was specific binding of FBL to ACE; mean FBL binding was 6.6 ± 5.2 PSL/mm2, compared with 3.4 ± 2.5 PSL/mm2 in segments incubated in solution containing cold, 10−6 M lisinopril (P < 0.0001) (Fig. 1). The volume fraction of collagen was higher in the infarcted (n = 28, 63% ± 15%) than in the periinfarcted (n = 39, 14% ± 5%) and remote, noninfarcted areas (n = 11, 11% ± 3%) of LV myocardium. Mean FBL binding was 6.3 ± 4.5 PSL/mm2 in infarcted, 7.6 ± 4.7 PSL/mm2 in periinfarcted, and 5.0 ± 1.0 PSL/mm2 in remote, noninfarcted segments. The difference in mean FBL binding was significant between remote, noninfarcted, and periinfarcted segments (P < 0.02). The binding of FBL to ACE was nonuniform in infarcted, periinfarcted, and remote, noninfarcted segments, and there was an apparent increase in ACE activity in the juxtaposed areas of replacement fibrosis (Fig. 2).
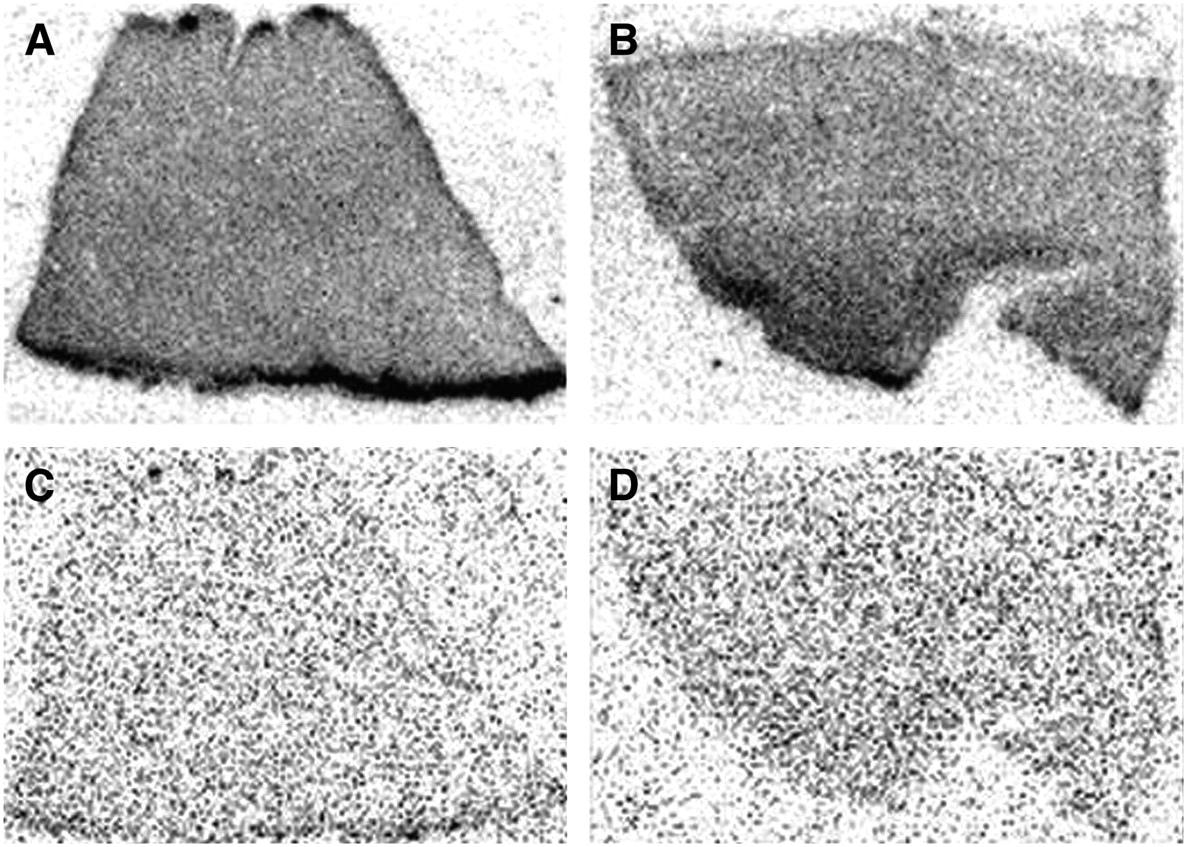
Autoradiographic evidence for specific binding of FBL to ACE in human heart tissue removed from cardiac transplant recipient with ischemic cardiomyopathy. Contiguous myocardial segments incubated in vitro with radiolabeled FBL are shown without (A and B) and with (C and D) nonradiolabeled 10−6 M lisinopril. The latter should block binding at ACE, as shown in C and D, whereas the former should allow binding at ACE and nonspecific sites, as shown in A and B. Binding as indicated in tissue slice shown in A and B by dark gray is not present in tissue slice in C and D, indicating binding of FBL specifically to ACE rather than to nonspecific sites.
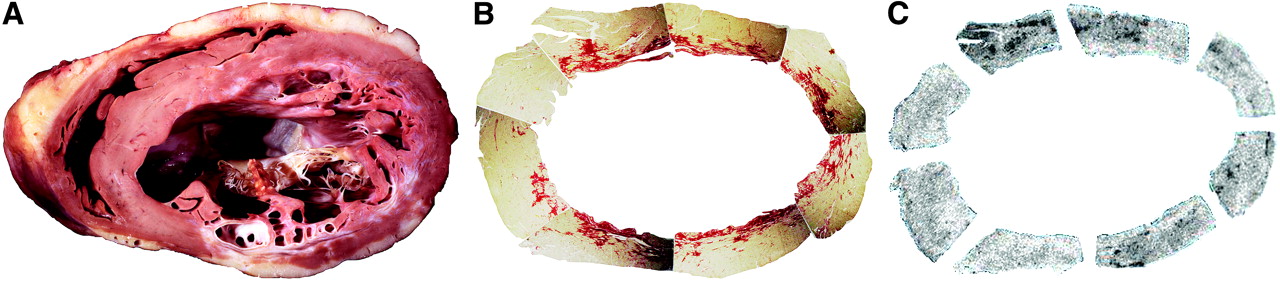
Presence and distribution of ACE activity in relation to collagen replacement as assessed by picrosirius red stain in human heart tissue removed from cardiac transplant recipient with ischemic cardiomyopathy. Gross pathology of midventricular slice (A), with corresponding contiguous midventricular slices stained with picrosirius red stain (B) and 18F-FBL autoradiographic images (C), is shown. FBL binding to ACE is nonuniform in infarcted, periinfarcted, and remote, noninfarcted segments. Increased FBL binding can be seen in segments adjacent to collagen replacement.
Immunohistochemical Evidence for Presence of Tissue ACE System and Angiotensin II Receptors
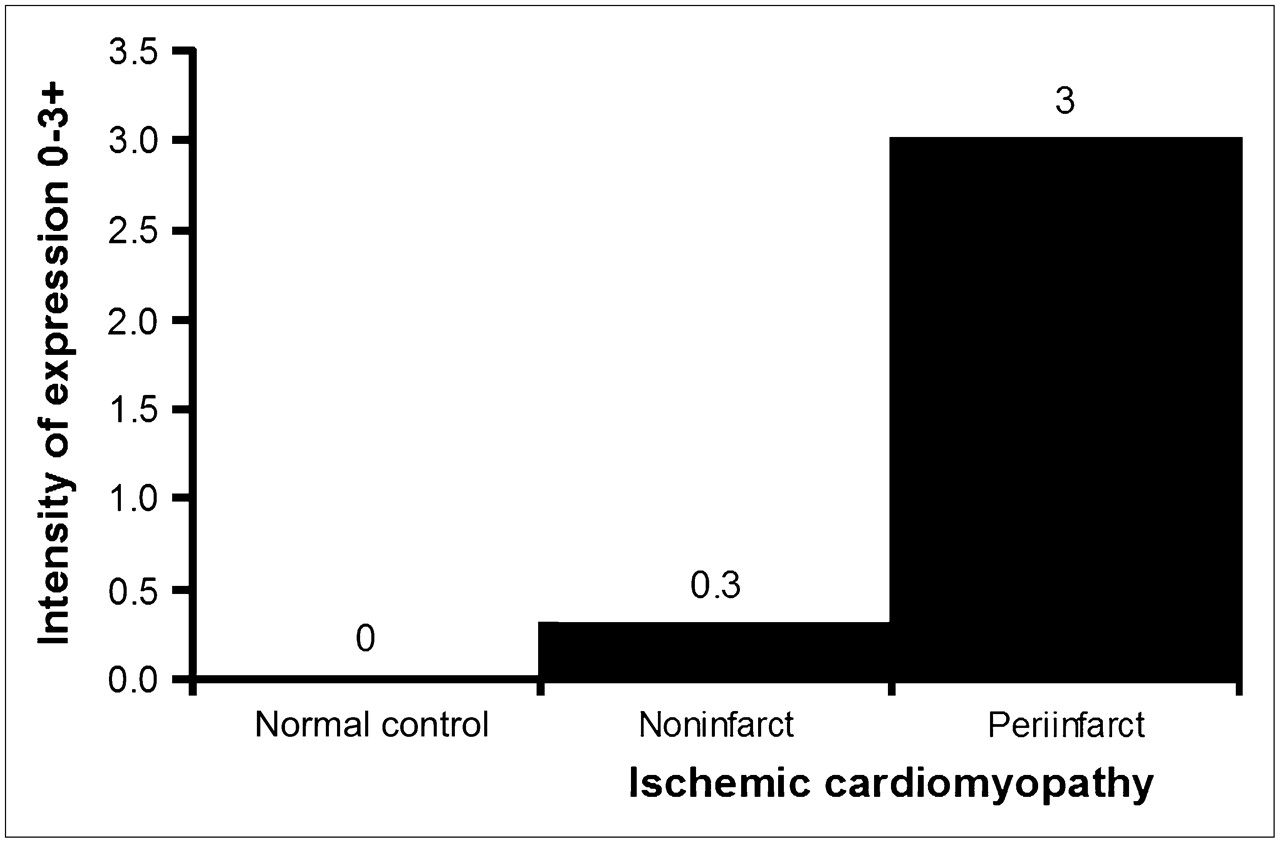
The overall distribution of tissue ACE by immunohistochemical staining was similar to that of autoradiographic FBL binding. Myocardial ACE distribution was similar to that determined by autoradiographic FBL binding. ACE immunoreactivity was primarily localized to myocytes and the media of small arteries. Some activity was also noted in endocardial cells and endothelial cells of intramyocardial arteries. Semiquantitative assessment of ACE reactivity showed no expression in normal control tissue, minimal expression (0−1+) in remote, noninfarcted segments, and marked expression (3+) in periinfarcted segments (P < 0.001) (Fig. 3).

Semiquantitative assessment of ACE immunoreactivity in normal myocardium and in remote, noninfarcted, and periinfarcted myocardium of patients with ischemic cardiomyopathy. Assessment is on a scale of 0–3+, where 0 = absent, 1+ = mild, 2+ = moderate, and 3+ = extensive.
Distribution of AT1R
In noninfarcted segments, AT1R immunoreactivity was confined to smooth muscle and endothelium. Myocytes were not reactive. In contrast, periinfarcted segments showed increased AT1R immunoreactivity, which was localized in the myocytes. Although some AT1R immunoreactivity was also seen in infarcted segments, it was localized predominantly in fibroblasts or within islands of surviving myocytes.
Distribution of Mast Cell Chymase
The number of mast cells in ischemic cardiomyopathy was significantly higher than that in the normal heart (4.2 ± 2.7 vs. 1.5 ± 1.2 mast cells/field, ×200, P < 0.001). Moreover, a higher number of mast cells was present in the remote, noninfarcted segments than in periinfarcted segments (5.1 ± 3.2 vs. 3.2 ± 22 mast cells per field, P < 0.001) in ischemic cardiomyopathy.
DISCUSSION
The renin–angiotensin system is activated early in patients with heart failure. Although serum renin–angiotensin activity often returns to relatively normal levels in well-compensated heart failure, tissue renin–angiotensin activity may remain elevated in the myocardium and vasculature and thereby contribute significantly to the progression of myocardial remodeling. ACE inhibition is now central to the treatment of patients with heart failure, LV dilatation, and remodeling after myocardial infarction. Large clinical trials using ACE inhibitors have proven them to reduce the risk of morbidity and mortality in patients with heart failure (3,4). However, significant interindividual variability in response to ACE inhibition has been recognized in such patients. Factors such as genetic differences, age, sex, race, comorbid conditions, concomitant medications, and renal and liver functions are important determinants of individual response to ACE inhibitors.
Presently, there is no reliable method that predicts the response of an individual patient to ACE inhibition. Both pharmacogenomics and molecular imaging have the potential of providing significant insight in this regard. Nuclear imaging techniques are especially suited for cardiac molecular imaging because of the large number of potentially available molecular targets, relatively high intrinsic sensitivity, and excellent depth penetration. PET is especially advantageous because of being quantitative and providing high spatial resolution. Therefore, a strategy aimed at assessment of tissue ACE upregulation would allow an optimal treatment with antiangiotensin agents while minimizing the likelihood of drug toxicity and adverse reactions.
The findings in this study suggest a role for ACE imaging. In patients with ischemic cardiomyopathy and heart failure, a specific binding of radiolabeled FBL to ACE was shown. Specific ACE binding was about twice as great as nonspecific binding. Furthermore, the binding of FBL was nonuniform in infarcted, periinfarcted, and remote, noninfarcted myocardial segments. ACE binding in periinfarcted segments was about 1.3-fold greater than binding in remote, noninfarcted segments. A similar pattern of nonuniform distribution was observed with AT1R immunoreactivity. There was increased ACE activity and AT1R immunoreactivity in the juxtaposed areas of replacement fibrosis, consistent with their observed roles in the development of scarring and remodeling of the collagen matrix in ischemic cardiomyopathy.
Angiotensin II, a potent vasoconstrictor, has been implicated in LV remodeling, interstitial fibrosis, and cell death in heart failure (17,18). In an experimental model where survival, LV remodeling, cardiac fibrosis, and gene expression were examined in AT1aR knockout mice and wild-type mice at 1 and 4 wk after a large, acute myocardial infarction, the findings in mice were remarkably similar to the protective effect of ACE inhibitors after myocardial infarction in humans (18). At 4 wk after myocardial infarction, control mice showed more marked LV remodeling and fibrosis than did AT1aR knockout mice. Despite producing a similar initial infarct size, the cumulative 4-wk mortality rate was reduced from 22.7% to 5.9% in AT1aR knockout mice, compared with controls. This genetic evidence in mice attests to angiotensin II as the responsible agent for interstitial fibrosis and LV remodeling in the clinical setting. In patients with hypertension, preliminary evidence indicates that ACE inhibition can result in regression of myocardial fibrosis as measured by LV collagen volume fraction and myocardial hydroxyproline concentration (19).
The development of a tissue-specific ACE radioligand represents a first step toward demonstrating the role of ACE and AT1R in directly regulating cardiac function and LV remodeling in heart failure. To date, the synthesis of 2 radiolabeled ACE inhibitors has been reported in the literature: 18F-captopril (8) and FBL (9). 18F-Captopril is complicated by the instability of the sulfhydryl moiety and the apparent conversion in vitro to an isomer form (20,21). FBL is a nonsulfhydryl, 18F-labeled ACE inhibitor. In 1991, radioiodinated MK351A, which is N-[(s)-1-carboxy-3-phenylpropyl]-l-lysyl-tyrosyl-l-proline, was prepared (22). In that case, the tyrosine moiety was reacted with the lysine in lisinopril to form a molecule that could easily be iodinated. The MK351A had a high affinity for ACE (dissociation constant [KD] = 2 nM), comparable to that for lisinopril (KD = 0.13 nM) and higher than that for captopril (KD = 22 nM). This higher affinity produced higher-resolution in vitro autoradiography when compared with 3H-captopril (23). This approach validates the ability to add significant bulk to the ε-amine of lysine in lisinopril without compromising ACE activity. Other analogs of lisinopril, with larger substituents on the ε-amino group of lysine, have also been reported (24).
In a subset of heart failure patients, ACE inhibition has been shown to be less effective. This is explained, in part, by the fact that angiotensin II may also be produced by ACE-independent mechanisms, such as chymase, a mast cell enzyme (25). Both upregulation of ACE and an increased number of mast cells have been described in heart failure (26). Our data support the presence of an alternative pathway for angiotensin II production. There was a significant increase in the number of mast cells in the myocardium of patients with ischemic cardiomyopathy, especially in the remote, noninfarcted segments of the LV. ACE immunoreactivity, on the other hand, was higher in the periinfarcted myocardium than in the remote myocardium and was lowest in segments with the highest number of mast cells. The distribution of angiotensin II, however, appeared similar in the periinfarcted and remote segments of the LV. These observations provide insight to why the addition of AT1R blockers to ACE inhibitors and aldosterone antagonists is beneficial in heart failure.
Based on these encouraging data, we believe that the development of an ACE inhibitor that binds specifically to myocardial ACE in the heart (at a high percentage injected dose per gram) will lead to a new generation of imaging probes for monitoring disease progression and the effectiveness of treatments for heart failure. The clinical implication of such imaging probes would be to identify patients with increased myocardial ACE activity, prospectively and in the early stages of heart failure, before the transition to replacement fibrosis and remodeling occurs. Imaging techniques that can identify patients with increased ACE, prospectively, before the transition to replacement fibrosis and remodeling occurs, may result in preserved LV function, thereby improving overall prognosis.
CONCLUSION
These data suggest that FBL binds specifically to ACE; that the binding is nonuniform in infarcted, periinfarcted, and remote, noninfarcted segments; and that there is an apparent increase in ACE activity in the juxtaposed areas of replacement fibrosis. On the other hand, the distribution of mast cell chymase appears nonuniform and disparate from ACE.
Acknowledgments
This research was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health.
References
- Received for publication July 6, 2006.
- Accepted for publication October 4, 2006.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Myocardial Fibrosis: Emerging Target for Cardiac Molecular Imaging and Opportunity for Image-Guided Therapy
- The Potential of Clinical Phenotyping of Heart Failure With Imaging Biomarkers for Guiding Therapies: A Focused Update
- Myocardial Viability: Survival Mechanisms and Molecular Imaging Targets in Acute and Chronic Ischemia
- Molecular Imaging of the Cardiac Extracellular Matrix
- Cardiac Positron Emission Tomography/Computed Tomography Imaging of the Renin-Angiotensin System in Humans Holds Promise for Image-Guided Approach to Heart Failure Therapy
- Targeted Molecular Imaging: Target Significance and Probe Validation
- Molecular Imaging of Human ACE-1 Expression in Transgenic Rats
- Multimodality Imaging of Myocardial Injury and Remodeling
- Integration of Infarct Size, Tissue Perfusion, and Metabolism by Hybrid Cardiac Positron Emission Tomography/Computed Tomography: Evaluation in a Porcine Model of Myocardial Infarction
- Cardiac Positron Emission Tomography
- Noninvasive Imaging of Angiotensin Receptors After Myocardial Infarction
- Nuclear Cardiac Stress Testing in the Era of Molecular Medicine
- Exposing ACE up the Sleeve...