


Abstract
Radiolabeled analogs of α-melanocyte-stimulating hormone (α-MSH) are potential candidates for the diagnosis and therapy of melanoma metastases. After our recent observation that a linear octapeptide α-MSH analog incorporating the metal chelator 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA) at the C-terminal lysine, [Nle4,Asp5,d-Phe7,Lys11(DOTA)]-α-MSH4–11 (DOTA-NAPamide), showed high accumulation in melanomas in a mouse model, low uptake in normal tissues, and moderate uptake in the kidneys, we attempted to identify the structural parameters influencing tumor uptake versus kidney uptake. Methods: We designed a series of novel DOTA-α-MSH analogs differing from DOTA-NAPamide by small alterations, such as the position of DOTA in the peptide, hydrophobicity, and charge, by modifying the C-terminal Lys11 residue. They were evaluated both for their melanocortin type 1 receptor (MC1R)–binding potency and for their biodistribution by use of the B16F1 melanoma mouse model. Results: When DOTA was shifted to the N terminus of the peptide, a 3-fold increase in kidney retention was obtained. However, when the ε-amino group of the Lys11 residue was acetylated in addition to the DOTA relocation, kidney uptake returned to the low values obtained with DOTA-NAPamide; this result indicated that neutralization of the ε-amino group positive charge of the Lys11 residue rather than the position of DOTA accounted for the low kidney retention. Unexpectedly, no further reduction in kidney uptake was obtained by the introduction of 1 or 2 negative charges on Lys11. Melanoma uptake was in accordance with MC1R affinity; the highest values were obtained for peptides bearing carboxy-terminal amidation and positioning of DOTA. Conclusion: The kidney uptake of DOTA-α-MSH analogs could be considerably reduced, without affecting MC1R affinity, by altering (neutralizing) the charge of the Lys11 residue. Accordingly, the resulting peptides exhibited a high ratio of tumor uptake to kidney uptake that is favorable for diagnostic and therapeutic applications. These structure–activity data may help to improve the performance of DOTA-α-MSH analogs and other radiopeptides.
- melanoma imaging
- melanocortin type 1 receptor
- α-melanocyte-stimulating hormone
- DOTA
- scintigraphy
- internal radiotherapy
Radiopeptides are attractive tools for cancer diagnosis and therapy because a variety of human tumors overexpress receptors for regulatory peptides or peptide hormones. The best examples illustrating the rationale of this strategy are radiolabeled somatostatin analogs, which are now routinely used in clinics to image tumors expressing somatostatin receptors and which promise to be of value for internal radiotherapy in patients (1). Nevertheless, the side effects of nonspecific retention in the kidneys limits the therapeutic efficacy of radiopeptides, as renal toxicity is the dose-limiting factor (2, 3). For the same reason, the diagnosis of tumors localized in the renal region is markedly compromised.
The renal accumulation of radiopeptides is not specific to somatostatin analogs but is also observed with other peptides carrying metal chelators, such as 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA) (4) and diethylenetriaminepentaacetic acid (DTPA) (5). It is most apparent with intracellularly retained isotopes, such as 111In, 99mTc (6), and many other radiometals with therapeutic potential. Although the use of radioisotopes, such as radioiodine, that are readily liberated from the peptide inside cells via specific dehalogenases may partly solve the problem, the use of such radioisotopes has a very detrimental effect on tumor retention (7). It is clear that the success of a therapeutic strategy with radiopeptides depends on a maximal reduction of renal uptake (8).
In recent years, many studies focused on the elucidation of the mechanism of retention of radiopeptides in the kidneys, and it is now relatively well established that radiopeptides are reabsorbed in the proximal tubules via luminal endocytosis after glomerular filtration. Then they are delivered to lysosomes, where they are hydrolyzed to a final radioactive metabolite that cannot leave the lysosomes, leading to long-term sequestration of the radioisotope in proximal tubular cells (8, 9). Interestingly, reabsorption of radiopeptides was shown to be reduced by systemic administration of cationic amino acids and their derivatives, both in animals and in human patients (9). However, careful evaluation of their toxicity still is needed. Radiopeptides exhibiting lower renal uptake clearly would represent a major step forward.
Over the past 2–3 y, we studied radiopeptide-based targeting systems for melanoma metastases. To this end, analogs of the tridecapeptide α-melanocyte-stimulating hormone (α-MSH) were developed and were expected to accumulate in melanoma lesions because the α-MSH receptor, also called the melanocortin type 1 receptor (MC1R), is overexpressed in melanoma cells (10–14). In a recent investigation, Froidevaux et al. developed a short linear DOTA-α-MSH analog, [Nle4,Asp5,d-Phe7,Lys11(DOTA)]-α-MSH4–11 (DOTA-NAPamide) (15), in which DOTA was conjugated to the C terminus of the peptide via the ε-amino group of the Lys11 residue. After labeling was done with 111In or 67Ga/68Ga, high melanoma uptake and strikingly low kidney uptake were observed in mice: 3.98 percentage injected dose/g (%ID/g) for 67Ga-DOTA-NAPamide at 4 h after injection. The diminution in kidney retention was not compensated for by a concomitant increase in uptake in other excretory organs, as is often observed. To our knowledge, the amount of radioactivity accumulated in the kidneys was the lowest reported so far with DOTA-α-MSH analogs (16–18). In comparison, 111In-labeled [βAla3,Nle4,Asp5,d-Phe7,Lys10(DOTA)]-α-MSH3–10 (DOTA-MSHoct), a DOTA-α-MSH analog sharing 6 of 8 amino acid residues with DOTA-NAPamide, exhibited kidney uptake of 13.5 %ID/g at the same time point (18). The reduced kidney uptake of DOTA-NAPamide makes this peptide a promising candidate for diagnosis and brings internal radiotherapy of metastatic melanoma a step nearer.
The DOTA-NAPamide structure may represent an interesting model for identifying the structural features that are important for the favorable biologic behavior of radiopeptides—in particular, low kidney uptake—and thereby serve to further improve the biologic behavior of DOTA-α-MSH analogs and radiopeptides in general. To this end, we have now designed a series of novel DOTA-α-MSH analogs differing from DOTA-NAPamide by some well-defined alterations that could influence biologic behavior. The modifications that we studied included the position of the DOTA chelator in the peptide, hydrophobicity, and charge, through modification of the C-terminal acid group (which is negatively charged at a physiologic pH) or the side-chain amino group of the Lys11 residue (which has a positive charge at a physiologic pH). These DOTA-α-MSH analogs were compared in vitro for their MC1R-binding potency and, after radiolabeling was done with 111In, for their tissue distributions in a melanoma mouse model. Their relative levels of performance—in particular, kidney uptake—are discussed in relation to their structural features.
MATERIALS AND METHODS
Peptides and Radioligands
α-MSH was a gift from Novartis, and (Nle4,d-Phe7)-α-MSH (NDP-MSH) was purchased from Bachem. The other α-MSH analogs (Fig. 1) were synthesized in our laboratory by use of continuous-flow technology and a 9-fluorenylmethoxycarbonyl strategy. The construction of DOTA-MSHoct by conjugation of partially protected DOTA to MSHoct was performed as previously described (18). Peptides were conjugated to DOTA by the addition of a deprotected peptide (4.5 μmol) that had been dissolved in N,N′-diisopropylethylamine (1.5 μL):N,N-dimethylformamide (DMF) (100 μL) to DOTA (4.5 μmol) and preincubated for 10 min with 0-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (5.4 μmol):DMF (300 μL). After 1 h of incubation at room temperature and precipitation of the peptide in ice-cold diethyl ether, deprotection of DOTA was performed by the addition of 3.6 mL of trifluoroacetic acid (TFA), 0.2 mL of thioanisole, 0.18 mL of water, and 0.02 mL of 1,2-ethanedithiol. After the mixture was stirred for 4 h, the deprotected DOTA-peptide was precipitated with ice-cold diethyl ether and resuspended in 10% acetic acid before purification by reversed-phase high-performance liquid chromatography (RP-HPLC). The major peak was collected and analyzed by electrospray ionization mass spectrometry.

Structures of compounds used in this study. Ac = acetyl group; Suc = succinyl group.
Succinylation of the side-chain amino group of the Lys residue was performed by slowly adding 170 μmol of succinic anhydride to 3.7 μmol of a peptide that had been dissolved in guanidine HCl (5 mol/L) while the pH was maintained at 8 with NaOH (2 mol/L). Hydroxylamine then was added to a final concentration of 1 mol/L, and the pH was adjusted to 10 with NaOH. After 1 h of incubation, the reaction mixture was loaded onto a small reversed-phase cartridge (Sep-Pak C18; Waters Corp.), and the peptide was eluted with ethanol before purification by RP-HPLC. The major peak was collected and analyzed by electrospray ionization mass spectrometry.
Acetylation of the side-chain amino group of the Lys residue was performed by reaction of a peptide (10 μmol in DMF) with N,N′-diisopropylethylamine (20 μmol), 4-nitrophenyl acetate (20 μmol), and 1-hydroxybenzotriazole (10 μmol). After 16 h of incubation, the peptide was precipitated with ice-cold diethyl ether, purified by RP-HPLC, and analyzed by electrospray ionization mass spectrometry.
Incorporation of 111In into DOTA-peptides was performed by the addition of 92.5 MBq of 111InCl3 (Mallinckrodt) to a DOTA-peptide (10 nmol) that had been dissolved in 53 μL of acetate buffer (0.4 mol/L, pH 5) containing 2 mg of gentisic acid. After 30 min of incubation at 95°C, the radiolabeled DOTA-peptide was purified on a small reversed-phase cartridge (Sep-Pak C18) with ethanol as the solvent. The purity of the radioligand was assessed by RP-HPLC with a Jasco PU-980 chromatography system connected to a Radiomatic 500TR LB506C1 γ-detector (Packard) and a Spherisorb ODS2 5-μm column (Waters) under the following conditions: eluent A, 0.1% TFA in water; eluent B, 0.1% TFA in acetonitrile; gradient, 0–2 min with 96% eluent A, 2–22 min with 96%–45% eluent A, 22–30 min with 45%–25% eluent A, 30–32 min with 25% eluent A, and 32–34 min with 25%–96% eluent A; and flow rate, 1.0 mL/min. The specific activity of the radioligand was always >7.4 GBq/μmol.
NDP-MSH was iodinated by the chloramine-T (Merck) method. NDP-MSH (3 μg) was mixed with Na125I (37 MBq; Amersham) in 50 μL of phosphate-buffered saline (0.3 mol/L, pH 7.2), and then 10 μL of chloramine-T (2 mg/mL) was added. After incubation for 5 min, the reaction was stopped with 500 μL of dithiothreitol (20 mg/mL), and the monoiodinated peptide was purified by use of a syringe packed with 0.25 g of Spherisorb ODS and 10-μm reversed-phase silica (Phase Separations) and a stepwise gradient of aqueous methanol containing 1% TFA. The fractions containing 125I-NDP-MSH were supplemented with dithiothreitol (1.5 mg/mL) and stored at −20°C. Before each binding experiment, an additional purification was performed by RP-HPLC.
Cell Culture
Mouse melanoma cell line B16F1 (19) was cultured in modified Eagle medium supplemented with 10% heat-inactivated fetal calf serum, l-glutamine at 2 mmol/L, 1% nonessential amino acids, 1% vitamin solution, penicillin at 50 U/mL, and streptomycin at 50 μg/mL (all from Gibco) at 37°C in a humidified atmosphere of 95% air and 5% CO2. For expansion or experiments, the cells were detached with 0.02% ethylenediaminetetraacetic acid in phosphate-buffered saline (150 mmol/L, pH 7.2–7.4).
In Vitro Binding Assay
Triplicates of 100-μL B16F1 cell suspensions adjusted to 5 × 106/mL were incubated in 96-well U-bottom microplates (Falcon 3077) for 3 h at 15°C with 50 μL of a series of dilutions (from 10−6 to 10−12 mol/L) of competitor peptides and 50 μL of 125I-NDP-MSH (50,000 cpm). The binding medium consisted of modified Eagle medium with Earle salts (Gibco), 0.2% bovine serum albumin, and 1,10-phenanthroline (0.3 mmol/L; Merck). The reaction was stopped by incubation on ice for 10 min, and the cell-bound radioactivity was collected on filters by use of a cell harvester (Packard). The radioactivity was counted by use of a microplate scintillation counter (TopCount; Packard), and the 50% inhibitory concentration (IC50) was calculated by use of Prism software (GraphPad Software).
Biodistribution in B16F1 Tumor–Bearing Mice
All animal experiments were performed in compliance with Swiss regulations for animal treatment.
Female B6D2F1 mice (C57BL/6 × DBA/2 F1 hybrids; breeding pairs obtained from IFFA-CREDO) were implanted subcutaneously with 0.5 million B16F1 cells. One week later, 200 μL containing 185 kBq of radioligand diluted in 0.9% NaCl and 0.1% bovine serum albumin (pH 7.5) were injected intravenously into the lateral tail vein. For the determination of nonspecific uptake, 50 μg of α-MSH were coinjected with the radioligand. The animals were sacrificed at 4, 24, or 48 h. Organs and tissues of interest were dissected, rinsed to remove excess blood, weighed, and assayed for radioactivity by use of a γ-counter. The %ID/g was calculated for each tissue. The total counts injected per animal were calculated by extrapolation from the counts for a standard taken from the injection solution for each animal.
Analysis of Data
Unless otherwise stated, results are expressed as mean ± SEM. Statistical evaluation was performed by use of the Student t test or a 1-way ANOVA. When significant overall effects were obtained with the ANOVA, multiple comparisons were made with the Bonferroni correction. A P value of <0.05 was considered statistically significant. The area under the curve (AUC) was calculated by use of Prism software for a particular time period by use of the mean tissue uptake value at each time point.
RESULTS
Synthesis of DOTA-α-MSH Analogs
Synthesis of α-MSH analogs and coupling to DOTA (Fig. 1) were performed as described in Materials and Methods. After RP-HPLC purification to >99% purity, conjugation of DOTA to the ε-amino group of Lys11 was achieved with a yield of 11%–18%, whereas acetylation and succinylation at this position resulted in yields of 26%–41% and 15%–41%, respectively. Mass spectrometry confirmed the expected molecular weights for NAPamide (experimental, 1,099.5; calculated, 1,099.27), DOTA-NAPamide (experimental, 1,485.5; calculated, 1,485.7), peptide I (experimental, 1,100.6; calculated, 1,100.25), peptide II (experimental, 1,141.7; calculated, 1,142.3), peptide III (experimental, 1,486.5; calculated, 1,486.65), peptide IV (experimental, 1,442.6; calculated, 1,443.9), peptide V (experimental, 1,485.9; calculated, 1,485.67), peptide VI (experimental, 1,544.5; calculated, 1,543.68), and peptide VII (experimental, 1,543.4; calculated, 1,542.7).
Receptor Binding
The newly designed α-MSH analogs (Fig. 1) and our standard peptide, DOTA-NAPamide, were assessed for their MC1R affinity by competition binding assays with the natural MC1R ligand α-MSH as an internal control. As shown in Table 1, all DOTA-peptides (peptides III–VII), except for peptide VI, displayed receptor affinities in the low range of nanomoles per liter, suitable for a tumor vector; however, DOTA-NAPamide remained the most potent MC1R ligand. C-terminal amidation invariably had a positive influence on the binding potency of up to 10-fold (DOTA-NAPamide vs. peptide III). In contrast, modification of the ε-amino group of Lys11 by DOTA conjugation, acetylation, or succinylation affected receptor affinity negatively, although to different degrees (2- to 23-fold variations). The introduction of DOTA at the N terminus of the peptide (peptides IV–VII) also decreased binding potency and might be more deleterious than attachment of the chelator on the side-chain amino group of Lys11 (compare IC50 of DOTA-NAPamide with those of peptides IV and V).
MC1R Affinities of α-MSH Analogs
Thus, this structure–MC1R affinity study demonstrates that both C-terminal amidation and C-terminal positioning of DOTA are important for maximum MC1R-binding potency, as has been observed with DOTA-NAPamide.
Biodistribution in Melanoma-Bearing Mice
Table 2 shows the tissue distributions of DOTA-NAPamide and the other structurally related DOTA-α-MSH analogs labeled with 111In. Tissues, including melanomas, were collected 4, 24, or 48 h after injection of the radioligand. All DOTA-α-MSH analogs accumulated in melanomas to various degrees. DOTA-NAPamide exhibited the highest uptake, followed by peptides IV, V, VII, III, and VI (Table 2 and Fig. 2). The rank order of melanoma uptake was in full accordance with that of MC1R affinity. The data were best fitted to an exponential model, as opposed to a linear model (data not shown), indicating that as the IC50 decreases (i.e., binding affinity increases), the melanoma uptake is augmented at an increasing rate. Melanoma uptake was found to be an MC1R-mediated process for all DOTA-α-MSH analogs, as it could be substantially reduced by coinjection of 50 μg of α-MSH (data not shown). Clearance from normal tissues was fast for all DOTA-peptides, the exception being the kidneys, which serve as excretory organs. At all time points, maximum renal retention was observed for peptide IV, whereas that for peptides III, V, and VII was similar to that for DOTA-NAPamide and that for peptide VI, somewhat lower retention being apparent only 4 h after injection. Accordingly, the kidney AUC calculated for the time period from 4–48 h was, on average, 3 times greater for peptide IV than for the other DOTA-peptides (Fig. 2). After radioligand uptake, the release of 111In from the kidneys was found to be a slow process because, as shown in Figure 2, on average, 58% of the radioactivity measured in the kidneys at 4 h after injection was still there 1 d later and 33% was still there 2 d later. In comparison, the release of radioactivity from melanomas was much faster, because the percentages reached only 34% and 18% on days 1 and 2, respectively. There was no difference in the rates of radioactivity released from the kidneys and melanomas for the DOTA-peptides tested, with the exception of peptide IV, which might lead to slightly slower radioactivity excretion from the kidneys.

Uptake of DOTA-α-MSH analogs. 111In-DOTA-α-MSH analogs were injected into melanoma-bearing mice, and radioactivity accumulated in tumor and kidney was measured 4, 24, and 48 h after injection. Results are expressed as tumor and kidney AUCs (A), ratio of tumor uptake to kidney uptake calculated from AUCs (B), and percent uptake at 4 h (C). Results are expressed as mean ± SEM (n = 4–12). Asterisk indicates P value of <0.05 vs. DOTA-NAPamide; dagger indicates P value of <0.05 vs. kidney.
Tissue Distributions of α-MSH Analogs 4, 24, and 48 Hours After Injection
Thus, the DOTA-α-MSH analogs were found to accumulate in melanomas to an extent matching their receptor affinity. They were all rapidly cleared from normal tissues, with the exception of the kidneys, where they accumulated in various amounts depending on their structures.
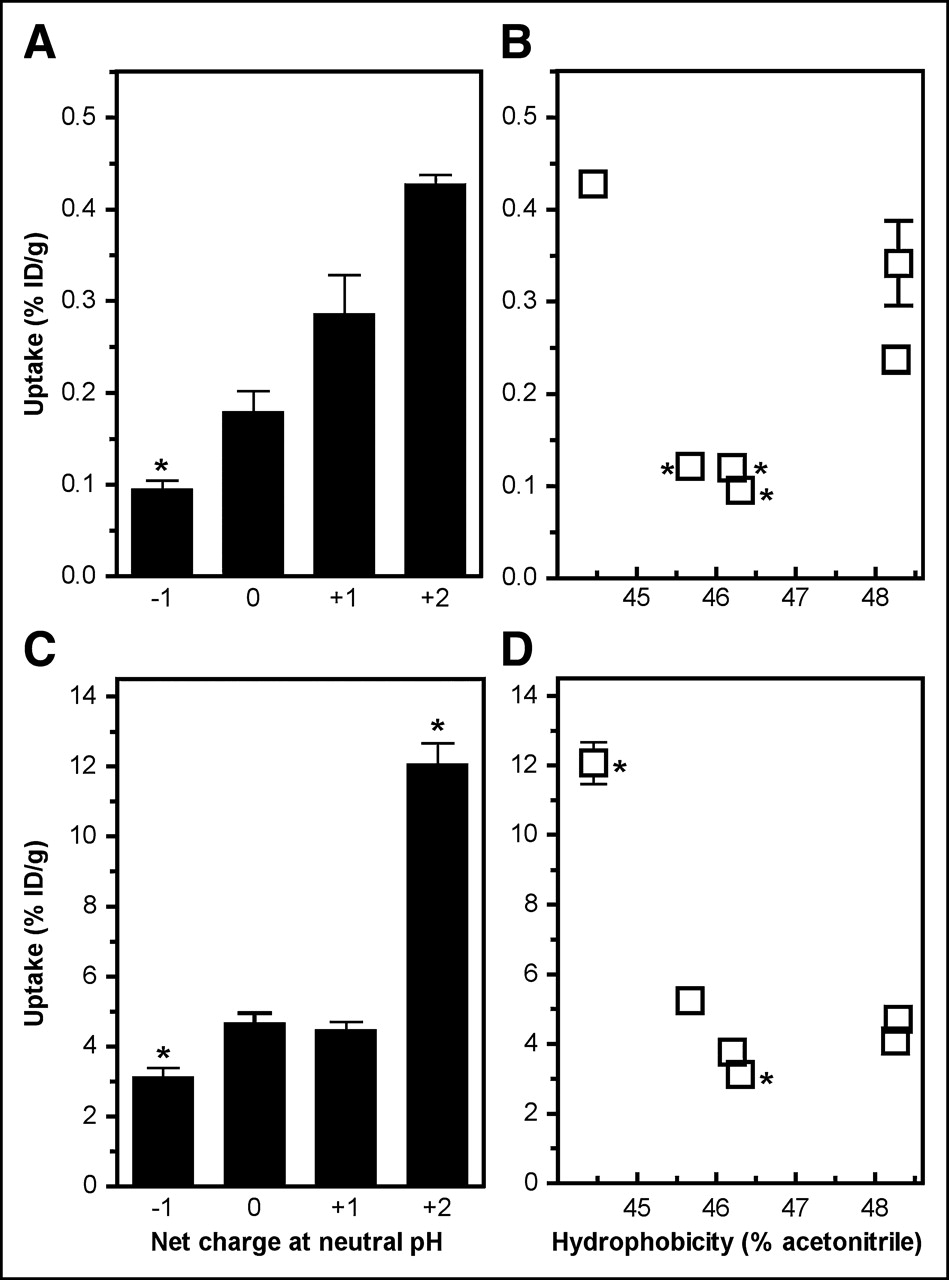
Effects of Charge and Hydrophobicity on Kidney Uptake and Liver Uptake
To address the contributions of charge and hydrophobicity to the biologic behavior of the DOTA-α-MSH analogs, kidney uptake and liver uptake were plotted as a function of the net charge of the peptide (calculated for a physiologic pH of 7) and its hydrophobicity (Fig. 3). The net charge (Table 3) was calculated on the basis of known pKa values for amino acid residues and functional groups. To avoid the influence of charge distribution within the peptide, charge variation was confined to the C-terminal Lys11 residue on amidation, succinylation, acetylation, or conjugation of DOTA. Hydrophobicity was measured by RP-HPLC analysis (Table 3). As clearly shown in Figure 3, peptide charge or hydrophobicity greatly influenced kidney uptake; an almost 3-fold decrease was observed between the most positively charged 111In-labeled DOTA-α-MSH peptide (peptide IV; net charge, +2) and peptides carrying 1 charge less (DOTA-NAPamide and peptide V; net charge, +1). A further decrease in charge either did not modify (peptides III and VII; net charge, 0) or slightly reduced (peptide VI; net charge, −1; 1.4-fold decrease) kidney retention. It is of interest that liver uptake showed a similar charge-dependent relationship, indicating that a decrease in kidney uptake was not compensated for by an increase in liver uptake. There was no correlation between peptide hydrophobicity and liver uptake.

Effect of hydrophobicity on uptake of DOTA-α-MSH analogs. 111In-DOTA-α-MSH analogs were injected into melanoma-bearing mice, and radioactivity accumulated in liver and kidney was measured 4 h after injection. (A and C) Liver uptake and kidney uptake, respectively, as function of net charge. (B and D) Liver uptake and kidney uptake, respectively, as function of hydrophobicity. Table 3 shows 111In-DOTA-α-MSH analog charge and hydrophobicity. Results are expressed as mean ± SEM (n = 4–12). Asterisk indicates P value of <0.05 vs. DOTA-NAPamide (B and D) or DOTA-α-MSH analogs carrying net charge of +1 (A and C).
Physical Properties of DOTA-α-MSH Analogs
Thus, neutralization of the positive charge of the ε-amino group of Lys11, either by acetylation or by coupling of 111In-DOTA, played a key role in the reduction of kidney uptake. No or only a marginal diminution in kidney retention was observed when a negative charge(s) was introduced on Lys11.
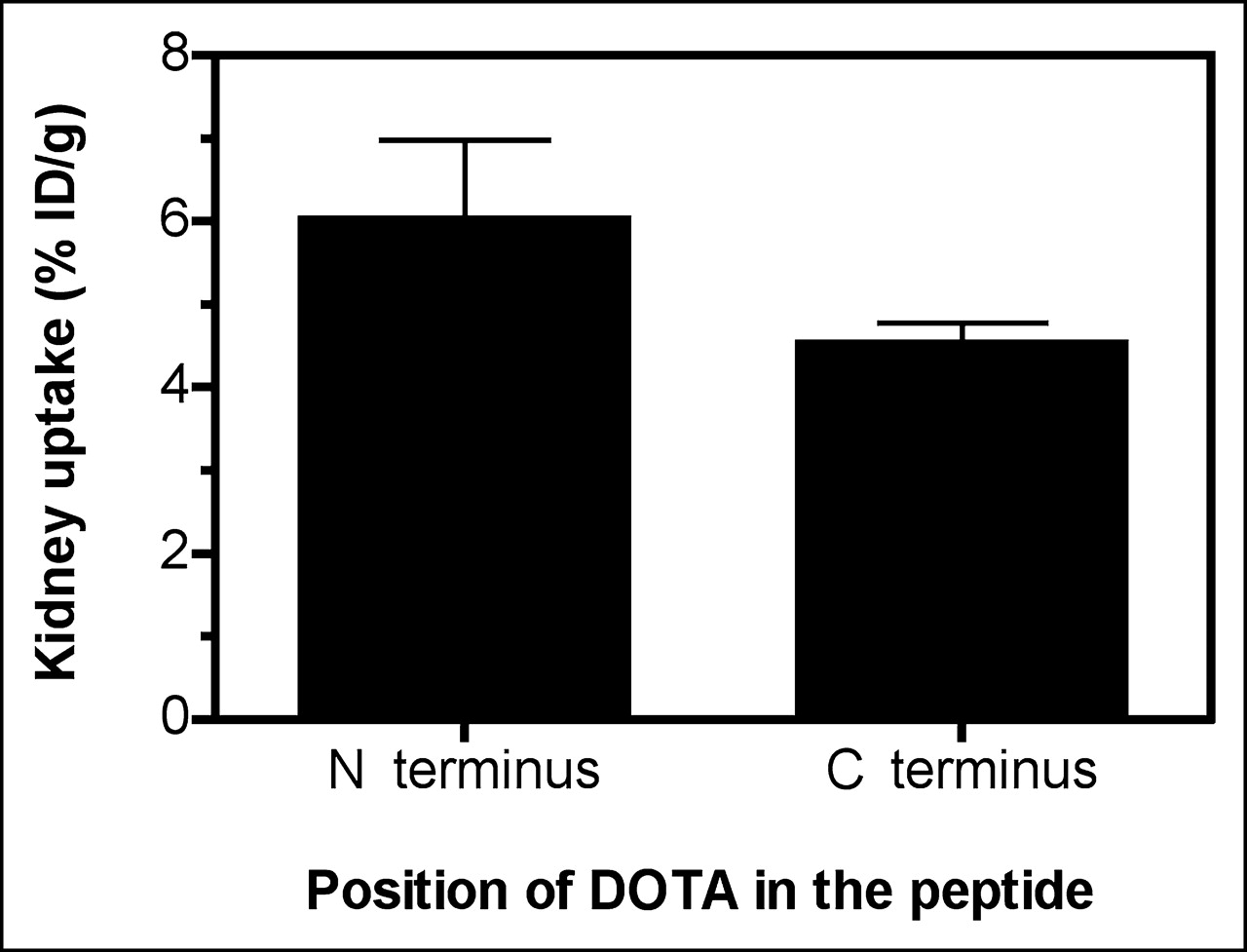
Effect of Position of DOTA in Peptide on Kidney Uptake
To investigate whether the position of DOTA in the peptide might influence kidney retention, kidney uptake values obtained for peptides with DOTA incorporated at their N termini (peptides VI, V, VI, and VII) were compared with those found for peptides bearing DOTA at their C termini via the ε-amino group of the C-terminal Lys11 residue (DOTA-NAPamide and peptide III). As shown in Figure 4, there was no statistically significant difference between the 2 groups, suggesting that DOTA can be coupled to either end. The apparent 3-fold decrease in kidney uptake when 111In-DOTA was shifted from the N terminus to the C terminus (compare DOTA-NAPamide and peptide IV in Fig. 2 and Table 2) was demonstrated to be attributable to neutralization of the charge by the coupling of 111In-DOTA to the ε-amino group of Lys11, because acetylation at this position (peptide V) induced a similar effect.

Effect of position of DOTA in peptide on uptake of DOTA-α-MSH analogs. 111In-DOTA-α-MSH analogs were injected into melanoma-bearing mice, and radioactivity accumulated in kidney was measured 4 h after injection. Values obtained for 111In-DOTA-α-MSH analogs bearing DOTA at their N terminus (n = 16) were compared with those obtained for 111In-DOTA-α-MSH analogs incorporating DOTA at their C terminus via ε-amino group of C-terminal Lys11 residue (n = 16). Results are expressed as mean ± SEM. No statistical difference between groups was noted.
Thus, the position of DOTA in the peptide did not affect kidney uptake apart from a charge neutralization effect.
DISCUSSION
Encouraging data on the use of a short linear DOTA-α-MSH analog, DOTA-NAPamide, as a melanoma vector have been reported. In mouse models, 111In- and 67Ga/68Ga-labeled DOTA-NAPamide exhibited high melanoma uptake and low kidney uptake; the ratios of tumor uptake to kidney uptake, calculated from the AUC for 4–48 h, were 1.11 and 1.82, respectively. Surprisingly, although sharing 6 of 8 amino acid residues with DOTA-NAPamide, DOTA-MSHoct showed considerably inferior biologic performance, with a ratio of tumor uptake to kidney uptake, calculated from the AUC for the same period of time, of as low as 0.24. These data suggested that a small alteration in the chemical structure of DOTA-NAPamide was responsible for its superior melanoma-targeting ability. A high ratio of tumor uptake to kidney uptake calculated from the AUC is important for diagnostic applications and imperative for therapeutic purposes, as nephrotoxicity is currently the dose-limiting factor that reduces the therapeutic index of radiopeptides. DOTA-NAPamide is therefore an interesting vector, as it brings achievement of internal radiotherapy for metastatic melanoma a step nearer and may also help in understanding the relationships between the chemical structures of radiopeptides and in vivo pharmacokinetics.
In this study, we focused on the identification of the structural feature(s) underlying the superior biologic performance of DOTA-NAPamide. Because the major difference between DOTA-NAPamide and DOTA-MSHoct lies in the conjugation of DOTA to the C terminus as opposed to the N terminus of the peptide, we hypothesized that this unusual position in a radiopeptide was responsible for the superior performance, in particular, the low kidney uptake, of DOTA-NAPamide. In accordance with this hypothesis, relocating DOTA to the N terminus (peptide IV) dramatically increased kidney uptake (3-fold increase). However, when the ε-amino group of Lys11, left free in peptide IV, was acetylated (peptide V), kidney uptake was shown to return to the low values obtained with DOTA-NAPamide. These data indicate that the low renal accumulation of DOTA-NAPamide most likely resulted from the neutralization of the positive charge of the ε-amino group of Lys11 by conjugation of DOTA which, after labeling with 111In, resulted in a final charge of 0 on the side chain of Lys11. Diminution of the positive charge of molecules has already been reported to lessen both glomerular filtration and tubular reabsorption (20–23). The influence of charge on renal uptake has also been illustrated for radiopeptides. Kidney accumulation of 111In-labeled DTPA-d-Phe1-octreotide was reported to be higher than that of 111In-labeled DTPA-l-Lys1-octreotide (24). Moreover, several studies showed that the kidney retention of radiopeptides could be significantly reduced by systemic administration of cationic but not neutral or anionic amino acids and their derivatives (5). These data suggested that tubular reabsorption might be facilitated by ionic interactions between positively charged peptides and negatively charged surfaces of proximal tubular cells; that is, renal uptake might be highest with positively charged peptides.
Because neutralization of the positive charge of the ε-amino group of Lys11 dramatically reduced kidney uptake, probably via diminution of tubular membrane–peptide interactions, it was tempting to speculate that the introduction of a negative charge(s) at this position might further reduce kidney accumulation via a repulsion mechanism. Succinylation (the addition of 1 negative charge; peptide VII) did not significantly affect kidney uptake. Similarly, replacement of the C-terminal amide group of Lys11 with a carboxylate group (peptide III), which also generated a negative charge on Lys11, did not modify kidney accumulation. Only the combination of the 2 alterations, succinylation and deamidation (the addition of 2 negative charges; peptide VI) resulted in a very slight reduction in kidney uptake. One possible explanation is that the Lys11 residue, normally fully exposed when bearing a positive or a neutral charge, was no longer accessible to the surfaces of tubular cells when carrying a negative charge(s) because of possible peptide refolding attributable to newly formed ionic interactions within the peptide. In this hypothesis, the charge location on the peptide rather than its net charge would play a major role in kidney uptake. Consistent with this idea are the findings that the addition of negative charges could either increase (1) or decrease (24) the kidney retention of radiopeptides.
After radioligand uptake, the subsequent release of radioactivity from melanomas and the kidneys was not dependent on the peptide structure. Collectively, DOTA-α-MSH analogs were found to be much more rapidly released from melanomas than from the kidneys, leading to a decrease in the ratio of tumor uptake to kidney uptake over time. The reason for this differential excretion rate is not clear, because DOTA-α-MSH analogs are assumed to undergo a similar process in both the kidneys and melanomas after cellular uptake. Indeed, 111In-DOTA-α-MSH analogs were shown to accumulate in the endosomal or lysosomal compartment of B16F1 cells after in vitro exposure (S.F., M.C.-C., H.T., and A.N.E., unpublished data, 2005), and a similar localization in renal tubular cells was documented after reabsorption of radiopeptides incorporating a metal chelator (9, 25). One hypothesis is that the rate of peptide hydrolysis is higher in melanoma lysosomes, resulting in the more rapid production of small radioactive metabolites exhibiting higher endosomal or cell release potential than larger metabolites or intact radiopeptides. The upregulation of lysosomal enzymes in melanomas has been documented (26).
CONCLUSION
In summary, our structure–activity study revealed that the charge of the ε-amino group of the Lys11 residue of DOTA-α-MSH analogs plays a critical role in kidney uptake. Neutralization of the positive charge at this position resulted in a dramatic decrease in kidney uptake without changing uptake in other normal organs, including other excretory organs, such as the liver. Amidation at the C terminus was associated with increased MC1R-binding potency and enhanced tumor uptake. A combination of both structural features led to DOTA-NAPamide, a promising DOTA-α-MSH analog for melanoma targeting in the clinic; this analog exhibited high melanoma uptake and low kidney retention. It is also interesting that DOTA could be coupled to either end of NAPamide and maintain MC1R affinity in the low range of nanomoles per liter. These data should make it possible to develop α-MSH analogs carrying 2 DOTA groups and thereby exhibiting higher specific activity after radiolabeling (2 radiometals per peptide). It is to be anticipated that enhanced specific activity will augment melanoma uptake, as previously documented (15).
Acknowledgments
We thank Dr. Joyce Baumann for critical review of the article. This work was supported by the Swiss Cancer League and the Swiss National Science Foundation.
Footnotes
Received Oct. 20, 2004; revision accepted Jan. 16, 2005.
For correspondence or reprints contact: Sylvie Froidevaux, PhD, Actelion Pharmaceuticals Ltd., Gewerbestrasse 16, CH-4123 Allschwil, Switzerland.
E-mail: sylvie.froidevaux{at}actelion.com
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Effects of the Amino Acid Linkers on the Melanoma-Targeting and Pharmacokinetic Properties of 111In-Labeled Lactam Bridge-Cyclized {alpha}-MSH Peptides
- Reduction of the Ring Size of Radiolabeled Lactam Bridge-Cyclized {alpha}-MSH Peptide, Resulting in Enhanced Melanoma Uptake
- PET of Malignant Melanoma Using 18F-Labeled Metallopeptides
- Small-Animal PET of Melanocortin 1 Receptor Expression Using a 18F-Labeled {alpha}-Melanocyte-Stimulating Hormone Analog
- Melanoma Imaging with Pretargeted Bivalent Bacteriophage
- Synthesis and Biologic Evaluation of 64Cu-Labeled Rhenium-Cyclized {alpha}-MSH Peptide Analog Using a Cross-Bridged Cyclam Chelator