


Abstract
18F-Labeled small synthetic peptides have emerged as attractive probes for imaging various molecular targets with PET. The α-melanocyte-stimulating hormone (α-MSH) receptor (melanocortin type 1 receptor [MC1R]) is overexpressed in most murine and human melanomas. It is a promising molecular target for diagnosis and therapy of melanomas. However, 18F compounds have not been successfully developed for imaging the MC1R. Methods: In this study, an α-MSH analog, Ac-Nle-Asp-His-d-Phe-Arg-Trp-Gly-Lys-NH2 (NAPamide), was radiolabeled with N-succinimidyl-4-18F-fluorobenzoate (18F-SFB). The resulting radiopeptide was evaluated as a potential molecular probe for small-animal PET of melanoma and MC1R expression in melanoma xenografted mouse models. Results: The binding affinity of 19F-SFB−conjugated NAPamide, 19F-FB-NAPamide, was determined to be 7.2 ± 1.2 nM (mean ± SD) using B16/F10 cells and 125I-(Tyr2)-[Nle4,d-Phe7]-α-MSH [125I-(Tyr2)-NDP] as a radioligand. The biodistribution of 18F-FB-NAPamide was then investigated in C57BL/6 mice bearing subcutaneous murine B16/F10 melanoma tumors with high expression of MC1Rs and Fox Chase Scid mice bearing human A375M melanoma with a relatively low number of MC1R receptors. Biodistribution experiments showed that tumor uptake values (percentage injected dose per gram of tumor [%ID/g]) of 18F-FB-NAPamide were 1.19 ± 0.11 %ID/g and 0.46 ± 0.11 %ID/g, in B16/F10 and A375M xenografted melanoma at 1 h after injection, respectively. Furthermore, the B16/F10 tumor uptake was significantly inhibited by coinjection with excess α-MSH peptide (P < 0.05), indicating that 18F-FB-NAPamide specifically recognizes the MC1R in living mice. Small-animal PET of 18F-FB-NAPamide in mice bearing B16/F10 and A375M tumors at 1 h after tail vein injection revealed good B16/F10 tumor-to-background contrast and low A375M tumor-to-background ratios. Conclusion: 18F-FB-NAPamide is a promising molecular probe for α-MSH receptor-positive melanoma PET and warrants further study.
Cutaneous malignant melanoma is one of the most lethal cancers. Its incidence is increasing rapidly, making it a significant public health problem in Europe and the United States (1,2). Although extensive research has been performed to develop novel strategies for systemic treatment of malignant melanoma, effective therapies are still not available. The most important approach for improvement of survival of patients with melanoma still remains early diagnosis, along with accurate staging of the extent of disease (3,4).
Many molecular imaging agents have been evaluated for detection of melanoma. Currently, 18F-FDG is widely used in staging melanoma. The sensitivity, specificity, and accuracy of 18F-FDG PET for detecting recurrent melanoma range from 70% to 100% (3). However, when imaging patients with early-stage melanoma, whole-body 18F-FDG PET shows only a low mean sensitivity of 17.3% (0%–40%) for detecting sentinel lymph node(s) (SLN). 18F-FDG PET can also miss micrometastatic lesions that are <1 cm in diameter and are located mainly in the lungs, liver, or brain (3). Furthermore, 18F-FDG provides only the metabolic activity of tumors, as the molecular targets for 18F-FDG are glucose transporters (e.g., GLUT1) and hexokinase (5). Therefore, there is a need to develop imaging agents with higher sensitivity and specificity for small lesions and detection of SLN metastases as well as molecular probes that can visualize the expression and activity of other molecular targets and biologic processes in melanoma.
The α-melanocyte-stimulating hormone (α-MSH) receptor (melanocortin type 1 receptor [MC1R]) plays an important role in the proliferation and differentiation of melanocytes as well as in the development and growth of melanoma cells. It is considered as a genetic link to skin cancer (6). The MC1R is overexpressed in most murine and human melanoma metastases (7,8), thus making it a promising molecular target for imaging and therapy of melanomas. Various α-MSH peptides radiolabeled with 18F (9), 99mTc (10,11), 111In (12–17), 125I (18), 67Ga (19), 86Y (20), and 64Cu (20–22) that can recognize the MC1R in vitro or in vivo have been prepared and evaluated for melanoma detection. Moreover, an α-MSH peptide, ReCCMSH(Arg11), radiolabeled with a therapeutic radionuclide—either 188Re or 212Pb—has shown promising therapeutic efficacy in mice bearing either B16F1 murine or TXM13 human xenografted melanoma (23,24). These results highlight the potential of using α-MSH analogs tagged with radionuclides for metastatic melanoma imaging or peptide receptor-targeted radionuclide therapy.
For different human and murine melanoma cell lines, the MC1R expression ranges from several hundred to around 10,000 receptors per cell (7). To stratify a patient population and identify those suitable for MC1R-targeted therapy, a noninvasive imaging tool for visualizing and quantifying the MC1R expression in living subjects will be very useful. An α-MSH analog, Ac-Nle-Asp-His-d-Phe-Arg-Trp-Gly-Lys-NH2 (NAPamide), can target the MC1R with excellent pharmacokinetic properties in tumor mice models (19). We prepared and evaluated a 64Cu-labeled 1,4,7,10-tetraazacyclododecane-N,N′,N′′,N′′′-tetraacetic acid (DOTA)-NAPamide (21,22) for imaging MC1R expression using small-animal PET. Our results indicate that 64Cu-DOTA-NAPamide is able to image MC1R expression imaging in living mice. However, the biodistribution and small-animal PET data show that 64Cu-DOTA-NAPamide has high accumulation and retention in the liver and kidneys, which has been repeatedly observed for other radiometal-labeled peptides (10,12,20,25). The significant hepatobiliary accumulation and retention of the 64Cu-labeled compound is primarily due to the release of uncoordinated 64Cu from the radiolabeled product by decomposition in the blood or transchelation in the liver (26). Further studies to optimize the biodistribution of 64Cu-DOTA-NAPamide and to achieve better imaging quality are needed.
It is well known that radiohalogenated peptides compared with radiometal chelated peptides exhibit different metabolic patterns and clearance routes (27–30). The fast clearance of radiohalogenated peptides from normal tissues is sometimes an advantage for the development of imaging probes. Therefore, in the current study, 18F-radiolabeled NAPamide (18F-FB-NAPamide) was prepared by conjugation with N-succinimidyl-4-18F-fluorobenzoate (18F-SFB) through the lysine side-chain ε-amino group (Fig. 1). 18F-FB-NAPamide was then evaluated in C57BL/6 mice bearing subcutaneous murine B16/F10 melanoma tumors with high levels of the MC1R and Fox Chase Scid mice bearing human A375M melanoma with low levels of the MC1R.
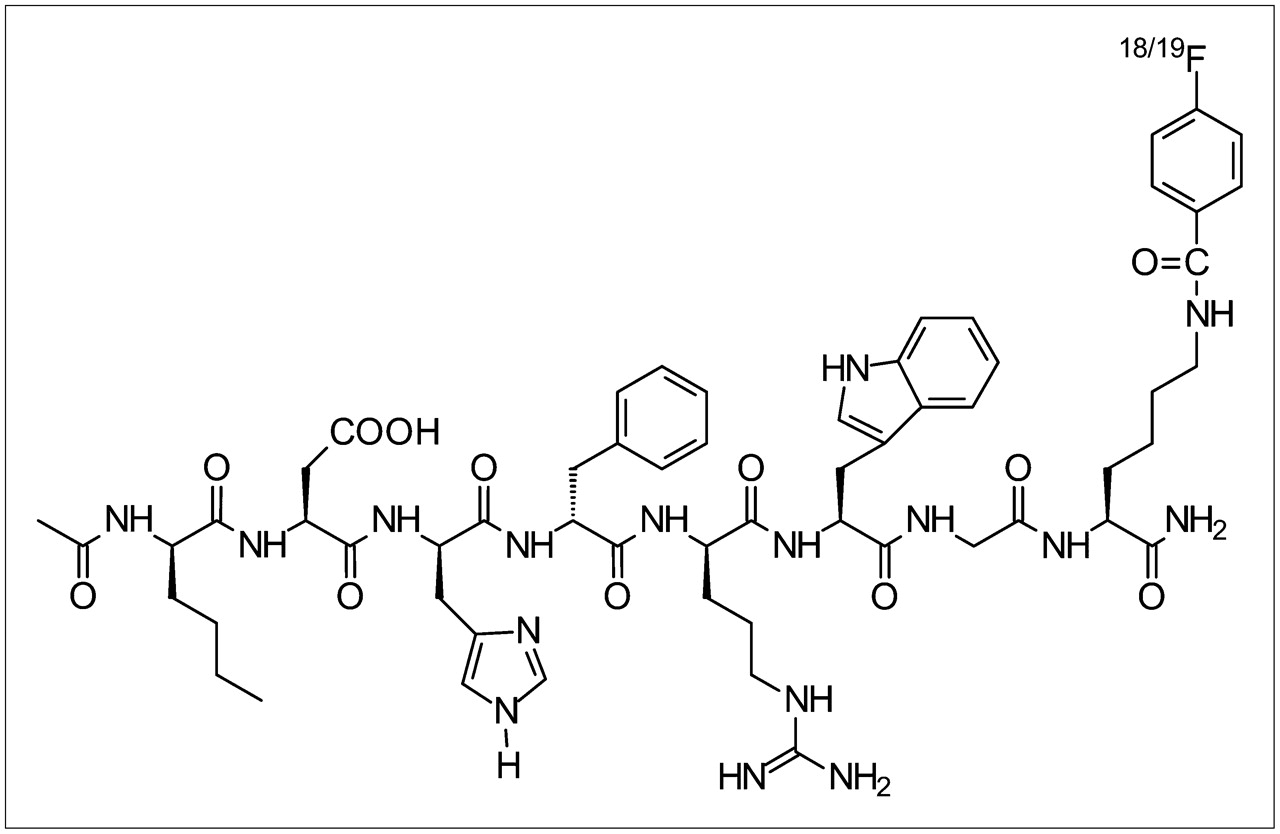
Schematic structure of 18/19F-FB-NAPamide.
MATERIALS AND METHODS
General
NAPamide was synthesized with >95% purity by the Stanford Protein and Nucleic Acid Biotechnology Facility, using standard 9-fluorenylmethyloxycarbonyl (Fmoc)/O-benzotriazolyl-tetramethyluronium hexafluorophosphate (HBTU) chemistry on rink amide 4-methylbenzhydrylamine (MBHA) resin. Ethyl 4-(dimethylamino)benzoate, methyl trifluoromethane-sulfonate, and O-(N-succinimidyl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TSTU), 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo-[8.8.8]hexacosan (K2.2.2), potassium carbonate (K2CO3), tetrabutylammonium hydroxide, and all other chemicals were obtained from Sigma-Aldrich Chemical Co. A CRC-15R PET dose calibrator (Capintec Inc.) was used for all radioactivity measurements. Reverse-phase high-performance liquid chromatography (RP-HPLC) was performed on a Dionex Summit HPLC system (Dionex Corp.) equipped with a 170U 4-Channel UV-Vis absorbance detector and radioactivity detector (model 105S; Carroll & Ramsey Associates). Ultraviolet (UV) detection wavelengths were 218, 254, and 280 nm for all experiments. Both semipreparative (Vydac; 218TP510-C18, 10 × 250 mm) and analytic (Dionex; Acclaim120 C18, 4.6 × 250 mm) RP-HPLC columns were used. The mobile phase was solvent A, 0.1% trifluoroacetic acid (TFA)/H2O, and solvent B, 0.1% TFA/acetonitrile. Matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS) was performed on a Perseptive Voyager-DE RP Biospectrometry instrument by the Stanford Protein and Nucleic Acid Biotechnology Facility. α-Cyano-4-hydroxy-cinnamic acid (α-CHCA, prepared as 10 g/L in 33.3% CH3CN:33.3% EtOH:33.3% H2O:0.1% TFA) was used as the solid matrix. A375M cell line was a generous gift from Dr. Michael Kolodny (University of California, Los Angeles, CA), and B16/F10 murine melanoma cells were obtained from the American Type Tissue Culture Collection. C57BL/6 and Fox Chase Scid mice were purchased from Charles River Laboratories.
Chemistry and Radiochemistry
The reference standard 19F-FB-NAPamide was prepared by reaction of NAPamide with N-succinimidyl 4-fluorobenzoate ([SFB] SFB was prepared as described earlier (31,32)). Briefly, NAPamide (11.6 mg, 10.6 μmol) dissolved in 100 μL water, SFB (5.0 mg, 21.1 μmol) dissolved in 100 μL acetonitrile, and 800 μL Na2HPO4 buffer (0.1 M, pH 9.0) were mixed and reacted for 2 h at room temperature. The reaction was then quenched by adding 50 μL TFA. The resulting FB-NAPamide conjugate was then purified by HPLC on a semipreparative C-18 column. The flow rate was 3 mL/min, with the mobile phase starting with 95% solvent A and 5% solvent B (0–3 min), going to 35% solvent A and 65% solvent B at 33 min, then going to 85% solvent B and 15% solvent A (33–36 min), maintaining this solvent composition for another 3 min (36–39 min), and returning to the initial solvent composition by 42 min. Fractions containing the product were collected and lyophilized. The identity of FB-NAPamide was confirmed by MALDI-TOF-MS.
The radiofluorination synthon 18F-SFB was prepared on the basis of the procedure reported previously (31,32). 18F-SFB (specific activity, 200–250 GBq/μmol) dissolved in 100 μL acetonitrile was added to the NAPamide peptide (100 μg) dissolved in Na2HPO4 buffer (900 μL, pH 8.0) and reacted for 2 h at 40°C. After adding 50 μL TFA to quench the reaction, the reaction solution was injected into a semipreparative HPLC column using the same elution gradient as the one used in the cold FB-NAPamide purification. The HPLC fractions containing the 18F-FB-NAPamide were then collected, combined, and evaporated with a rotary evaporator to dry the product. The radiolabeled peptide was reconstituted in phosphate-buffered saline (PBS) and passed through a 0.22-μm Millipore filter into a sterile vial for in vitro and animal experiments.
Octanol/Water Partition Coefficient
To determine the lipophilicity of 18F-FB-NAPamide, approximately 370 kBq of radiolabeled peptide in 500 μL of PBS (pH 7.4) was added to 500 μL of octanol in an Eppendorf microcentrifuge tube. The resulting biphasic system was mixed vigorously for 10 min and left at room temperature for another 60 min. The 2 phases were then separated by centrifugation at 2,000g for 5 min (model 5415R Eppendorf microcentrifuge; Brinkman). From each layer, an aliquot of 100 μL was removed and counted in a γ-counter (Packard Instruments). The partition coefficient (log P) was then calculated as a ratio of counts in the octanol fraction to the counts in the water fraction. The experiment was repeated 3 times.
Cell Assays
B16/F10 murine and A375M human melanoma cells were cultured in Dulbecco's modified Eagle high-glucose medium, which was supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, and 48 mg of gentamicin. The cells were expanded in 75-cm2 tissue culture flasks and kept in a humidified atmosphere of 5% CO2 at 37°C, with the medium changed every other day. A confluent monolayer was detached with trypsin and dissociated into a single cell suspension for further cell culture.
The in vitro cell-binding assays were performed with the murine melanoma cell line B16/F10 as previously described (7–9). Briefly, cells were seeded at a density of 0.2 million per well in 24-well tissue culture plates and allowed to attach overnight. After a wash with the binding medium (modified Eagle media with 25 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid), 0.2% bovine serum albumin [BSA], and 0.3 mM 1,10-phenanthroline), the cells were incubated at 25°C for 2 h with FB-NAPamide (peptide concentration varying from 10−12 to 10−6 M) and approximately 50,000 counts per minute (cpm) of 125I-(Tyr2)-[Nle4,d-Phe7]-α-MSH [125I-(Tyr2)-NDP] in 0.5 mL of binding media. The cells were rinsed twice with 0.01 M PBS (pH 7.4)/0.2% BSA and lysed in 0.5 mL of 1.0 M NaOH for 5 min, and the radioactivity of the cells was measured. The data were analyzed using the ORIGIN6.0 computer program, and the concentration of competitor required to inhibit 50% of the radioligand binding (IC50 value) of the FB-NAPamide was calculated.
Animal Biodistribution Studies
All animal studies were performed in compliance with federal and local institutional rules for the conduct of animal experimentation. C57BL/6 female mice, 7- to 8-wk old, were inoculated subcutaneously in the right shoulder with 1 × 106 cultured B16/F10 murine melanoma cells. Nine to 10 d after inoculation, the tumors had grown to a weight of ∼500 mg, and the tumor-bearing mice were subjected to in vivo biodistribution and imaging studies. Fox Chase Scid mice, 6-wk-old, were inoculated with 4 × 106 A375M human melanoma cells. Three weeks to 4 wk after inoculation, the mice bearing A375M tumors were ready for further studies.
For biodistribution studies, the C57BL/6 mice bearing B16/F10 murine melanoma allografts and Fox Chase mice bearing A375M human melanoma xenografts (n = 4 for each group) were injected with about 740 kBq (20 μCi) of 18F-FB-NAPamide with (blocking group, n = 3) or without 200 μg NDP ([Nle4,d-Phe7]α-MSH) through the tail vein and were sacrificed at different time points from 1 to 4 h after injection. Melanoma-bearing mice were also injected with 18F-FDG (3.7–7.4 MBq) under the same experimental conditions as that for 18F-FB-NAPamide (no fasting and no anesthesia, n = 3). At different times after the injection of radioactivity, the mice were sacrificed using carbon dioxide. Tumor and normal tissues of interest were removed and weighed, and their radioactivity was measured in a γ-counter. The radioactivity uptake in the tumor and normal tissues was expressed as a percentage of the injected radioactive dose per gram of tissue (%ID/g).
microPET Imaging
PET of tumor-bearing mice was performed on a microPET R4 rodent model scanner (Siemens Medical Solutions USA, Inc.). The mice bearing B16/F10 allografts and A375M melanoma xenografts were injected with about 3.7 MBq (100 μCi) of 18F-FB-NAPamide via the tail vein. At 120 min after injection, the mice were anesthetized with 2% isoflurane and placed in the prone position near the center of the field of view of the microPET scanner. Ten-minute static scans were obtained, and the images were reconstructed by a 2-dimensional ordered-subsets expectation maximization algorithm.
Statistical Method
Statistical analysis was performed using the Student t test for unpaired data. A 95% confidence level was chosen to determine the significance between groups, with P < 0.05 being significantly different.
RESULTS
Chemistry and Radiochemistry
The synthesis of the 19F-FB-NAPamide conjugate was achieved through coupling of SFB with the ε-amino group of the lysine residue in the NAPamide. The desired product was purified by semipreparative HPLC, and the purity of the target compound was generally obtained in a 30%–40% yield and >95% purity. The retention time on analytic HPLC for 19F-FB-NAPamide was found to be 19.2 min. The purified 19F-FB-NAPamide was characterized by MALDI-TOF-MS. The measured molecular weight (MW) was consistent with the expected MW: m/z = 1220.56 for [M+H]+ (C59H78FN16O12, calculated MW = 1221.59). Similarly, 18F-FB-NAPamide was prepared by conjugation of NAPamide with radioactive 18F-SFB (specific activity of 200–250 GBq/μmol, as estimated by radio-HPLC). Its retention time on analytic HPLC was also found to be 19.2 min. The total time for radiosynthesis of radiopeptide took about 3 h. The maximum overall radiochemical yield with decay correction was 37%. The radiochemical purity of the labeled peptide was >95%, as verified by analytic HPLC analysis (Fig. 2). From the octanol/water partition coefficient measurement, the log P value of the tracer was determined to be –0.78 ± 0.05 (mean ± SD), indicating intermediate hydrophilicity.

HPLC radiochromatogram of purified 18F-FB-NAPamide.
In Vitro Receptor-Binding Assay
The receptor-binding affinity study of 19F-FB-NAPamide for the MC1R was performed using B16/F10 cells with high MC1R expression. The IC50 value of the peptide was 7.2 ± 1.2 nM (mean ± SD) using a competitive receptor-binding assay (Fig. 3, n = 3), suggesting that 18F-FB-NAPamide is worthy of further in vivo evaluation.

In vitro inhibition of 125I-(Tyr2)-NDP binding to MC1R on murine B16/F10 melanoma cell line by FB-NAPamide (IC50 = 7.2 ± 1.2 nM). Results expressed as percentage of binding are mean ± SD of triplicate measurements.
Biodistribution Studies
The in vivo biodistribution of 18F-FB-NAPamide was examined in either B16/F10 murine allograft or A375M human xenograft melanoma-bearing mice. Biodistribution of the radiolabeled peptide with or without coinjection of 200 μg NDP at 1 and 4 h was obtained in the B16/F10 tumor model, whereas only 1-h biodistribution data were obtained in the A375M melanoma model (Table 1). In both tumor models, the radiofluorinated peptide displayed rapid blood clearance; 0.65 ± 0.07 and 0.38 ± 0.18 %ID/g blood uptake was observed at 1 h after injection for B16/F10 and A375M tumor-bearing mice, respectively. The difference of blood uptake in these 2 tumor models was not significant (P = 0.06). Moderate activity accumulation in the high MC1R-expressing B16/F10 tumor was also observed at early time points (1.19 ± 0.11 %ID/g at 1 h after injection), and the activity decreased to 0.25 ± 0.05 %ID/g at 4 h after injection (Table 1). For the A375M tumors with low MC1R expression, tumor uptake was 0.46 ± 0.11 %ID/g at 1 h after injection (Table 1), which was significantly lower than the uptake of 18F-FB-NAPamide in the B16/F10 tumor model (P < 0.05). Furthermore, the in vivo specificity of 18F-FB-NAPamide tumor uptake was investigated by coinjection of 200 μg of NDP with the radiofluorinated peptide in both tumor models. In the B16/F10 tumor model, coinjection of NDP specifically reduced the tumor uptake of 18F-FB-NAPamide from 1.19 ± 0.11 to 0.61 ± 0.09 %ID/g (P < 0.05) without changing the distribution of 18F-FB-NAPamide in most normal tissues studied. However, coinjection of NDP with radiolabeled peptide did not significantly reduce the uptake of 18F-FB-NAPamide in the A375M tumor (0.46 ± 0.11 %ID/g for nonblocking group vs. 0.33 ± 0.02 %ID/g for blocking group) at 1 h after injection (Table 1).
Biodistribution Data for 18F-FB-NAPamide in C57BL/6 Mice Bearing Subcutaneously Xenotransplanted B16/F10 Murine Melanoma and Fox Chase Scid Mice Bearing A375M Human Melanoma
For both tumor models, 18F-FB-NAPamide displayed relatively low accumulation and retention in the liver. For example, the liver uptake in the B16/F10 tumor model was only 0.79 ± 0.25 and 0.13 ± 0.14 %ID/g at 1 and 4 h after injection, respectively. However, high renal activity was also found at all times after injection in both tumor models (Table 1).
The biodistribution of 18F-FDG was also studied in 2 melanoma mouse models. In the B16/F10 melanoma model, 18F-FDG showed high accumulation in tumors. B16/F10 tumor uptake (mean ± SD) of 10.86 ± 3.85 and 6.18 ± 1.04 %ID/g was observed at 2 and 4 h after injection, respectively. Moreover, 18F-FDG showed rapid clearance from the blood. A high tumor-to-blood ratio (29.20 ± 4.99 %ID/g) was achieved even at 1 h after injection. However, high muscle uptake of 18F-FDG (4.52 ± 2.27 and 2.27 ± 0.37 %ID/g at 2 and 4 h after injection, respectively) resulted in a low tumor-to-muscle ratio at both times points (Table 2). In the A375M melanoma-bearing mouse model, 18F-FDG showed low tumor accumulation (1.61 ± 0.29 %ID/g), moderate blood uptake (0.53 ± 0.30 %ID/g), and high muscle uptake (3.89 ± 1.14 %ID/g) at 1 h after injection, respectively. Poor tumor-to-blood and tumor-to-muscle ratios were obtained for 18F-FDG in the A375M model.
Biodistribution Data for 18F-FDG in C57BL/6 Mice Bearing Subcutaneously Xenotransplanted B16/F10 Murine Melanoma at 2 and 4 Hours After Injection and Fox Chase Scid Mice Bearing A375M Human Melanoma at 1 Hour After Injection
microPET Imaging
Decay-corrected coronal (Fig. 4, top) and transaxial (Fig. 4, bottom) microPET images of mice bearing B16/F10 tumors on the right shoulder (left and middle images) and mice bearing A375M tumors (right images) at 1 h after tail vein injection of 18F-FB-NAPamide (3.7 MBq [100 μCi]) are shown in Figure 4. The B16/F10 tumor was clearly visualized with good tumor-to-contralateral background contrast at 1 h after injection (left images in Fig. 4). Moreover, coinjection of NDP with radiolabeled peptide reduced the uptake of 18F-FB-NAPamide in the B16/F10 tumor at 1 h, resulting in a lower tumor-to-background ratio (middle images in Fig. 4). Finally, lower uptake and poor tumor to normal tissue contrast in A375M tumors was also observed (right images in Fig. 4).

Decay-corrected coronal (top) and transaxial (bottom) microPET images of C57BL/6 mouse bearing B16/F10 tumor (left and middle images) and Fox Chase Scid mouse bearing A375M tumor (right images). Images were acquired 1 h after tail vein injection of 18F-FB-NAPamide (3.7 MBq [100 μCi]) with or without coinjection of 200 μg NDP peptide. Arrows indicate location of tumors.
DISCUSSION
Small synthetic peptides have emerged as attractive carriers for the delivery of imaging moieties (radionuclides, fluorescence dyes, or paramagnetic metal ions) to diseased tissues for molecular imaging (33). This type of molecule possesses several distinct advantages: (a) easy synthesis and modification, with various established labeling approaches available; (b) favorable pharmacokinetics; (c) high-affinity and high-specificity peptides available for different molecular targets (receptors, antigens, enzymes, and so forth); and (d) low toxicity and immunogenicity. (34–36). Radiolabeled peptides usually show a rapid blood clearance and high target-to-background ratios at an early time after injection because of their small size. Therefore, short-lived radioisotopes (18F, 99mTc, and so forth) are great candidates for labeling peptides. Currently, 18F appears to be a favorable PET radionuclide of choice for labeling bioactive peptides by virtue of its excellent physical properties and wide availability (34). Various techniques have been developed to radiofluorinate peptides efficiently and site specifically. Some 18F-labeled peptides hold great potential in clinical applications for quantitatively detecting and characterizing molecular targets in human diseases using PET (34,37). The α-MSH analog NDP was labeled with 18F-SFB (9). The resulting radiopeptide displays high in vitro binding affinity to α-MSH receptors in B16/F1 cells (IC50 = 112 ± 22 pM). It also shows rapid clearance from normal tissues in normal BALB/c mice. However, to our knowledge, the use of 18F-SFB-labeled NDP and any other 18F-labeled α-MSH peptides for PET imaging of melanoma has not yet been reported (9).
We recently synthesized a 64Cu-labeled α-MSH peptide, 64Cu-DOTA-NAPamide, and successfully demonstrated its use for microPET imaging of melanoma and MC1R expression (21,22). In this research, we further develop 18F-labeled NAPamide as a molecular imaging probe for MC1R microPET imaging. The radiofluorination of NAPamide can be easily and efficiently achieved through site-specific conjugation of the radiosynthon 18F-SFB with the lysine side-chain ε-amino group. 18F-FB-NAPamide was prepared in reasonable yield (∼37%) and specific activity (200–250 GBq/μmol) at the end of synthesis. As determined by the competition receptor-binding assay, the fluorinated NAPamide 19F-FB-NAPamide displays high binding affinity with MC1R in B16/F10 cells (7.2 ± 1.2 nM). The binding affinity of unmodified peptide, NAPamide, is reported to be 0.27 ± 0.07 nM using B16/F1 cells and 125I-(Tyr2)-NDP as radioligand (9). These data suggest that the lysine residue in NAPamide tolerates modification, to some extent, and may be suitable for conjugation with different labeling moieties. The high affinity of nonradioactive 19F-FB-NAPamide further warrants in vivo evaluation of its radioactive counterpart.
Subsequently, 2 melanoma cell lines—B16/F10 with high MC1R capacity (21,687 ± 4,171 sites/cell) and A375M with low receptor density (400 ± 93 sites/cell) (21,22)—were used to inoculate female C57BL/6 and Fox Chase Scid mice, respectively. The in vivo biodistribution of 18F-FB-NAPamide indicates that the tracer has higher tumor uptake values and tumor-to-muscle ratios at 1 h after injection in B16/F10 mice than those in A375M mice (P < 0.05) (Table 1). Furthermore, the B16/F10 tumor uptake can be significantly inhibited by coinjection with cold NDP peptide (P = 0.002), whereas no significant inhibition of A375M tumor uptake was found (P = 0.12) (Table 1). The similar uptake of the blocking and nonblocking groups can be explained by the very low MC1R number in A375M cells (400 sites/cell). These data clearly indicate that 18F-FB-NAPamide specifically recognizes the MC1R in vivo. microPET of 18F-FB-NAPamide in mice bearing A375M or B16/F10 tumors at 1 h after tail vein injection reveals a good B16/F10 tumor-to-background contrast, whereas a lower A375M tumor-to-background ratio is observed (Fig. 4). The reduced B16/F10 tumor-to-background ratio with coinjection of NDP supports the specificity of the tracer in vivo (Fig. 4). Overall, the PET data are consistent with the finding obtained from the biodistribution studies. 18F-FB-NAPamide shows a great potential to image melanoma with different MC1R densities in living subjects using quantitative microPET imaging technology. Future research is needed to correlate the imaging signal with the target receptor levels and to evaluate the tracer in more melanoma models. Such studies will further elucidate the use of the 18F-FB-NAPamide for imaging MC1R expression.
A biodistribution study of 18F-FDG in B16/F10 and A375M tumor-bearing mice was also performed. 18F-FDG shows a high B16/F10 tumor uptake and tumor-to-blood ratio at 2 and 4 h after injection, whereas a low A375M tumor accumulation and tumor-to-blood ratio were observed. Moreover, the high uptake of 18F-FDG in muscle also results in low tumor-to-muscle ratios for both B16/F10 and A375M tumor models. The results described earlier suggest that the glucose metabolic rate for murine melanoma B16/F10 and human melanoma A375M is different. It was also found that 18F-FB-NAPamide showed higher tumor-to-muscle ratios in B16/F10 mice at 4 h after injection (7.46 ± 2.68 vs. 2.85 ± 1.01, P < 0.05) and in A375M mice at 1 h after injection (2.07 ± 0.08 vs. 0.44 ± 0.15, P < 0.05) (Tables 1 and 2). Note that for the 18F-FDG biodistribution studies, mice were neither fasted nor anesthetized before and after 18F-FDG administration. This could have explained the high uptake of 18F-FDG in muscle. Perhaps, a more optimal tumor or normal tissue 18F-FDG uptake could have been obtained if precautions had been taken in handling the mice. Whether 18F-FB-NAPamide might have some advantages over 18F-FDG in identifying melanoma lesions under certain clinical conditions remains to be determined.
Compared with 64Cu-DOTA-NAPamide (21,22), 18F-FB-NAPamide shows reduced accumulation and retention in liver and kidney (Table 1), but the tumor uptake and retention are also reduced in B16/F10 mice. For example, the B16/F10 tumor uptake at 1 and 4 h after injection was 1.19 ± 0.11 and 0.25 ± 0.05 %ID/g, respectively. This phenomenon is common and has been previously reported (18). It is usually caused by a fast clearance from normal tissues and a rapid release of radioactivity from tumors after internalization of radiohalogenated peptides. Similarly, 18F-FB-NAPamide displays much lower murine melanoma uptake than that of 99mTc-CCMSH (10). However, considering the high sensitivity of PET over SPECT, the 18F-labeled α-MSH analog may still possess some advantages over SPECT radionuclide-labeled α-MSH peptide. Several strategies can be applied to design a better MC1R-targeted radiofluorinated α-MSH peptide—for example, use of a receptor-targeting component that resists proteolytic degradation, incorporation of a d- or nonnatural amino acid such as d-Lys to serve as a residue for label conjugation, and selection of radiofluorination synthons with high enzymatic stability and long cellular retention, and so forth. The highly stable rhenium cyclized α-MSH peptide Ac-d-Lys-ReCCMSH(Arg11), reported previously (18), may also be a good candidate for developing a 18F-labeled peptide for MC1R imaging.
CONCLUSION
The α-MSH analog 19/18F-FB-NAPamide with high affinity to the MC1R was successfully synthesized. Biodistribution and microPET studies demonstrate that the tracer can differentiate B16/F10 and A375M with high and low MC1R expression, respectively. 18F-FB-NAPamide is a promising PET molecular probe for imaging MC1R-positive melanoma and MC1R expression in vivo.
Acknowledgments
This work was supported, in part, by National Cancer Institute (NCI) Small Animal Imaging Resource Program grant R24 CA93862 and by NCI In Vivo Cellular Molecular Imaging Center grant P50 CA114747.
Footnotes
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication January 4, 2007.
- Accepted for publication March 16, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- 52Mn-labelled Beta-cyclodextrin for Melanoma Imaging: A Proof-of-concept Preclinical Study
- 18F-PFPN PET: A New and Attractive Imaging Modality for Patients with Malignant Melanoma
- 4-11C-Methoxy N-(2-Diethylaminoethyl) Benzamide: A Novel Probe to Selectively Target Melanoma
- Theranostics of Malignant Melanoma with 64CuCl2
- Melanoma Imaging with Highly Specific PET Probes: Ready for Prime Time?
- PET of Malignant Melanoma Using 18F-Labeled Metallopeptides
- Melanin-Targeted Preclinical PET Imaging of Melanoma Metastasis