


The concept introduced by Thimister et al. (1) in their investigation of acute myocardial infarction, reported in this issue of The Journal of Nuclear Medicine, is the difference in the mechanism of localization of the radiopharmaceutical annexin A5, compared with that of antimyosin or pyrophosphate (2,3). Both antimyosin and pyrophosphate require loss of integrity of the cell membrane to achieve localization. Annexin A5 localizes to the outer leaflet of the intact membrane of cells expressing phosphatidyl serine (PS). The nanomolar affinity of 99mTc-annexin A5 toward cell membrane-bound PS provides high contrast in the images. Unlike irreversibly damaged myocardium (detected by antimyosin imaging), the apoptotic process may be amenable to intervention, allowing these severely injured cells to survive and continue to contribute to overall ventricular function (2,3). Indeed, significant therapeutic implications can be gleaned from the article of Thimister et al.
APOPTOSIS, PS EXTERIORIZATION, AND ANNEXIN A5-BASED IMAGING
Apoptosis is associated with organized cell disintegration associated with activation of cytoplasmic enzymes, nuclear fragmentation, and destruction of DNA (4,5). The intracellular components of the process are difficult to image with intravenously administered agents. However, the extracellular expression of PS, as an integral component of the process, permits ready access of ligand for annexin binding. Normally, anionic phospholipids such as PS and phosphatidyl ethanolamine are actively recruited to the inner leaflet of the lipid bilayer by an energy-dependent enzyme called translocase; PS is virtually absent on the surface of normal cells (6,7). In contrast, a second adenosine triphosphate-dependent enzyme, “floppase,” actively pumps cationic phospholipids such as phosphatidyl choline and sphingomyelin to the outer leaflet of the lipid bilayer to maintain an asymmetric distribution of phospholipids across the plasma membrane. During apoptosis, translocase and floppase enzymes are deactivated and the asymmetric pattern of distribution is lost. The random bidirectional distribution is further exaggerated by activation of “scramblase” enzyme.
As apoptosis proceeds, evaginations develop in the cell membrane. These are brought about by random distribution of phospholipids in the lipid bilayer and alterations in cytoskeletal proteins. Blebs develop in the cell membrane, and the fragments of cytoplasmic proteins and nuclear material are packaged as apoptotic bodies in the remnants of the cell membrane. The apoptotic bodies also express PS and are removed by macrophages that express PS receptors (8).
Annexin A5 (35 kDa) has a high affinity for PS exposed on the cell membrane (9). Recombinant human annexin A5 has been radiolabeled with 99mTc (9), and the feasibility of imaging apoptosis has been demonstrated both in experimental and in clinical cardiovascular disease states (10–12). In all these diseases, areas of myocardial damage demonstrate a mixture of apoptotic and necrotic cell death (2,3).
CONNOTATION OF PS EXTERNALIZATION IN MYOCARDIAL INFARCTION
In an earlier publication from this group of investigators, Hofstra et al. demonstrated annexin A5 uptake in patients with acute myocardial infarction (12). The uptake of annexin A5 was clearly confined to the region of perfusion loss as demonstrated by 201Tl perfusion imaging. In an accompanying editorial, we struggled to explain the annexin A5 positivity in the myocardial region that was expected to predominantly contain necrotic myocytes (13). We presumed that the cells subjected to prolonged ischemic insult may lose the capacity to provide the energy needed for (translocase and floppase) enzymes to maintain PS asymmetry, allowing the PS to appear on the outer leaflet of nonviable cell membrane. Alternatively, annexin may access residual inner-leaflet PS through sarcolemmal defects (the hallmark of myocyte necrosis). However, the most logical assumption was that the apoptotic and necrotic cell deaths may form a continuum and that annexin A5 may actually target the apoptotic cells (with exteriorized PS) that undergo secondary necrosis because of continued ishemic injury. We proposed that annexin A5 should simply be accepted as the (exteriorized) PS-seeker and that annexin targeting should not dwell on the blurry boundaries between apoptosis and necrosis. Such pathologic states that express PS could be considered phosphatidyl serinopathies because of their amenability to noninvasive detection by annexin A5 imaging (13).
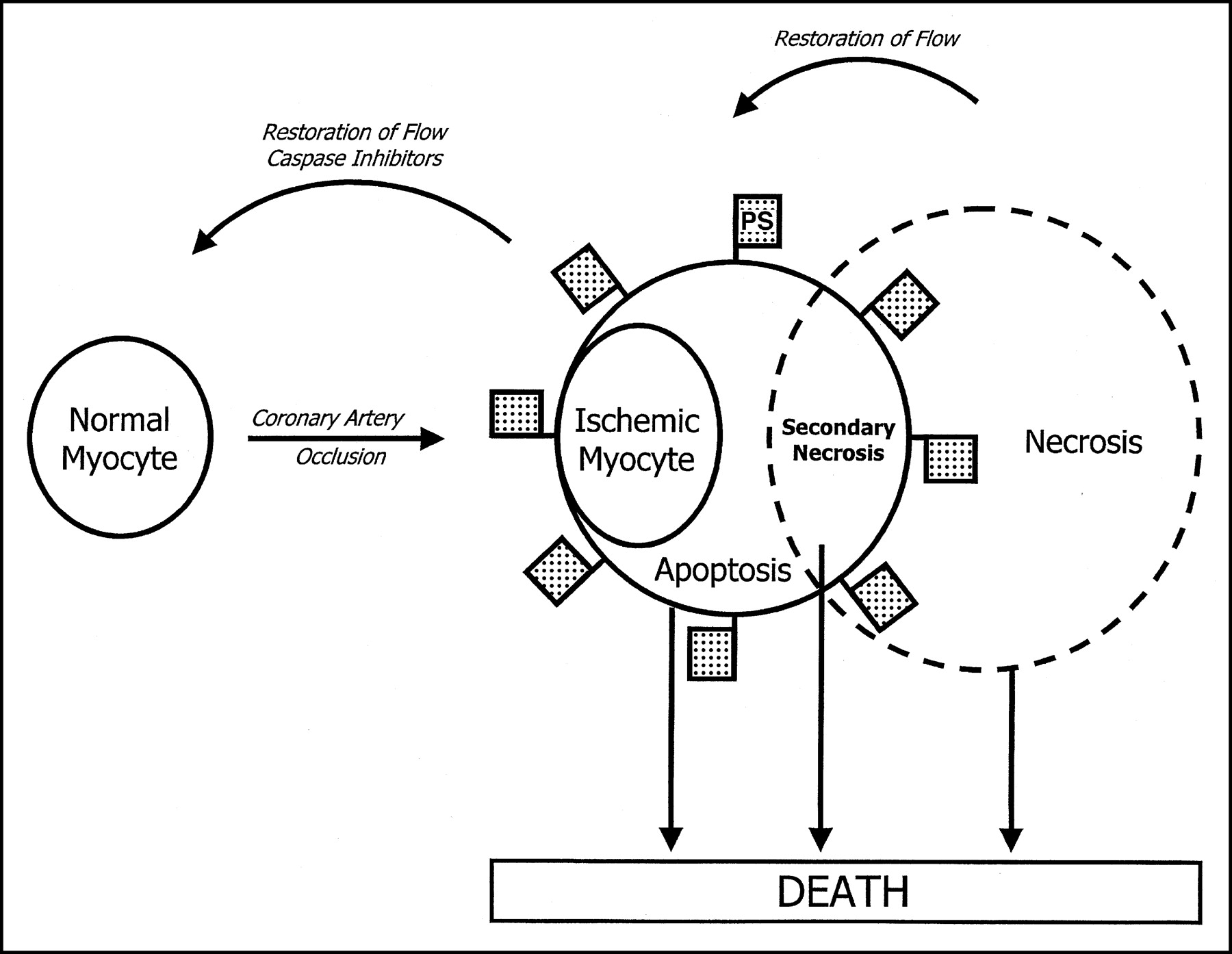
However, there may be much more to the story. The present study (1) and one of our recent studies (14) suggest that annexin localization may not always represent cell death. Limited data from the study of Thimister et al. (1) demonstrate that annexin localization in regions of infarction is partially resolved in 3–4 d and completely resolved by 8 d after reperfusion, suggesting either removal of these cells from the ischemic zone or recovery of the cells in which annexin A5 had concentrated. The reduction in the perfusion deficit in the subacute stage, associated with restoration of regional wall motion in the involved territory, favors the latter explanation of salvage of the cells that were once annexin positive. If so, and if we may be allowed to stretch our imagination, we reason that a subcellular pathologic substrate in ischemia is early apoptosis (Fig. 1). Barring terminological nuances, ischemia and apoptosis should be amenable to complete recovery. In contrast, continued ischemia and consequent loss of energy should lead to secondary necrosis.

Hypothesizing an ischemia-apoptosis-necrosis spectrum. Coronary artery occlusion leads to ischemic changes. Annexin A5 imaging data support that ischemia is indeed equivalent to early phase of apoptosis, which is represented by death receptor upregulation, PS exteriorization, and caspase-3 activation (14,18). In cardiomyocytes, cell death by apoptosis is unlikely since reduction in blood supply depletes adenosine triphosphate and death may ensue by secondary necrosis. PS-flagged death mechanisms may be amenable to annexin A5 imaging. Pathologic states leading to sarcolemmal interruption should be detectable by antimyosin/pyrophosphate imaging (2,3).
We have provided evidence of annexin uptake in briefly and persistently ischemic myocytes (14). In a rabbit model of severe ischemia produced by left circumflex coronary artery occlusion for either 10 min or 40 min followed by 30 min of reperfusion, 99mTc-labeled annexin A5 was administered intravenously and the animals were sacrificed at 3 h. Ex vivo imaging demonstrated significant annexin A5 uptake in the initial zone of ischemia, with a maximum uptake of 0.27% ± 0.16% of the injected dose per gram in the once-ischemic area, compared with 0.03% ± 0.01% in normal myocardium and 0.55% ± 0.30% in infarcted tissue. No histopathologic evidence of necrosis or apoptosis was observed in the tissue subjected to brief, severe ischemia, whereas a mixture of apoptotic and necrotic cells was seen in the infarcted specimens. Separation of subcellular components from the cell membrane of the once-ischemic cells revealed that 53% ± 6% of radioactivity had been internalized. These results suggest that PS exposure does not necessarily indicate irreversible damage (14). A delay and diminution in the extent of injury (depicted by optical imaging with fluorescent annexin A5) has been observed by preadministration of caspase inhibitors in a murine model of myocardial infarction (16). Caspase inhibition also led to a reduction in the eventual infarct size in a rabbit model (17).
These studies have opened a Pandora’s box. Can ischemia be subsumed within the spectrum of apoptosis? Could annexin-positive myocytes constitute an indication for intervention? What is the temporal window for treating PS-expressing ischemic cells? Is it possible to develop a model of annexin uptake vis-à-vis the extent of troponin leakage as an index of reversibility? Can inhibitors of apoptosis abrogate ischemic injury beyond reperfusion? If annexin positivity identifies a significant fraction of salvageable myocytes, we can expect a major shift in the management of acute coronary syndromes, somewhat similar to the one that began 2 decades ago with the advent of thrombolytic therapy (18).
Footnotes
Received and accepted Nov. 25, 2002.
For correspondence or reprints contact: Jagat Narula, MD, PhD, Heart Failure/Transplant Center, Hahnemann University Hospital, 245 N. 15th St., MS115-NT742, Philadelphia, PA 19102-1192.
E-mail: Jagat.Narula{at}drexel.edu
↵* P.S. = phosphatidyl serine; the commentary hypothesizes on and emphasizes the enormous potential of detection of PS expression on myocytes during acute ischemic events.




{kind=link}