


Abstract
Dialkylamino-alkyl-benzamides possess an affinity for melanin, suggesting that labeling of such benzamides with 18F could potentially produce melanin-targeted PET probes able to identify melanotic melanoma metastases in vivo with high sensitivity and specificity. Methods: In this study, N-[2-(diethylamino)ethyl]-4-18F-fluorobenzamide (18F-FBZA) was synthesized via a 1-step conjugation reaction. The σ-receptor binding affinity of 19F-FBZA was determined along with the in vitro cellular uptake of radiofluorinated 18F-FBZA in B16F10 cells. In vivo distribution and small-animal PET studies were conducted on mice bearing B16F10 melanoma, A375M amelanotic melanoma, and U87MG tumors, and comparative studies were performed with 18F-FDG PET in the melanoma models. Results: In vitro, uptake of 18F-FBZA was significantly higher in B16F10 cells treated with l-tyrosine (P < 0.001). In vivo, 18F-FBZA displayed significant tumor uptake; at 2 h, 5.94 ± 1.83 percentage injected dose (%ID) per gram was observed in B16F10 tumors and only 0.75 ± 0.09 %ID/g and 0.56 ± 0.13 %ID/g was observed in amelanotic A375M and U87MG tumors, respectively. Lung uptake was significantly higher in murine lungs bearing melanotic B16F10 pulmonary metastases than in normal murine lungs (P < 0.01). Small-animal PET clearly identified melanotic lesions in both primary and pulmonary metastasis B16F10 tumor models. Coregistered micro-CT with small-animal PET along with biopsies further confirmed the presence of tumor lesions in the mouse lungs. Conclusion: 18F-FBZA specifically targets primary and metastatic melanotic melanoma lesions with high tumor uptake and may have translational potential.
Malignant melanoma is one of the most lethal cancers because of its high systemic metastatic potential. The incidence of this disease has doubled over the past 2 decades and is continually increasing, making it a significant public health problem in Europe and the United States (1,2). Currently, although regimens for effective treatment of melanoma are still not available, increased surveillance with early diagnosis and accurate staging of the disease is an important approach to increasing survival. PET offers the promise of noninvasively imaging micrometastases (3,4) but must be coupled with an appropriate probe to provide oncologists with highly sensitive detection of metastases and accurate staging of high-risk melanomas.
Through imaging of different tumor molecular targets and pathways, several PET probes, including 18F-FDG (3–5), 6-18F-fluoro-l-dihydroxyphenylalanine (6), l-[methyl-11C]methionine (7), 3′-18F-fluoro-3′-deoxy-l-thymidine (8), 18F-galacto-RGD peptide (9), and others, have been evaluated for melanoma detection in patients. However, reports suggest that the overall detection rate has been “extremely low” for occult metastatic lesions in patients with stage IB or II melanoma using 18F-FDG PET/CT (5). 18F-FDG also failed to identify metastatic lesions smaller than 1 cm in diameter located mainly in common sites for melanoma metastases: the lungs, liver, or brain (3). Moreover, the molecular targets for 18F-FDG are glucose transporters (e.g., glucose transporter 1) and hexokinase, which relate to the glycolytic activity of tumors (10). Novel PET probes with a higher specificity and sensitivity for other molecular targets and biologic processes in melanoma are still highly desired for visualizing and monitoring their expression and activity or for detecting small lesions and metastases.
Melanin is an amorphous, irregular polymer comprising mixtures of 2 separate but biogenetically related pigments, eumelanins and phenomelanins (11,12). Melanin biosynthesis is an essential metabolic pathway regulated by tyrosinase activity in melanocytes (13). In malignant melanoma, melanin formation is highly increased because tyrosinase activity is significantly elevated (12,14). Taking advantage of the unique physiologic process of melanin synthesis, many studies have developed melanin-targeted radiotherapeutic and chemotherapeutic (15–17) agents for melanoma treatment. Benzamide analogs possess selective affinities with melanin and, over the past 2 decades, have been extensively investigated for development of SPECT agents for melanoma detection. Many benzamide analogs exhibit excellent in vivo tumor-targeting profiles. It was reported that N-(2-diethylaminoethy1)-4-125I-iodo-benzamide (125I-BZA) displayed uptake of 6.75 ± 0.67 percentage injected dose per gram (%ID/g) at 1 h in the tumors of C57BL6 mice bearing B16 melanoma. At 24 h, a tumor-to-blood ratio of as high as 37.3 ± 6.9 was attained (18). Several iodinated benzamide derivatives, including 123I-BZA (19), N-(2-diethylaminoethyl)-2-123I-iodobenzamide (20), 123/131I-N-(2-diethylaminoethyl)-3-iodo-4-methoxybenzamide (21), and 123I-2-hydroxy-3-iodo-6-methoxy-N-[(1-ethyl-2-pyrrolidinyl)methyl] benzamide (22), have been further studied in patients for melanoma imaging. These SPECT probes have shown promising results for clinical detection of melanoma lesions. Additionally, based on the structural elements of these benzamides, several 99mTc-complexes have also been designed and also display high uptake in melanoma tumors and excellent in vivo melanoma-imaging characteristics with SPECT in preclinical models (23,24). Very recently, a benzamide analog labeled with 18F was evaluated in a subcutaneous melanoma model and a biodistribution study demonstrated its promising tumor-targeting ability (25). Thus, the benzamide analogs are a reasonable starting point for the further development of melanin-targeted 18F-labeled probes.
18F is an ideal PET probe (half-life, 110 min; β+ particles emitted at an energy of 635 keV; 97% abundant). In this study, an 18F-labeled benzamide analog, N-[2-(diethylamino)ethy1]-4-18F-fluorobenzamide (18F-FBZA), was synthesized and evaluated for melanin-targeted melanoma imaging.
MATERIALS AND METHODS
General
N-succinimidyl-4-fluorobenzoate (SFB) was purchased from ABX GmbH. All other chemicals, including N,N-diethylenediamine (DEDA), trifluoroacetic acid, N,N′-diisopropylethylamine, and acetonitrile (CH3CN), were purchased from Sigma-Aldrich Chemical Co. The tumor cell lines and all instruments, including the electrospray ionization mass spectrometry, nuclear magnetic resonance, reverse-phase high-performance liquid chromatography (HPLC), and PET dose calibration equipment, were the same as described in our previous publication (26).
Synthesis of 18/19F-FBZA
The nonradioactive reference standard 19F-FBZA was prepared by reaction of DEDA with SFB. Briefly, DEDA (9.8 mg) and SFB (5.3 mg) dissolved in 300 μL of dimethyl sulfoxide and 5 μL of N,N′-diisopropylethylamine were mixed and reacted for 80 min at 50°C. The reaction solution was injected into a semipreparative HPLC column for purification. The flow rate was 3 mL/min, with the mobile phase starting with 95% solvent A and 5% solvent B (0–3 min), going to 35% solvent A and 65% solvent B for 33 min, and then changing to 15% solvent A and 85% solvent B, which was maintained for another 3 min (36–39 min), followed by a return to the initial solvent composition by 42 min. Fractions containing the product were collected (retention time, 15.5 min) and yielded 70% of the desired compound, which was subsequently lyophilized and characterized by electrospray ionization mass spectrometry or nuclear magnetic resonance. The measured molecular weight was consistent with the expected molecular weight: m/z = 239.08 measured for [M+H]+ (C13H20FN2O calculated molecular weight = 239.15); 1H-nuclear magnetic resonance (400 MHz, dimethyl sulfoxide-d6): δ 0.97 (quartet, 8H), 2.5 (s, 15H), 3.32 (t, 18H), 7.3 (dd, 2H), 7.9 (dd, 2H), 8.5 (t, 1H).
The radiofluorination synthon, 18F-SFB, was prepared according to a previously reported procedure (26). 18F-SFB (specific activity, 200–250 GBq/μmol) dissolved in 100 μL of dimethyl sulfoxide was added to the DEDA (100 μg) and N,N′-diisopropylethylamine (5 μL) and reacted for 30 min at 50°C. The reaction solution was injected into an analytic HPLC column using the same elution gradient (flow rate, 1 mL/min) as for the synthesis of nonradioactive 19F-FBZA. The HPLC fractions containing the radiolabeled product 18F-FBZA were collected, combined, and evaporated with a rotary evaporator. The 18F-FBZA was reconstituted in phosphate-buffered saline and passed through a 0.22-μm Millipore filter into a sterile vial for in vitro and in vivo animal experiments. The total radiosynthesis time for 18F-FBZA was 3 h, with an overall decay-corrected yield of 50% at the end of synthesis.
Serum Stability
The in vitro stability of 18F-FBZA was evaluated by incubation of 7.4 MBq (∼200 μCi) of probe with mouse serum (1 mL) at 37°C. At different times (30, 60, 120, and 150 min), the solution was filtered using a NanoSep 10 K centrifuge (Pall Corp.) to isolate low-molecular-weight metabolites. The filtrates were analyzed by reverse-phase HPLC under conditions identical to those used for analyzing original 18F-FBZA.
In Vitro Cell Uptake Studies
B16F10, A375M, and U87MG cells were cultured in Dulbecco's modified Eagle high-glucose medium (Gibco Life Sciences) supplemented with 10% fetal bovine serum with penicillin and streptomycin. The cells were regularly maintained in a 37°C, 5% CO2 humidified incubator. The cellular uptake studies were performed on B16F10 cells. Briefly, about 1 × 106 B16F10 cells were plated in a 12-well plate and pretreated with 2 mM l-tyrosine for 24 h. The cells were then incubated with advanced modified Eagle's medium containing 25 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid), 0.2% bovine serum albumin, and 0.3 mM 1,10-phenanthroline containing 3.7 kBq (0.1 μCi) of 18F-FBZA for 30 min, and for 120 min under either 37°C or 4°C. Nontreated cells were used as a control. The cells were then washed 3 times with ice-cold phosphate-buffered saline and lysed with 0.5 N NaOH for 5 min at room temperature. The radioactivity of the cell lysate was counted by a Wallac 1480 automated counter (Perkin Elmer). The counts per minute and percentage of uptake were plotted as a function of time using Prism 5.0 (GraphPad).
σ-Receptor Binding Assays
σ-receptor binding assays were conducted using standard methods detailed previously (27). The in vitro σ1-binding affinity of 19F-FBZA was determined in a competition assay using guinea pig brain membranes and the high-affinity σ1-ligand 3H-(+)-pentazocine. The σ2-receptor binding affinity of 19F-FBZA was determined using rat liver membrane preparations along with 3H-1,3-di-o-tolylguanidine as the radioligand in the presence of 10 μM pentazocine to mask σ1-receptors. Competition assays were performed with 12 concentrations of 19F-FBZA ranging from 1 × 10−10 to 1 × 10−3 M and protein samples (0.15 mg of membrane protein) in Tris-HCl (50 mM), pH 8.0, for 120 min at 25°C in a 0.25-mL volume.
Biodistribution
All animal studies were performed in compliance with the federal and local institutional rules for the conduct of animal experimentation. The subcutaneous tumor models were created with 5- to 6-wk-old male C57BL/6 mice (Charles River) for B16F10 and Nu/Foxn1 mice (Harlan) for A375M or U87MG. Pulmonary melanoma metastases were established in C57BL/6 mouse lungs via tail vein injection of 0.2 × 106, 0.4 × 106, or 0.8 × 106 B16F10 cells.
Biodistribution studies were conducted 1 or 2 h after tail vein injection of 18F-FBZA in tumor-bearing mice (B16F10, A375M and U87MG; n = 3 each). The mice were sacrificed at 1 or 2 h after injection. Tumors, blood, and other major organs of interest were harvested, weighed, and counted in a Wallac 1480 automated counter. The results were expressed as %ID/g. To compare the melanoma-imaging abilities of 18F-FBZA with 18F-FDG, B16F10 mice were kept fasting overnight before the experiment. 18F-FDG (3.7 MBq, 100 μCi) was injected via the tail vein of the B16F10 tumor–bearing mice, and small-animal PET and biodistribution experiments were preformed 1 h after injection (n = 5).
In Vivo Imaging Procedure
PET of tumor-bearing mice was performed on an R4 rodent model scanner (Concord Microsystems). A group of mice (n = 3) bearing melanotic B16F10 was injected with 18F-FBZA (3.31–3.86 MBq [89.4–104.3 μCi]) via the tail vein. For the amelanotic A375M tumors (n = 3), 0.87−0.95 MBq (23.6–25.8 μCi) of the probe was injected, whereas for U87MG tumors (n = 3), 0.29–0.4 MBq (7.95–10.93 μCi) of 18F-FBZA was injected. At 1 and 2 h after injection, the mice were anesthetized with 2% isoflurane (AErrane; Baxter) and placed prone near the center of the field of view of the scanner. For 18F-FDG studies, a 3.7-MBq (100-μCi) dose of 18F-FDG was injected via the tail vein. Five-minute static scans were obtained, and the images were reconstructed using a 2-dimensional ordered-subsets expectation maximum algorithm.
Respiratory gated micro-CT images were acquired using an in vivo scanner (eXplore Locus; GE Healthcare). Immediately after the PET scan, the mice were transported to and positioned in the CT scanner while still fixed to their polystyrene bed containing 4 fiducial markers at different positions. The mice were sedated with 2% isoflurane during the scan. The micro-CT images were acquired with the x-ray source set at 70 kVp and 40 μA and synchronized by respiratory gating on a Biovet system. CT images were reconstructed using a fanbeam re-sorting algorithm with a standard ramp filter. Images were reconstructed on a 512 × 512 pixel grid with a pixel size of 49 × 49 μm. No radiographic contrast medium was used.
PET images were imported using ASIPro VM (Concorde Microsystems). Regions of interest (ROIs) were drawn manually over the tumor or organ of interest on decay-corrected whole-body coronal images. The mean counts per pixel per minute were obtained from the ROIs and converted to counts per milliliter per minute using a calibration constant. No attenuation correction was performed. CT images were imported using MicroView (version 2.1.2; GE Healthcare). For coregistration with PET datasets, the fiducial markers were aligned using a wizard in the nonproprietary AMIDE software, version 0.9.1 (28).
Statistical Methods
Statistical analysis was performed using the Student t test for unpaired data. A 95% confidence level was chosen to determine the significance of differences between groups, with a P value of less than 0.05 indicating a significant difference.
RESULTS
Chemistry and Radiochemistry
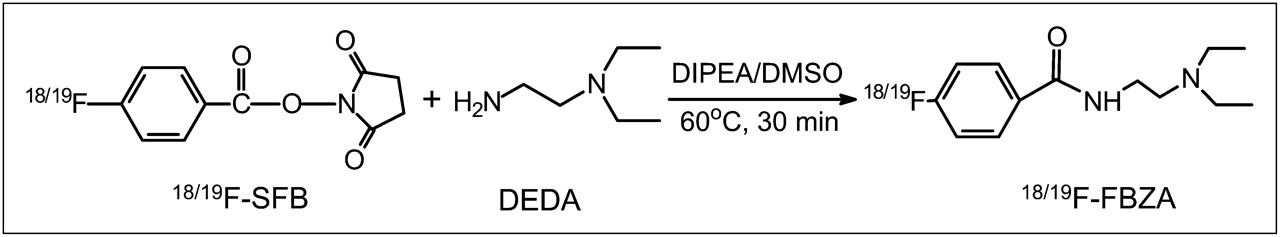
The nonradioactive 19F-FBZA was synthesized in a 1-step coupling reaction between DEDA and SFB (Fig. 1). HPLC purification of the nonradioactive 19F-FBZA yielded approximately 70% of the desired product with a 15.5-min retention time. The identity of the isolated compound was subsequently verified and confirmed by electrospray ionization mass spectrometry and nuclear magnetic resonance.

Synthetic scheme for preparation of 18/19F-FBZA.
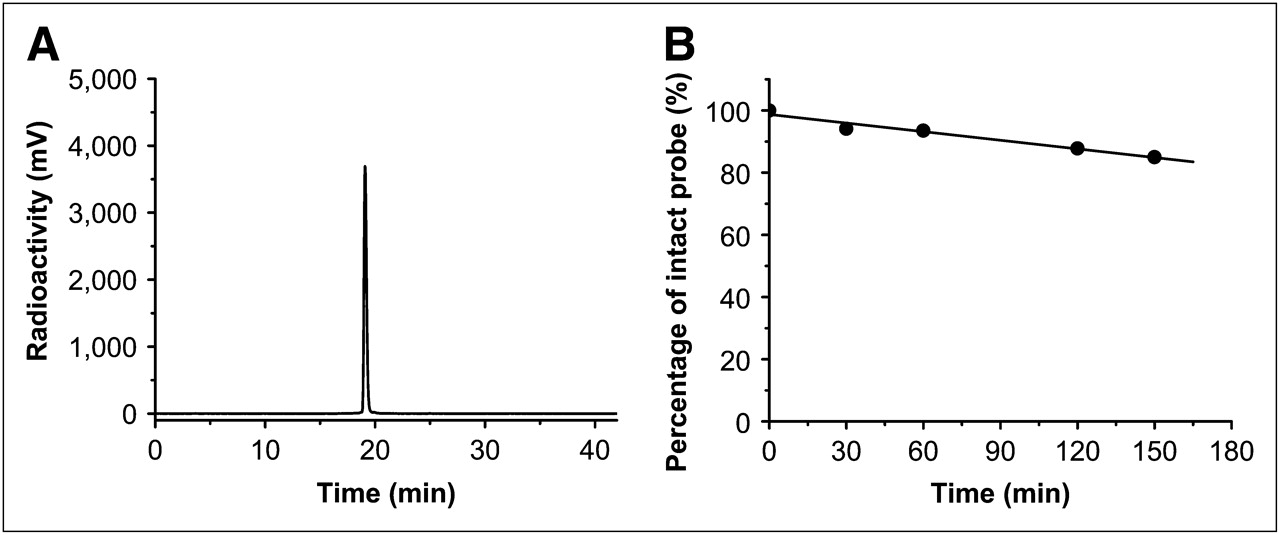
Similarly, radiosynthesis of 18F-FBZA was achieved through coupling of the radiosynthon, 18F-SFB, with the amino group of DEDA. The procedure and radiochemical synthetic module for production of the radiosynthon 18F-SFB have been well established in our radiochemistry facility, and 18F-SFB has been successfully used and described in our earlier work for 18F-labeling of a peptide for melanoma imaging (26). The total time for radiosynthesis of 18F-FBZA was approximately 3 h. The maximum overall radiochemical yield with decay correction was 50%, and the specific activity of 18F-FBZA was estimated to be 132–166 GBq/μmol at the end of synthesis. The radiochemical purity of the product was greater then 95% as verified by analytic HPLC analysis (Fig. 2A). The authenticity of 18F-FBZA was verified by coinjection with the previously characterized nonradioactive 19F-FBZA. Both cold and radioactive FBZA compounds displayed similar HPLC retention times.
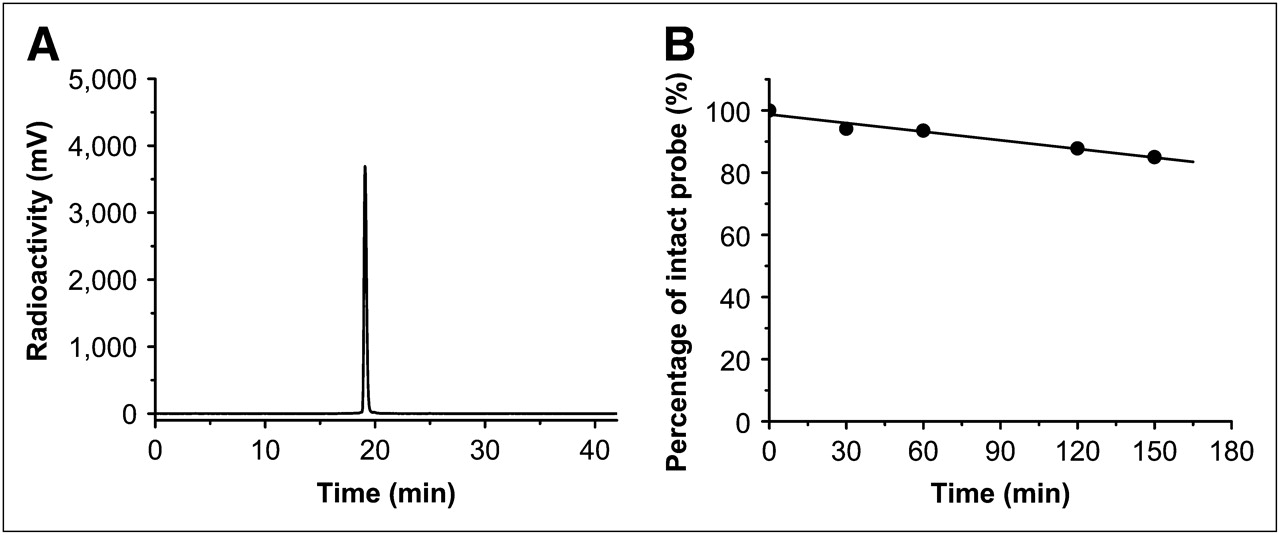
HPLC radiochromatogram of purified 18F-FBZA (A), and in vitro mouse serum stability study of 18F-FBZA (B).
Serum Stability of 18F-FBZA
18F-FBZA displayed good stability in mouse serum (Fig. 2B). The percentage of intact probe was 94.1%, 93.5%, 87.8%, and 85.1% at 30, 60, 120, and 150 min of incubation, respectively. Defluorination was not observed for 18F-FBZA incubated with mouse serum up to 150 min. Overall, 18F-FBZA can be reliably produced and demonstrates good in vitro stability.
Cell Uptake of 18F-FBZA
The B16F10 cell uptake of 18F-FBZA over a 2-h incubation period at 4°C and 37°C (Fig. 3) was 0.49% ± 0.03% and 0.52% ± 0.1%, respectively. Pretreatment of the B16F10 cells with l-tyrosine (2.0 mM) for 24 h significantly darkened B16F10 cells, compared with control cells. These tyrosine-stimulated cells displayed a significantly enhanced accumulation of 18F-FBZA at both 4°C and 37°C (P < 0.001). At 37°C (Fig. 3B), within 30 min of incubation the tyrosine-stimulated cells maximized at 10.7% ± 0.8% uptake of 18F-FBZA and remained at 8.1 ± 1.9 at 2 h, which amounts to an 18- to 25-fold increase compared with uptake by the nontreated B16F10 cells. At 4°C (Fig. 3A), the uptake maximized at 30 min with 4.5% ± 0.5% accumulation, which was 58% less than uptake at 37°C (P < 0.05).

In vitro cell uptake of 18F-FBZA. Digital photo is shown of B16F10 cell pellet with (right) or without (left) l-tyrosine treatment for 24 h at 4°C (A) and 37°C (B). Data are expressed as mean ± SD, with each data point representing triplicate study.
σ-Receptor-Binding Studies of 19F-FBZA
Benzamide analogs have previously been shown to possess high binding affinity with σ-receptors, which are normally overexpressed in melanoma cells (27,29). To gain a better understanding of the involvement of σ-receptor binding of 18F-FBZA in the uptake of FBZA in melanoma cells, we further measured the binding affinity of the nonradioactive 19F-FBZA in established σ-receptor assays using guinea-pig brain (σ1-receptor) and rat liver membrane (σ2-receptor) preparations (24). These assays showed that 19F-FBZA displays low affinity toward either σ1-receptor (inhibition constant, 8.90 μM) or σ2-receptor (inhibition constant, 0.12 mM).
Biodistribution
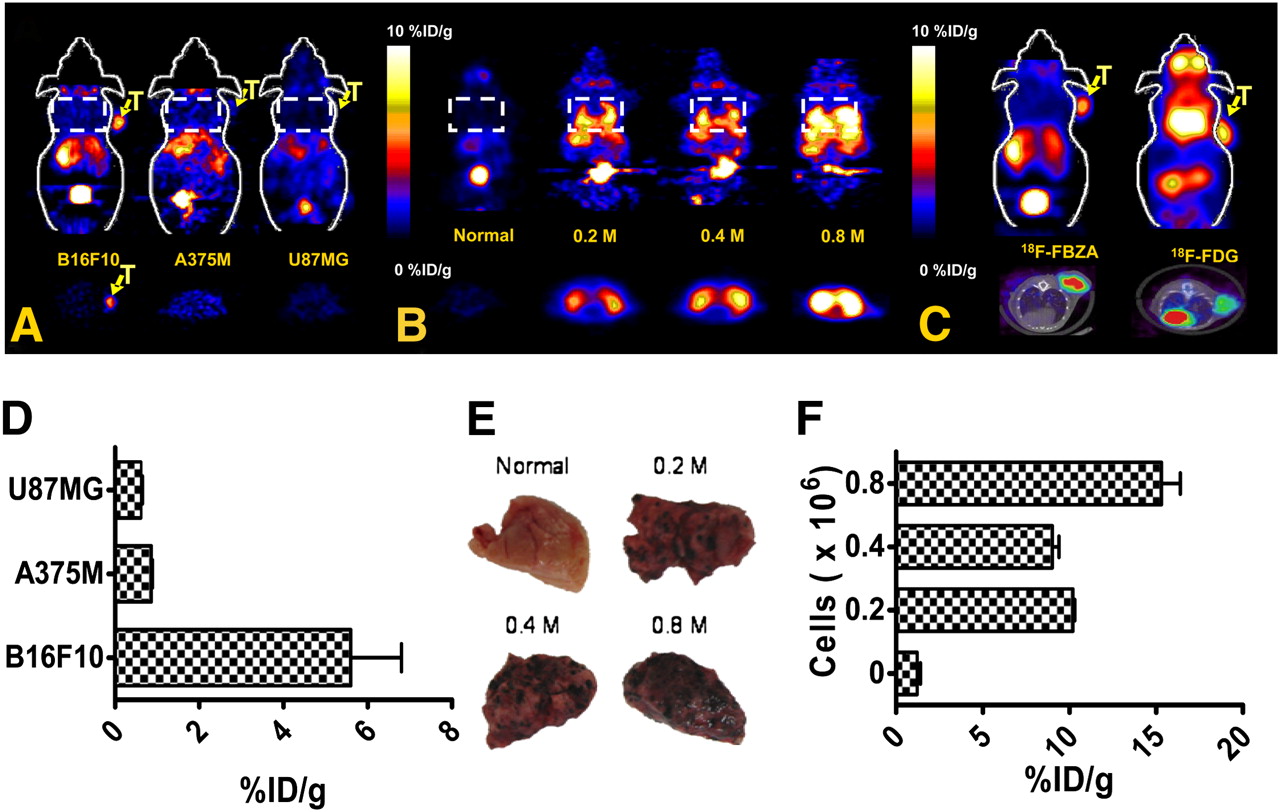
The in vivo biodistribution of 18F-FBZA was examined in B16F10 murine allografts, as well as in A375M and U87MG xenografts. In biodistribution studies of 18F-FBZA performed at 1 and 2 h in the B16F10 model, 18F-FBZA displayed a significant uptake in the melanotic B16F10 melanoma allograft, with 6.47 ± 2.16 and 5.94 ± 1.83 %ID/g at 1 and 2 h, respectively (Table 1; Supplemental Fig. 1 [supplemental materials are available online only at http://jnm.snmjournals.org]). At 2 h, the tumor-to-blood ratio was 34.0 ± 13.2 and the tumor-to-muscle ratio was 23.3 ± 10.1. The in vivo biodistribution of 18F-FBZA in the A375M and U87MG mouse models, however, was low, with tumor uptake of 0.75 ± 0.09 and 0.56 ± 0.13 %ID/g and tumor-to-blood ratios of 4.70 ± 0.78 and 3.57 ± 1.34, respectively (Table 1; Supplemental Fig. 1).
Biodistribution Results of Different Tumor-Bearing Mice
In the B16F10 pulmonary metastasis model, the mice were sacrificed 13 d after intravenous inoculation of B16F10 cells. In the 2 groups of mice (n = 3 each) (0.2 and 0.4 × 106 B16F10 tumor cells), biodistribution and small-animal PET studies showed that the radioactivity in melanoma lung metastases reached 10.0 ± 3.92 and 7.87 ± 3.56 %ID/g, respectively, at 2 h after injection (Table 2). In normal lung tissue, the probe accumulation was 0.99 ± 0.04 %ID/g at 2 h after injection (Table 2)—a value that was similar to that observed in the B16F10 subcutaneous model, in which the normal-lung uptake was 0.85 ± 0.23 %ID/g at 2 h. The lung-to-blood ratios for the pulmonary metastasis model were 24.7 ± 13.5 and 38.9 ± 10.3, respectively. The lung-to-blood ratio for the B16F10 subcutaneous model was 4.72 ± 0.46, whereas 4.23 ± 0.66 was observed in control C57BL6 mice at 2 h. Both absolute lung uptake and lung-to-blood ratio were significantly higher for the pulmonary metastasis model than for the subcutaneous model or for control C57BL6 mice (P < 0.01).
Biodistribution Results of 18F-FBZA in Normal Mice and B16F10-Bearing Mice at 2 Hours After Injection
To further explore the translational potential of 18F-FBZA in melanoma imaging, we compared 18F-FBZA and 18F-FDG (Table 1). 18F-FDG uptake by the B16F10 tumor was high at 1 h and was not significantly different from 18F-FBZA uptake in the same tumor model. Lung uptake was 7.61 ± 1.53 and 2.92 ± 0.40 %ID/g for 18F-FDG and 18F-FBZA, respectively, suggesting much higher normal-lung uptake of 18F-FDG (P < 0.01). Heart uptake of 18F-FDG was significantly higher than that of 18F-FBZA (P < 0.01). However, liver uptake of 18F-FDG was lower than that of 18F-FBZA at 1 h after injection (P < 0.05).
Imaging Studies
In static small-animal PET images (Fig. 4A), B16F10 tumors were clearly visualized at 1 h with good tumor-to-background contrast (see Supplemental Fig. 2 for dynamic scan), whereas for A375M and U87MG, tumor uptake was hardly visible. Liver and kidney uptake was visualized in all animals. ROI analysis of tumor uptake showed that B16F10 had significantly higher tumor uptake (5.6 ± 1.2 %ID/g) than did the other 2 tumor types (0.86 ± 0.03 and 0.61 ± 0.04 %ID/g for A375M and U87MG, respectively) (Fig. 4D) (P < 0.01).
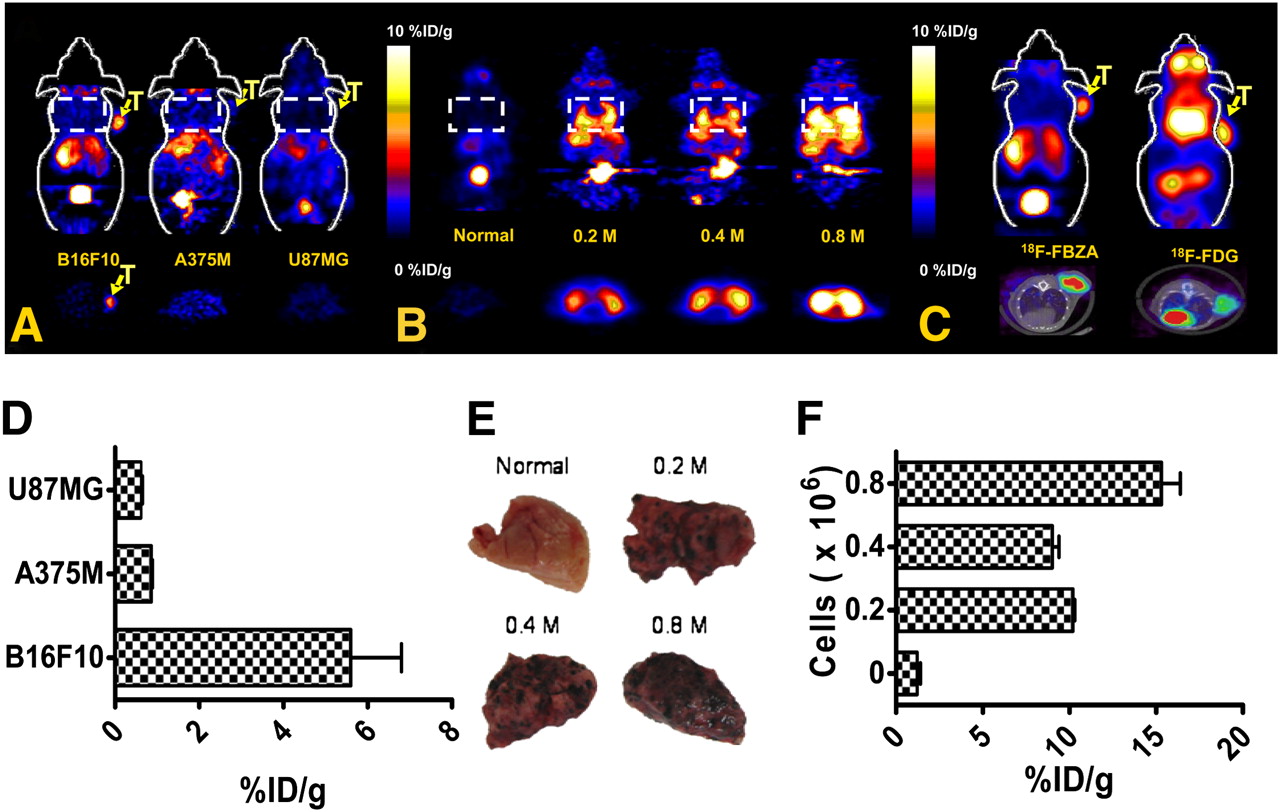
(A) Representative decay-corrected coronal (top) and transaxial (bottom) small-animal 18F-FBZA PET images of B16F10, A375M, and U87MG subcutaneous tumor models. Tumor locations (T) and lung regions (rectangles) are indicated. (B) Representative decay-corrected coronal (top) and transaxial (bottom) small-animal PET images of B16F10 melanoma lung metastasis model that was established 13 d after tail vein injection of 0.2 × 106 (n = 3), 0.4 × 106 (n = 3), or 0.8 × 106 (n = 2) B16F10 cells. (C) Representative decay-corrected small-animal PET coronal images (top) and small-animal PET/CT fusion transaxial images (bottom) of C57BL/6 mice bearing B16F10 tumors. (D) Quantification analysis of tumor uptake of 18F-FBZA in different models for comparison. (E) Photographs of biopsy samples of lung metastases. (F) Quantification analysis of 18F-FBZA uptake by melanoma lung metastases.
In vivo studies with the lung metastasis model, compared with the normal lung, clearly showed a region of symmetric uptake greater than the background level in the chest of mice bearing pulmonary metastases (Fig. 4B). ROI analysis showed that normal-lung uptake of 18F-FBZA was 1.2 ± 0.2 %ID/g whereas uptake of 10.2 ± 0.1, 9.0 ± 0.4, and 15.3 ± 1.1 %ID/g was observed for lungs harboring metastases resulting from tumor doses of 0.2, 0.4, or 0.8 × 106 B16F10 cells, respectively (Fig. 4F).
In vivo small-animal PET of the subcutaneous B16F10 melanoma using 18F-FBZA or 18F-FDG showed that the 2 agents have different biodistribution patterns (Fig. 4C). For 18F-FBZA, radioactivity accumulation in tumor and liver is observed as washout through the kidneys into the bladder. 18F-FDG, on the other hand, accumulated in the heart, eyes, (Harderian glands), and tumor, whereas liver activity was low. PET ROI analyses showed that tumor uptake for 18F-FBZA was 5.6 ± 1.2 whereas that for 18F-FDG was 6.31 ± 0.61 %ID/g (P > 0.05). PET/CT fusion images clearly demonstrated the tumor anatomy and specific tumor uptake of the different probes.
DISCUSSION
Malignant melanoma is well known for its aggressiveness and strong metastatic potential, and early detection and identification of metastasis can improve management and prognosis. The lethality of malignant melanoma is caused mostly by metastasis to distant organs, mainly the lungs, liver, brain, and soft tissues. Monoclonal antibodies against high-molecular-weight melanoma-associated antigens (30,31) or peptides targeting melanocortin receptor 1 (26,32) have been labeled with various radioisotopes for radioimmunodetection and radioimmunotherapy of malignant melanoma and its metastases. Melanin-binding peptides and antibodies are also used to target melanin and have achieved therapeutic effects in melanoma animal models (17,33,34). Though antibody and peptide-based approaches successfully target the primary tumor in many melanoma models, limitations include relatively slow and low tumor uptake, high kidney uptake, and in vivo instability, among others (17).
Compared with peptide-based probes, a group of molecules with coplanar fused rings has been shown to bind strongly with melanin (35). Particularly, benzamide-based small molecules can specifically target the melanotic melanomas and their metastases, as evidenced by numerous studies of radioiodinated benzamide analogs (19). In a limited clinical trial, the sensitivity and specificity of 123I-BZA were 100% and 81%, respectively (36). Very recently, a 125I-labeled BZA derivative was developed and showed high specificity and long tumor retention times—16-fold higher than for 125I-BZA at 72 h after injection—making it a promising radiopharmaceutical for targeted radionuclide therapy of melanoma (37). With the development of diagnostic PET and its high sensitivity, an 18F PET probe based on BZA molecules may improve the sensitivity of detection and diagnosis of melanotic melanoma and its metastasis.
An 18F-labeled benzamide, 18F-FBZA, was thus successfully synthesized and evaluated in cultured cells and tumor-bearing mice. In vitro cellular uptake studies showed that treatment of B16F10 cells with l-tyrosine (2 mM) substantially increased 18F-FBZA uptake from 0.32% ± 0.04% to 8.1% ± 1.9% at 37°C at 2 h, indicating that maximal 18F-FBZA uptake is associated with melanin content (Fig. 3). Earlier reports on cell culture studies and in vivo scintigraphic imaging with 123I-N-(2-diethylaminoethyl)-4-iodobenzamide suggested that σ1- and σ2-receptors might play an important role in uptake of benzamides, possibly attributable to the neuroectodermal origin of melanoma (38); however, the low σ-receptor affinity displayed by 19F-FBZA suggests that σ-receptors do not play a role in B16F10 uptake of this probe. This observation, coupled with the observation that cellular uptake for the 18F-FBZA probe at 4°C was significantly increased after tyrosine pretreatment, cannot be explained by receptor-mediated endocytosis. Alternatively, uptake of radioiodinated benzamides by melanoma has previously been observed and shown to be related to the melanin content of the cells (39). Labarre et al. have also shown that a complex interaction between BZA and melanin involves both ionic and hydrophobic binding sites (39). In addition, studies with melanoma-targeting 99mTc-complexes designed to mimic and contain the structural elements of BZA have also been shown to target melanoma on the basis of the melanin content of the tumors (29,40). Taken together, these findings indicate that uptake and accumulation in melanotic B16F10 cells of the neutral, lipophilic 18F-FBZA are caused by its passive diffusion through cell membranes into cytoplasm, followed by binding with melanin structures and trapping within cells.
In vivo studies used the B16F10 melanotic melanoma along with the amelanotic A375M melanoma and the human glioblastoma U87MG as control tumors. The in vivo tumor uptake of 18F-FBZA in B16F10 reached 6.47 ± 2.16 %ID/g at 1 h and remained high up to 2 h after injection. In contrast, the in vivo tumor uptake of 18F-FBZA by A375M attained only 0.75 ± 0.09 %ID/g, and U87MG also displayed a low tumor uptake of 0.56 ± 0.13 %ID/g at 2 h, both being significantly lower than the uptake observed for the melanotic B16F10 tumors (P < 0.01) (Table 1). Taken together with the in vitro cellular uptake assay, the in vivo results further prove that the target of 18F-FBZA binding is melanin within the melanoma.
Analysis of the in vivo results also reveals no significant difference in the uptake and distribution of 18F-FBZA in other nontarget organs among B16F10, A375M, and U87MG in vivo tumor models (Table 1) (P > 0.05). Given the higher tumor uptake in B16F10 melanotic melanoma, the tumor–to–normal-organ ratios are thus significantly higher for the B16F10 model (P < 0.01), indicating that 18F-FBZA has an excellent in vivo tumor targeting ability for melanotic melanoma. In view of the high incidence (88%) of melanotic malignant melanoma, 18F-FBZA represents a potentially viable PET probe for clinical studies of metastasis of malignant melanoma (41).
A goal of these studies was to explore the feasibility of using 18F-FBZA in the early detection of melanoma metastases. This goal was accomplished using the preclinical pulmonary metastasis melanoma model, in which we found that melanotic pulmonary lesions of B16F10 tumors specifically accumulated 18F-FBZA (Table 2; Fig. 4). Lung uptake in this B16F10 model was significantly higher than lung uptake of 18F-FBZA in the B16F10 subcutaneous model lacking pulmonary metastases or in control C57BL/6 mice (P < 0.05). Regarding 18F-FDG uptake in the chest, the values were 7.61 ± 1.53 %ID/g in the lung and more than 50 %ID/g in the heart, both of which can interfere with the delineation of melanotic lung lesions and thus limit the use of 18F-FDG for detection of pulmonary metastasis in melanoma (Table 1; Fig. 4C). In small-animal PET images, there was significantly higher lung uptake, shown by symmetric hot regions in the mouse chest bearing B16F10 pulmonary metastases. The individual metastatic lesions were smaller than the resolution of the small-animal PET scanner. That consideration, plus motion of the lung, prevented single micrometastasis from being discernable using small-animal PET (Fig. 4B).
Interestingly, brain uptake in the B16F10 model reached about 1.71 ± 0.06 and 0.35 ± 0.05 %ID/g at 1 and 2 h, respectively, after injection (Table 1), suggesting that the probe could pass the blood–brain barrier. The tumor-to-brain ratio at 1 h for 18F-FBZA reached 5.14 ± 1.10, compared with the lower value of 2.86 ± 0.35 for 18F-FDG that is due to the high normal-brain uptake of 18F-FDG. These results also demonstrated the potential of using 18F-FBZA for imaging melanoma brain metastases. However, uptake in melanized neurons remains to be addressed. Additionally, 18F-FBZA accumulates in the melanin-containing eyes (retinas) of normal C57BL/6 mice but not in the pink eyes of nude mice (Table 1; Supplemental Fig. 3), further suggesting the capability of cellular penetration and binding to melanin structures by 18F-FBZA.
In further comparing 18F-FBZA with 18F-FDG, we found that subcutaneous tumor uptake at both 1 and 2 h for 18F-FBZA was comparable to that for 18F-FDG at 1 h (P > 0.05). However, given the presence of phosphatases in the liver, the liver uptake at 1 h was much lower for 18F-FDG than for 18F-FBZA (P < 0.05). Kidney uptake is another major concern for radiopharmaceuticals, the kidney being a radiosensitive organ. Although there was no significant difference in kidney uptake of 18F-FBZA and 18F-FDG at 1 h, kidney uptake of 18F-FBZA decreased from 1 to 2 h—from 6.99 ± 3.13 to 0.93 ± 0.26 %ID/g—suggesting faster clearance of the 18F-FBZA probe.
CONCLUSION
A novel 18F-labeled PET probe, 18F-FBZA, was successfully synthesized via a 1-step conjugation reaction between the radiosynthon, 18F-SFB, and DEDA. 18F-FBZA specifically targeted both primary and pulmonary melanotic metastatic lesions with high tumor uptake and good tumor–to–normal-tissue ratios. These findings, taken together, show that 18F-FBZA represents a potential PET probe for imaging melanotic malignant melanoma and its metastases.
Acknowledgments
This work was supported, in part, by the Melanoma Research Alliance, grant R24 CA93862 from the Small Animal Imaging Resource Program of the National Cancer Institute, and grant P50 CA114747 from the In Vivo Cellular Molecular Imaging Center of the National Cancer Institute. We thank Carsten Nielsen for help with CT data acquisition.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication May 19, 2009.
- Accepted for publication June 30, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Melanin-Targeting Radiotracers and Their Preclinical, Translational, and Clinical Status: From Past to Future
- Whole-organism 3D Quantitative Characterization of Zebrafish Melanin by Silver Deposition Micro-CT
- Ultrasensitive detection of malignant melanoma using PET molecular imaging probes
- N-(2-(Dimethylamino)Ethyl)-4-18F-Fluorobenzamide: A Novel Molecular Probe for High-Contrast PET Imaging of Malignant Melanoma
- 4-11C-Methoxy N-(2-Diethylaminoethyl) Benzamide: A Novel Probe to Selectively Target Melanoma
- PET Imaging of Very Late Antigen-4 in Melanoma: Comparison of 68Ga- and 64Cu-Labeled NODAGA and CB-TE1A1P-LLP2A Conjugates
- Theranostics of Malignant Melanoma with 64CuCl2
- Melanoma Imaging with Highly Specific PET Probes: Ready for Prime Time?
- High-Contrast PET of Melanoma Using 18F-MEL050, a Selective Probe for Melanin with Predominantly Renal Clearance