


Abstract
After several decades of debate, it is now widely acknowledged that apoptosis, also known as programmed cell death, is central to homoeostasis and normal development and physiology in all multicellular organisms, including humans. The dysregulation of apoptosis can lead to the destruction of normal tissues in a variety of disorders, including autoimmune and neurodegenerative diseases (too much apoptosis) or the growth of tumors (too little apoptosis). In addition, effective therapy of tumors requires the iatrogenic induction of programmed cell death by radiation, chemotherapy, or both. Given the central role of apoptosis, it would be desirable to have a noninvasive imaging method to serially detect and monitor this process in cancer patients undergoing conventional radiation and chemotherapy treatments as well as for the development and testing of new drugs. In this article, the latest modalities and contrast agents described in the literature for the imaging of apoptosis in vivo are reviewed. First, the most recent developments in the biochemical characterization of the many intracellular pathways involved in this complex process are discussed. Next, a variety of new radionuclide tracers, including radiolabeled annexin V and caspase inhibitors for PET and SPECT, are described. Finally, the use of MRI, MR spectroscopy, and ultrasound as possible alternative imaging modalities for the imaging of apoptosis is addressed.
The biologic significance of apoptosis, or programmed cell death, in the regulation of normal development and homeostasis of multicellular organisms cannot be understated. One of the goals of apoptosis on a cellular level is the organized self-disassembly of an unnecessary or senescent cell into small membrane-bound packets called “apoptotic bodies.” These bodies can then be rapidly ingested by so-called professional phagocytes such as monocytes and macrophages or at a slower pace by adjacent stromal cells such as fibroblasts and vascular smooth muscle cells (VSMCs). Unlike necrosis, the net result of apoptosis is the safe removal of unwanted cells without release of toxic intracellular substances or the local incitement of inflammation. In the early phases of apoptosis, before the start of autodigestion of DNA and the self-packaging of intracellular contents, cells can also be directly engulfed by phagocytes. Phagocytes identify their targets by the prolonged extensive exposure of the anionic membrane phospholipid phosphatidylserine (PS) on the surface of unwanted cells. It is unclear at the present time which of these modes of apoptotic cell removal dominates in vivo. The mode of removal may in fact depend on the actual local conditions surrounding an unwanted cell(s).
Dysregulation of apoptosis results in the accumulation of unwanted cells, as in cancer or the premature removal of needed cells (such as in Alzheimer's disease or rheumatologic disorders). The formation of a cancer (dysregulation, cell accumulation) and its successful treatment (iatrogenic modification, cell removal) both represent opposite sides of the apoptosis coin.
In this article, the biochemical and morphologic changes observed during apoptosis are briefly reviewed. In addition, how multimodality imaging can be used to visualize these processes in vivo is discussed.
BASIC BIOCHEMICAL AND MORPHOLOGIC CHANGES OF APOPTOSIS
It has been over 40 years since Kerr et al. first coined the term “apoptosis” (1). The word “apoptosis” was derived from 2 Greek roots, “apo,” which means from, and “ptosis,” which means falling. In his Ph.D. thesis, Kerr sought to understand the rapid involution of hepatic parenchyma in response to the acute surgical interruption of the portal venous blood supply of the liver (2). He found that the ischemic hepatic lobes, interspersed between islands of normal tissue, transformed into small round vesicles of cytoplasm that often contained specks of condensed nuclear chromatin. These vesicles were then taken up and ingested by surrounding cells as well as by specialized (professional) mononuclear phagocytes. Although it was clear that these cells were dying, the process was different from necrosis, since there was no adjacent inflammation. In addition, the mitochondria and ribosomes contained within round vesicles remained intact throughout the process. Furthermore, the round vesicles occasionally occurred in clusters, which strongly suggested the “budding” or formation of these structures from the surface of the dying cell. Kerr's description of this new type of cell death contrasted with the findings of typical necrosis, in which cells lose membrane integrity and begin to swell up, followed by the spillage of their contents into surrounding tissues and the incitement of local inflammation. Since the cells that Kerr observed in the liver actually decreased in size as they died, he referred to this type of cell death as “shrinkage necrosis.”
Working with Searle, Kerr also found evidence of apoptosis within histologic specimens of basal cell carcinoma (3). This group of investigators reasoned that the abundance of apoptotic cells was the explanation of the slow net growth rate of these tumors despite the presence of great numbers of mitotic figures (dividing cells). They also found increased rates of apoptosis within cancer specimens obtained from sites of radiotherapy. As a visiting professor in Brisbane, Curry recounted the observation of apoptosis within the adrenal cortex of rats treated with prednisolone, by his colleague, Wylie (4). Curry then went on to demonstrate apoptosis associated with regression of experimental rat breast carcinoma after removal of the ovaries (5).
Despite the elegant description of apoptosis laid out by Kerr and his colleagues much still remains unknown about the biologic mechanisms and pathways that drive apoptosis. Continued research has shown that the histologic changes of apoptosis detailed by Kerr are preceded by an initiation stage called the “lag or trigger phase” (6–9). Multiple triggers of apoptosis are known, such as withdrawal of growth factors, DNA damage, immune reactions, ionizing radiation, chemotherapy, and ischemic injury (10–14). These and other triggers can start a cascade of events that lead to the morphologic changes of apoptosis. The lag time between exposure to the trigger(s) and the time of observable morphologic signs of apoptosis is highly variable depending heavily on cell type, type of trigger(s), intensity and exposure, duration of trigger, and the local environmental conditions of the cell (15). Most apoptotic pathways, however, converge on a common cascade of cysteine aspartate–specific proteases collectively known as the “caspases” (Fig. 1). The caspases when activated cross-link and cleave specific intracellular proteins involved with apoptosis.

Biochemical events in apoptosis.
Caspases are commonly grouped into either the initiator (caspases 8 and 10) or the execution set of caspases (caspases 3, 6, and 7) (16). Activation of the executioner set of caspases occurs via the extrinsic or intrinsic pathway of apoptosis. The extrinsic pathway is mediated by specific death receptors that bind specific ligands, including tumor necrosis factor (TNF), TRAIL (a TNF-related, apoptosis-inducing ligand that binds to the DR4 and DR5 death receptors), and FasL (a ligand that binds to the Fas receptor). The association of a ligand and its cognate death receptor led to the recruitment of adapter molecules such as FADD (Fas-associated death domain) or TRADD (tumor-associated death domain), thereby activating the initiator caspases 8 and 10 that in turn cleave and activate the executioner caspases 3, 6 and 7. The intrinsic or mitochondrial pathway is initiated by the release of cytochrome c from the mitochondria into the cytosol. Cytochrome c then interacts with Apaf-1 (apoptosis-activating factor 1), adenosine triphosphate (ATP), and pro-caspase 9 to form a structure known as the apoptosome. The apoptosome then cleaves and activates caspase 9, which leads to caspase 3, 6, and 7 activity stimulating apoptosis. Activation of caspase 9 without involvement of the apoptosome has also been described (17).
Each caspase is also associated with a specific inhibitor, allowing the system to be strictly regulated by several positive and negative feedback mechanisms. The final enzyme activated within the cascade is caspase 3. After activation of caspase 3 the morphologic events of apoptosis quickly follow resulting in the orderly breakdown of cellular proteins, including the cytoskeleton and nuclear matrix, and the activation of poly-ADP-ribose polymerase (PARP), an enzyme that facilitates the degradation of nuclear DNA into 50- to 300-kilobase-sized pieces (DNA ladder formation). These morphologic events are collectively called the “execution phase.” The hallmark of the start of the execution phase is the redistribution and exposure of PS on the cell surface (18). PS is normally restricted to the inner surface (inner leaflet) of the lipid bilayer by an ATP-dependent enzyme called “translocase” (19,20). Translocase in concert with a second ATP-dependent enzyme, “floppase,” which pumps cationic phospholipids such as phosphatidylcholine (PC) and sphingomyelin to the cell surface, maintains an asymmetric distribution of different phospholipids between the inner and outer leaflets of the plasma membrane (21–23). The rapid redistribution of PS and PC across the cell membrane (measured in minutes) at the beginning of the execution phase of apoptosis is facilitated by a calcium ion–dependent deactivation of translocase and floppase and activation of a third enzyme, called “scramblase” (19,20).
Although the previously mentioned series of events are believed to be largely correct there are still many new and surprising discoveries being made in the study of apoptosis. For instance, PS expression despite being regulated by calcium-dependent phospholipid scramblase activity (coupled to inactivation of the aminophospholipid translocase) is not necessarily triggered and controlled by apoptotic regulatory mechanisms (24). Until now, it has been generally accepted that PS externalization during apoptosis occurs downstream from cytochrome c release from the mitochondria. However, Balasubramanian et al. found that PS externalization is inducible, reversible, and independent of cytochrome c release, caspase activation, and DNA fragmentation. Furthermore, these investigators discovered that the exposure of plasma membrane PS required a sustained elevation in cytosolic ionized calcium, an event that could be inhibited by calcium channel blockers. Just as intriguing, they also observed that the endoplasmic reticulum (ER), the major storehouse and regulator of intracellular calcium, was a key player in the sustained release of ionized calcium needed to achieve the complete scrambling of lipids across the bilayer. Modification of the thiol groups with the ER with sulfhydryl reagents also caused the ER to lose its ability to retain calcium, thereby increasing cytosolic calcium ion concentration to levels sufficient for full PS exposure (i.e., from nanomolar to millimolar).
Another group of investigators (25) found the ER under certain conditions can also be a trigger for apoptosis (i.e., ER stress–induced cell death). Normally, the ER is the site of secretory protein synthesis, conformational maturation, and quality control for correctly folded proteins. Proteins failing to adopt a stable conformation are dislocated into the cytosol, where they are targeted for ubiquitylation (a tag to identify a protein for elimination) and proteasomal degradation. Proteasomal degradation of poorly folded ER-directed proteins is known as ER-associated degradation (ERAD), which is a cellular mechanism for achieving ER quality control. Conditions or certain drugs that lead to the abnormal accumulation of unfolded proteins result in ER stress. When challenged with ER stress, cells can reachieve homeostasis by initiating a series of orchestrated events known as the unfolded protein response (UPR). If unsuccessful, a proapoptotic series of events occur, including the ER stress–induced degradation of antiapoptotic Bcl-2 family proteins, cytochrome c release, caspase maturation, PS exposure, and finally apoptotic cell death.
In another set of experiments, Rosado et al. found that caspase 3 activation is not necessarily unique to apoptosis (26) and is essential for full activation of several cellular functions, including the aggregation of platelets and the secretion of enzymes from pancreatic acinar cells. The early physiologic activation of caspase 3 (although at a lower level than that associated with apoptosis) was found to be independent of cytosolic calcium ion levels, mitochondrial cytochrome c release, and the subsequent activation of caspase 9 and PS exposure.
Finally, even PS exposure, once considered the hallmark of the execution phase of apoptosis, can be observed in mitogen- or antibody-stimulated but otherwise nonapoptotic cell lines, including B and T lymphocytes, granulocytes, and mast cells (27–29). Viable PS-expressing VSMCs have also been observed in tumor but not normal vessels (30). In addition, PS exposure has been found by Elliott et al. (31) to be necessarily preceded by cell shrinkage and increased lipid mobility (decreased packing). This decrease in lipid packing was seen at the same time as cell shrinkage/membrane distortion, both of which were potently inhibited by blockers of volume-regulatory K+ and Cl− ion channels. Therefore, changes in plasma membrane organization and cell volume precede PS translocation. These investigators also postulated that PS redistribution in nonapoptotic cells may occur by a translocase-independent mechanism at energetically favorable sites of membrane perturbation where lipid packing focally is decreased (membrane lipid rafts).
How all of the new and seemingly contradictory experimental data fit in to the evolving picture of apoptotic cell death is unclear. What is clear is that the definition and the understanding of the biochemical events of apoptosis are in a state of flux. Therefore, any specific method to image and monitor apoptosis needs to be evaluated carefully as no one biochemical feature (except DNA ladder formation, a very late feature) appears to be uniquely associated with apoptotic cell death.
On the other hand there are several approaches to specifically stain cell suspensions and histologic specimens to detect apoptosis. The most widely used are the terminal deoxynucleotidyltransferase-dUTP nick end labeling (TUNEL) assays that were originally introduced by Gavrieli et al. in 1992 (6). TUNEL is based on the specific binding of terminal deoxynucleotidyltransferase (TdT) to 3′-OH ends of fragmented DNA. After proteolytic treatment of histologic sections, TdT incorporates X-dUTP (X = biotin, DIG, or fluorescein) at sites of DNA breaks. Terminally modified nucleotide avidin-peroxidase can then amplify the signal and allows for examination of labeled cells under light or fluorescent microscopy, flow cytometry, or via immunohistochemistry. Another DNA-based method is the detection of the internucleosomal fragmentation produced by endonucleases at expected intervals of 180 base pairs to 200 base pairs (16). Standard DNA extraction techniques are used to obtain the nucleic acids from either cells or homogenized tissue. DNA is then electrophoresed in an agarose gel that demonstrates the characteristic DNA ladder pattern.
The most widely used nonnuclear method involves fluorescent or biotin-labeled annexin V, a human protein with nanomolar affinity for cell membrane bound PS (7). An alternative approach uses radiolabeled forms of annexin V for PET or SPECT of apoptosis in vivo. This review will initially focus on the most recent history of radiolabeled annexin V imaging in both animals and humans followed by a discussion of other imaging modalities such as lipid proton MR spectroscopy that have been used to study apoptosis.
PHYSIOLOGY OF ANNEXIN V BINDING AND INTERNALIZATION
Annexin V (molecular weight [MW] ≈ 36,000) is an endogenous human protein that is widely distributed intracellularly, with very high concentrations in the placenta and lower concentrations in endothelial cells, kidneys, myocardium, skeletal muscle, skin, red cells, platelets, and monocytes (32). Although the precise physiologic function of annexin is uncertain, the protein has several well-studied functions, including inhibition of coagulation (annexin was originally discovered because of its ability to trap calcium [annex calcium] and prevent clotting); inhibition of phospholipase A2, an enzyme responsible for the release of arachidonic acid from the cell membrane (a component of the inflammatory process); and inhibition of protein kinase C, a system responsible for intracellular signaling.
More recent work has shown that annexin V may also have a significant role in the immunomodulatory effects of dying (apoptosis) and dead (necrotic) cells (33). As mentioned previously, apoptotic cells expressing sufficient PS on their surface are recognized and ingested without inciting an inflammatory response via a specialized set of anti-inflammatory events. Specifically, the recognition of exposed PS triggers the release of immunosuppressive cytokines (34), which retard inflammation and prevent the activation (maturation) of antigen-presenting dendritic cells (DC). Conversely, necrotic or lysed cells, as defined by a primary irreversible injury to the plasma cell membrane, induce local inflammation and an immune response. Alternatively, cells undergoing apoptosis may also become leaky if they are not cleared before they deplete their own intracellular ATP stores that are necessary to maintain plasma cell membrane integrity. Energy-depleted dying cells convert from apoptosis to necrosis in a process known as secondary necrosis. One example of secondary necrosis is PARP-mediated cell death (35). This process occurs when there is massive activation of PARP secondary to extensive DNA damage (strand breaks) caused by free radical ions. Free radical ion formation with subsequent PARP activation has been shown with radiation treatment, drugs such as doxorubicin, and cerebral ischemic reperfusion injury. Massive activation of PARP (normally a DNA repair enzyme) causes the depletion of its substrate, NAD+. As NAD+ stores can only be regenerated by the conversion of ATP to ADP, massive PARP activation places a further strain on the limited energy stores in the dying cell, causing it to become necrotic. Specific inhibition of PARP with drugs (or genetic engineering) can reduce or prevent ischemic cerebral and myocardial injury in animal models and increase rates of apoptosis in several cell lines (e.g., through decreased DNA repair of genetically unstable cancer cells) (36).
Regardless of the particular pathway, necrotic cells generate a local immune response that can greatly aggravate postischemic injury in the heart and brain (34). Once bound on the cell surface, annexin V can reduce the clearance of PS-expressing cells that are dying from either apoptosis or necrosis. This reduction increases the proinflammatory cytokine profile of the late clearance, and endogenous danger signals released from cells, which have lost their membrane integrity, build a proinflammatory microenvironment. DCs may then pick up antigens derived from the dying cells in a proinflammatory milieu and present the cell-derived antigens together with costimulation. In summary, this new evidence suggests that annexin V, a naturally occurring ligand specific for PS, interferes in vivo with the immunosuppressive effects of apoptotic cells.
The binding of annexin V to sites of PS expression in vivo has been found to be extremely complex and difficult to model (37,38). Although annexin V is a relatively large protein (about half the molecular weight of albumin), it was shown early on that the protein can be internalized at sites of ischemic injury both in the heart and in the brain and cross the intact blood–brain barrier (39). The transport of exogenously administered annexin V, a protein normally found almost exclusively within cells, also must occur across a protein gradient, suggesting the existence of an energy- dependent pump mechanism.
Kenis et al. (37,38) have found evidence that annexin V binds to rafts of PS exposed on the cell surface and forms a protein crystal followed by rapid internalization via a unique pathway of pinocytosis. This pathway consists of first the disassembly of the cortical actin network underlying the PS-exposing membrane patch. Annexin A5 then binds to PS and crystallizes on the cell surface as closely packed trimers that cause the underlying membrane to bend inward. The invaginated membrane patch then closes on itself and is transported into the cytosol in a microtubule-dependent manner. This pathway apparently is not related to clathrin- or caveolin-mediated endocytosis as it is neither actin driven nor preceded by membrane ruffling.
Other investigations of annexin V binding have found that PS can be expressed at low levels in a reversible fashion under conditions of cell stress that do not necessarily commit a cell to apoptotic cell death (40–48). These studies showed that intermediate levels of PS exposure could be found in cells with no other morphologic features of apoptosis. These relatively low levels of PS exposure could also be readily reversed on removal of physiologic stressors such as nitric-oxide, p53 activation, allergic mediators, and growth factor deprivation. Therefore, PS expression can be used to define tissues at risk for cell death that may recover or be amenable to prompt therapeutic intervention. Radiolabeled annexin V imaging has proven to be far more sensitive to regions of cellular injury in both animal models and human trials than one would expect simply based on TUNEL or other histologic assays, as both stressed (probably the majority of annexin V–positive cells) and dying cells can bind the tracer.
Better understanding of PS expression in vivo along with the mechanisms of exogenous annexin localization may lead to use of the annexin V pinocytic pathway to introduce drugs and other molecules into cells expressing PS that could halt or even reverse a wide variety of cellular injuries and stress.
PET AND SPECT WITH RADIOLABELED ANNEXIN V
In the early 1990s, 99mTc-labeled annexin V was proposed as a new method to detect clots in the atria of patients with atrial fibrillation (49,50). Although the human results were disappointing, these studies demonstrated that 99mTc-recombinant human (rh)–annexin V could be safely given at doses sufficient for clinical radionuclide imaging. Two clinical trials were later performed with 99mTc-N2S2-rh annexin for the imaging of apoptosis, the same formulation used in the clot detection trial (51,52). In the first study, Narula et al. (51) used radiolabeled annexin V to image acute rejection in 18 cardiac allograft recipients. Thirteen patients had negative and 5 had positive myocardial uptake of annexin V, as seen by electrocardiography-gated SPECT. Endocardial biopsies obtained within 1–4 d of the scan demonstrated histologic evidence of at least moderate transplant rejection.
In the second trial, Belhocine et al. (52) studied 15 cancer patients in late stage small and non–small cell lung cancers (SCLC and NSCLC, respectively), Hodgkin's and non-Hodgkin's lymphomas (HL and NHL, respectively), and metastatic breast cancers (BC). Annexin V SPECT was performed immediately before starting chemotherapy (day −2 and day −1) and immediately after the first course of treatment (day +1 and day +2). A negative annexin V study after therapy, that is, no change in tumor uptake of tracer from pretreatment baseline, correlated well with no tumor response in 6 of 8 patients. Two women (2 BC) of the 8 total patients with negative posttreatment annexin V studies actually had a clinically significant response to paclitaxel-based chemotherapy. On the other hand, all 7 patients with increased tumor uptake over baseline (positive annexin V study) had an objective tumor response. Five of these patients showed increased annexin V uptake at 40–48 h after chemotherapy (1 NHL, 1 HL, 1 SCLC, and 2 NSCLC), and 2 patients had increases in annexin V uptake observed at 20–24 h after treatment (1 NSCLC and 1 SCLC). Taken together, these results suggest a variability of the optimal timing with regard to the cancer type and emphasize the need to determine the best time in which to administer radiolabeled annexin V to determine therapeutic response in the design of all future imaging trials of tumor response to therapy regardless of modality or contrast agent (53), with an example shown in Figure 2 of the change in annexin V uptake with time after doxorubicin treatment of a transgenic murine syngeneic luc-expressing lymphoma.

Changes in tumor uptake over time for murine lymphoma. (A and B) Evaluation of tumor regression (in spleen) after doxorubicin treatment (single dose of 10 mg/kg) by serial bioluminescent imaging (BLI) 15 min after intraperitoneal injection of luciferin (150 mg/kg) into BALB/c mice with hematogenously disseminated BCL-1 gfp/luc murine lymphoma cells. (A) Representative example of tumor regression in single mouse over 4 d. (B) Quantitation of tumor regression as measured by BLI (4 mice per group). (C) SPECT images were obtained by use of LumaGEM system (Gamma Medical Instruments) with 1-mm pinhole collimator and 64 × 64 imaging matrix for projection data. Animals received 37–74 MBq of 99mTc-labeled annexin V-117. After 1 h, mice underwent SPECT. (D) Time course of 99mTc-annexin V-117 biodistribution after doxorubicin treatment. Mice were treated with doxorubicin for indicated times. At 1 h before each time point, 99mTc-annexin V was injected intravenously. Indicated organs were removed, and level of annexin uptake was measured by scintillation counting with γ-counter. Note that second highest peak and greatest nadir of annexin V uptake in diseased spleen occurred at 5 and 7 h, respectively, well before any loss of tumor cells, as determined by BLI. Kid = kidney.
The N2S2 labeling method, however, is cumbersome (and had a high degree of nonspecific excretion of tracer into bowel via excretion into bile). Even the simplified 99mTc-BTAP-Anx V kit contains 11 components with a series of very elaborate and time-consuming multiple reaction steps at different temperature and pH conditions and a rather complex purification procedure. For these reasons, including a very low labeling efficiency (30%–40%), an improved labeling method using the bifunctional agent hydrazinonicotinamide (HYNIC) was selected for further clinical trials (54). Like the penthioate radioligand (N2S2 method), 99mTc-HYNIC-annexin V shows the greatest uptake in the kidneys, liver, and urinary bladder. The biodistribution of 99mTc-HYNIC-annexin, however, is devoid of any bowel excretion, resulting in excellent imaging conditions in the abdominal region. In this procedure, HYNIC [succinimidyl (6-hydrazinopyridine-3-carboxylic acid)], also known as [succinimidyl (6-hydrazinonicotinic acid)], is used to randomly modify the accessible N-terminal groups in the lysine residues of rh-annexin V. The resultant compound can then be lyophilized and stored for labeling with 99mTc indefinitely. 99mTc labeling of reconstituted HYNIC-annexin V is performed by simply reacting the conjugate with 99mTc-pertechnetate in the presence of stannous tricine for 5–10 min at room temperature. Unfortunately, although 99mTc-HYNIC-annexin V is not concentrated in the liver or excreted in the bowel (unlike N2S2-labeled annexin V), it does concentrate in the cortex of the kidney, limiting visualization of any paranephric structures (55).
Despite this shortcoming, multiple clinical trials have confirmed the clinical utility of HYNIC-annexin V in determining the efficacy of chemotherapy in oncology patients (56–60), for the detection of apoptosis in areas of acute myocardial infarction (61,62), for defining the activity of rheumatoid arthritis (Roland Hustinx and Catherine Beckers, written communication, October 2003), for monitoring the effects of ischemic preconditioning (63–65), for detecting vulnerable plaque (66), for imaging acute stroke (67,68), and finally for the imaging of Alzheimer's dementia (69).
The most recent results in oncology suggest that for patients with late stage (IIIB and IV) SCLC and NSCLC (70), a single scan 24 h after the start of treatment may be sufficient to identify the subset of patients with a partial response to platinum-based chemotherapy. In patients who responded to therapy, annexin V uptake either decreased below baseline (n = 4) or increased (n = 1) as shown in Figure 3. Another key observation was the presence of a low level of 99mTc-annexin V uptake in the lesions at baseline that later had a tumor response. These data implied that the lesions with ongoing apoptosis have an increased likelihood of an apoptotic response to chemotherapy. An alternative explanation is increased expression of PS on vascular endothelial cells of tumor vessels during neoangiogenesis in tumors destined to respond to chemotherapy (71).
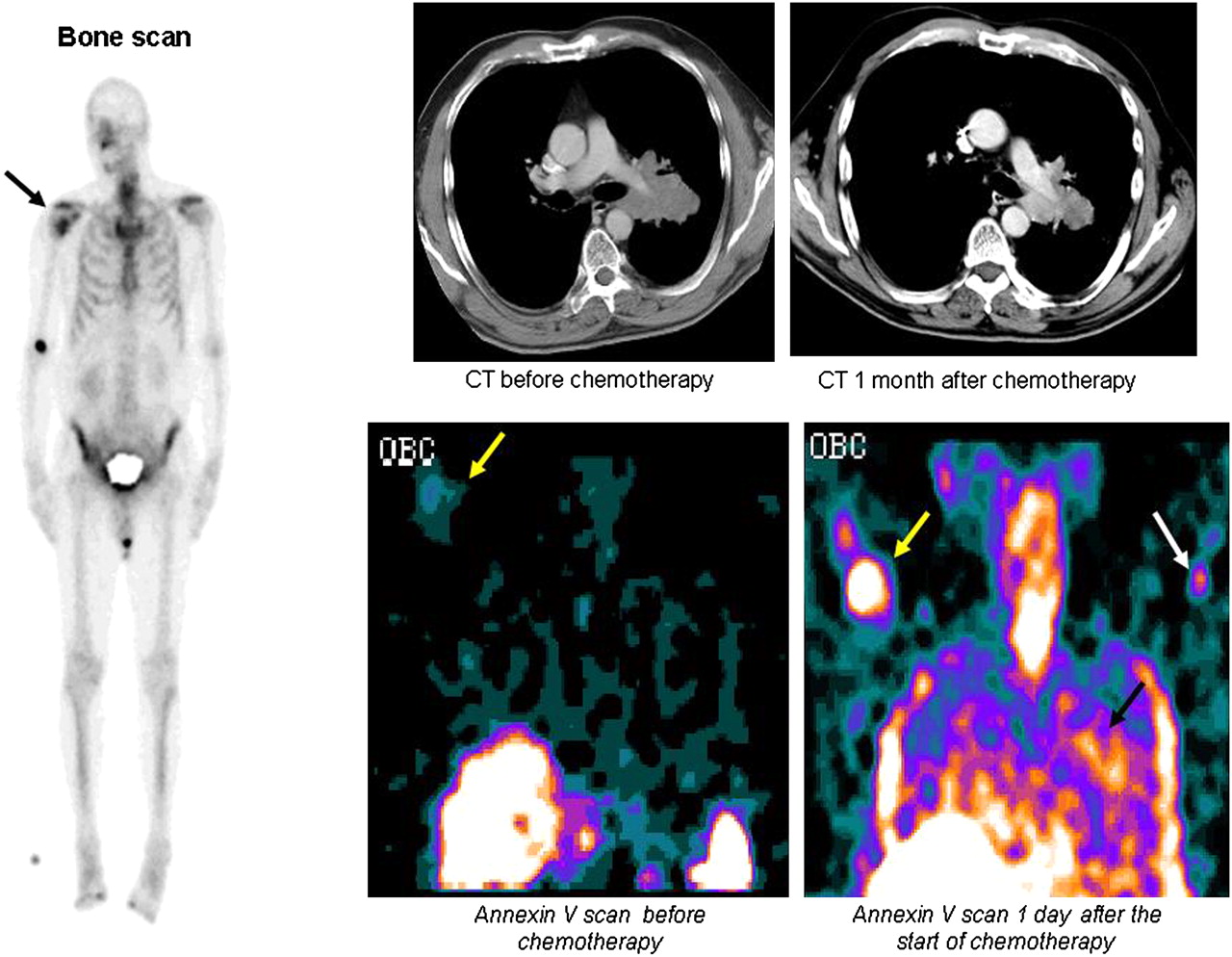
Early detection of tumor response in NSCLC with radiolabeled annexin. (Left) New right humeral metastasis was demonstrated on bone scan (black arrow) in 70-y-old man with stage IV NSCLC. (Right, top row) Chest CT (axial or transverse plane) showed response to treatment at 1 mo after treatment, with decrease in largest tumor diameter from 7.5 to 6.5 cm. (Right, bottom row) 99mTc-HYNIC-annexin V assessment (in coronal plane) revealed faint uptake at level of bony metastasis 2 h before first course of chemotherapy (yellow arrows), which significantly increased 1 d after start of therapy. In addition, increased tracer uptake was detected within primary lung tumor as early as 1 d after initiation of cis-platinum-based chemotherapy (black arrow). White arrow shows intravenous injection site in left arm. This patient was still alive after 126 d. This clinical case, which was part of a phase II/III clinical trial (NAS 2021; Middelheim Hospital, Antwerp, Belgium), illustrates capability of 99mTc-HYNIC-annexin V to localize at tumor sites undergoing spontaneous and chemotherapy-induced apoptosis. (Courtesy of Tarik Z. Belhocine and Johan Vandevivere.)
There are alternative methods for radiolabeling annexin V, including the use of self-chelating annexin V mutants that have lower concentrations in the kidneys of rodents than HYNIC-annexin V (72). The best-studied annexin V mutant with an endogenous site for 99mTc chelation is known as V-117. The protein contains 6 amino acids added at the N terminus, followed by amino acids 1–320 of wild-type annexin V. Amino acid Cys-316 is also mutated to serine in this molecule. 99mTc chelation is thought to occur via formation of an N3S structure involving the N-terminal cysteine and the immediately adjacent amino acids. The purified protein is then reduced and can be stored for later labeling with 99mTc using glucoheptonate as the exchange reagent.
Another self-chelating annexin V mutant, V-128, is a fusion protein with an endogenous technetium chelation site (Ala-Gly-Gly-Cys-Gly-His) added to the N terminus of annexin V (73,74). Both V-117 and V-128 have major advantages over the HYNIC chelator with regard to renal retention of 99mTc, with an attendant decrease in abdominal background and renal radiation dose.
It would be helpful to quantify radiolabeled annexin V concentration at lesion sites. Since PET has major advantages for quantitative imaging, several approaches to label annexin V with 18F have been developed. Two laboratories have used N-succinimidyl 4-fluorobenzoate (75,76) to synthesize 18F-annexin V. The fluorine-labeled agent has lower uptake in the liver, spleen, and kidneys than HYNIC-annexin V.
RECENT DEVELOPMENTS IN ASSAYS OF MODIFIED FORMS OF ANNEXIN V
Most imaging investigations have used radiolabeled annexin V that has been randomly modified with bifunctional agents attached to the accessible amino groups of the protein as outlined earlier. In addition, researchers have tested chemically modified annexin V with a variety of different in vitro assays and have generally concluded that the protein can withstand up to average derivatization stoichiometry of 2 mol/mol without a loss in PS binding affinity (77–81). However, these results have been called into question by the work of Bazzi and Nelsestuen (82), who found that the binding of annexins to membranes is negatively cooperative (i.e., a negative Hill effect) with respect to protein. This means that binding measurements made by titrating PS-expressing cells with labeled annexin V until full saturation will overestimate the binding affinity measured at higher protein concentrations. To make matters worse, many in vitro binding measurements have been made with calcium at 1.8 or 2.5 mmol/L rather than 1.25 mmol/L. Because the affinity of annexin V binding to cells declines greatly over a calcium range of 2.5–1.25 mmol/L, these results also may not accurately predict in vitro and in vivo binding of modified protein (83).
Tait et al. (83) have recently developed a newer method of measuring the membrane-binding affinity of annexin V by titrating with calcium (an ion necessary for PS binding) instead of protein. In addition, site specifically labeled forms of fluorescent or radioactive labels (as opposed to the standard random amino group labeling approach) are used for the competition with modified annexin. Finally, the assay is performed under conditions of very low occupancy (≤1% of membrane-binding sites occupied at saturation), thus avoiding the confounding effects seen as the membrane becomes more crowded (i.e., the negative Hill effect). These conditions best simulate the situation in vivo in which annexin V binding occurs at a calcium concentration of 1.25 mmol/L, the typical value for ionized calcium in vivo, and very low membrane occupancy with respect to the protein.
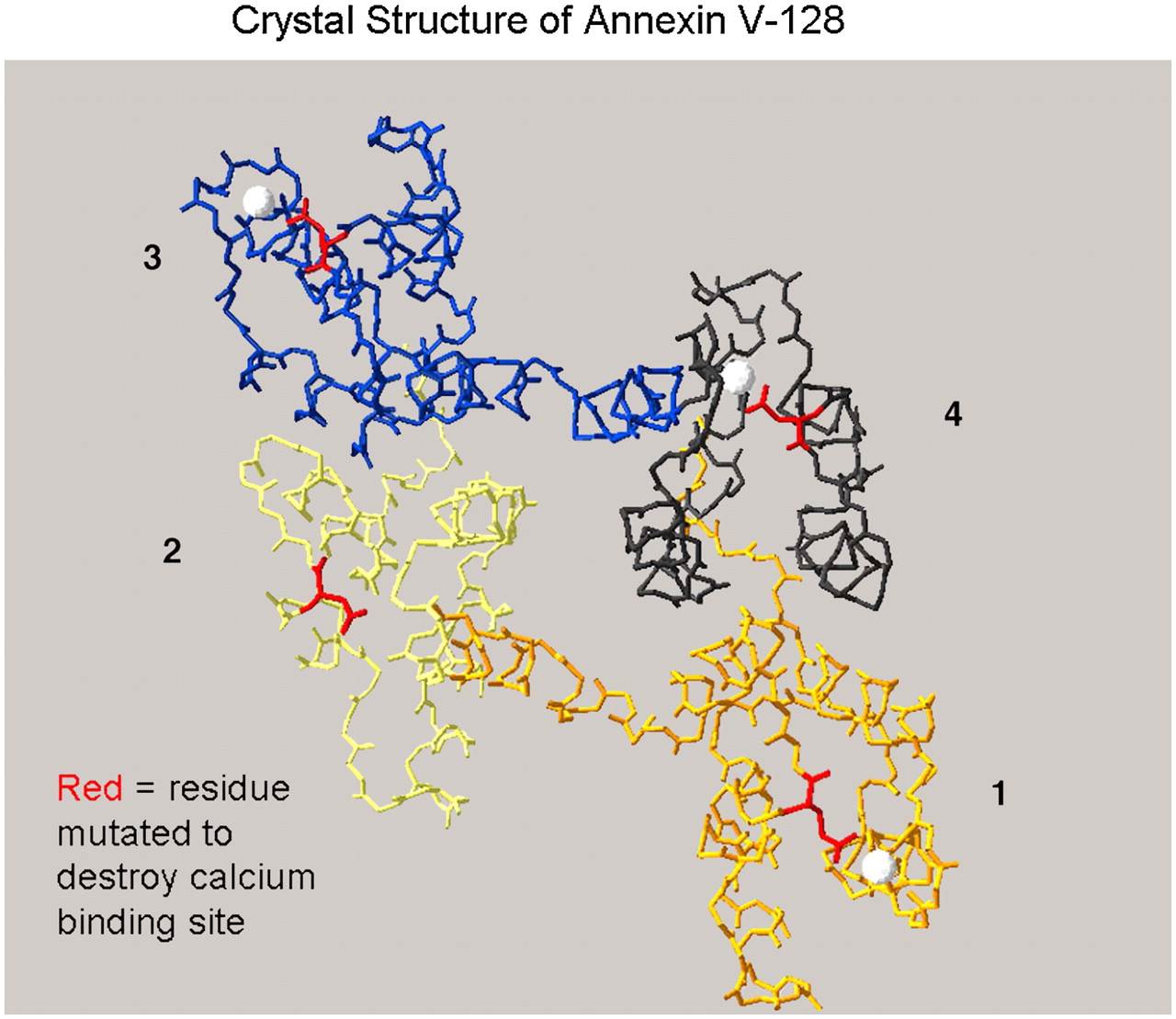
In related work, Tait et al. (84) constructed and systematically tested a set of self-chelating mutants of annexin V, including annexin V-128, that have various numbers of calcium binding sites both in vitro (with the new assay system) and in vivo (in mice undergoing cycloheximide-induced liver apoptosis) (Fig. 4). It was found that all 4 calcium binding sites are needed for full in vitro and in vivo binding of annexin V. Mutation (loss of function) of any 1 of the 4 calcium binding sites decreased the in vivo location of the tracer by 25%, and any 2 site mutations resulted in a 50% decline. These results indicate that mini forms of annexin with only 1 calcium binding site are unlikely to be useful as imaging agents (85). Comparison of HYNIC, mercaptoacetyltriglycine (MAG3) (86), fluorescein isothiocyanate (FITC), and biotin-labeled annexin V with annexin V-128 showed a 50% decrease in liver uptake of the tracer with randomly modified as opposed to self-chelating (site-specific) protein (84). Furthermore, modification of annexin V with as little as 0.5 mol of HYNIC per mole of protein is sufficient to lower in vitro and in vivo bioactivity substantially.
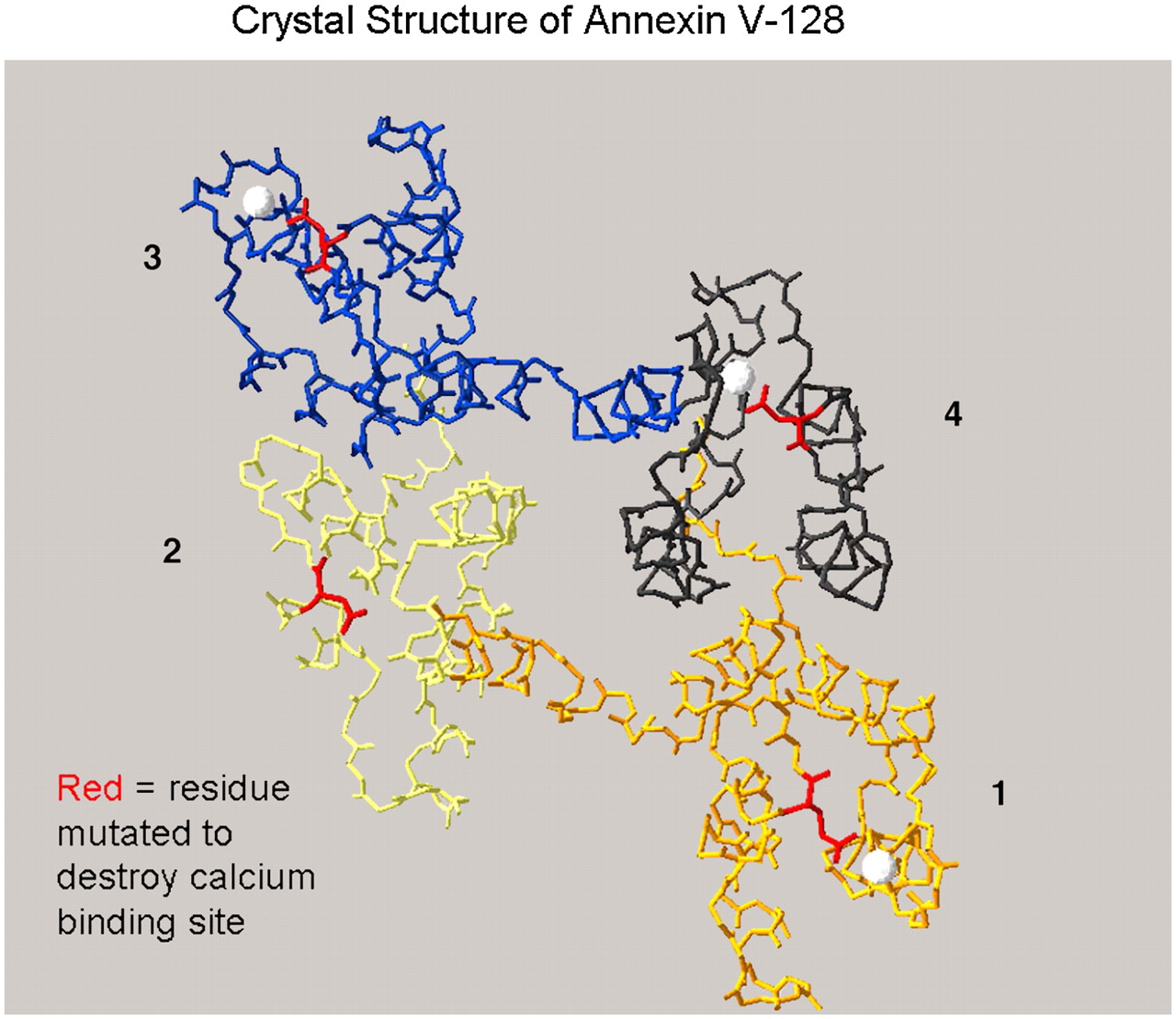
Structure of annexin V-128 and sites of mutation. Three-dimensional view of annexin V-128 is downward onto membrane-binding face of protein. Four domains (colored black, blue, yellow, or orange) are numbered. White spheres indicate approximate locations of calcium ions bound to AB-helix calcium-binding sites that have been mutated by Tait et al. (74). Polypeptide backbone is shown as a continuous line of peptides. Location of N-terminal technetium chelation site cannot be seen in this projection because it is on opposite face of molecule, beneath plane of page. (Structure is based on preliminary coordinates provided by Barbara Seaton, written communication, January 2004.)
The adverse effects of the random modification of annexin V have also been observed with 111In-diethylenetriaminepentaacetic acid (DTPA)-polyethylene glycol (PEG)-annexin V (87). Despite its prolonged circulation time, the uptake of 111In-DTPA-PEG-annexin V in a mammary carcinoma was not increased relative to a nonspecific control protein pegylated in the same manner. Therefore, any specificity for PS in the tumor had been lost, probably because the annexin V was heavily modified on amino groups to attach enough PEG. As has been shown, this type of chemical modification is damaging to the bioactivity of annexin V. Therefore, this approach is not likely to be fruitful for preparing highly bioactive forms of annexin with long circulation times. Other labeling methods, such as with 1,4,7,10-tetraazacyclododecane-N,N′,N″,N″′-tetraacetic acid (68Ga or 64Cu) and ethylene dicysteine (99mTc), have been attempted with native annexin V, but neither has proven to be superior to HYNIC-annexin V due to poor biodistribution characteristics or cumbersome labeling chemistry, respectively (88). Annexin V has also been proven to be quite heat labile and loses most of its activity even with heating at 56°C for 10 min (89) (while being quite stable at 37°C), precluding the use of many different types of labeling chemistries. The adverse effects of heating annexin V can be seen with the preformed chelate approach, in which 99mTc is sequestered by reacting with succinic dihydrazide (SDH) along with tricine and nicotinic acid, followed by conjugation to the annexin V by heating at 90°C, as proposed by Subbarayan et al. (90).
In summary, it appears that the self-chelating mutant annexin V-128 with site-specific modification is the best approach for the development of annexin V–based SPECT, PET, and fluorescent probes in the near future.
OTHER POTENTIAL TRACERS
Several annexin V derivatives have been developed for negative (T2* decay–cross-linked iron oxides [CLIO]) (91) and positive (T1 shortening, Gd-containing, annexin V–coated liposomes) (92) contrast or both (93) for MRI. Bimodality contrast-based annexin V agents have been tested as well, including AnxCLIO-Cy5.5, a magneto-optical (iron) nanoparticle that can be used as a bifunctional tracer in MRI and fluorescence imaging (94), and annexin V-conjugated quantum dots with a paramagnetic lipidic coating (Gd-DTPA) for MRI and fluorescent imaging (95).
Other radionuclide derivatives of annexin V have also been developed, including annexin V labeled for SPECT or PET with 123I (96) or 124I (97), respectively; 64Cu-labeled streptavidin for PET after pretargeting of PS with biotinylated annexin V (98); and another self-chelating (with 99mTc) annexin subfamily of proteins called annexin B1 (99). All of these tracers, with the exception of the self-chelating annexin B1, however, rely on the random modification of annexin V with bifunctional molecules; the result is a dramatic loss of in vivo PS binding affinity compared with unmodified wild-type (or self-chelating mutant) protein, as mentioned previously. As for annexin B1, it appears to have a high renal retention (50% injected dose), like HYNIC-modified forms of the protein, for reasons that are still unclear. Furthermore, in vitro assays to validate each tracer rely on the use of fluorescent forms of annexin V that also have been randomly modified and used in relatively high protein concentrations that can greatly overestimate the measured binding affinities.
Another class of possible alternative tracers for the imaging of apoptosis are those that can target the early loss of membrane asymmetry and exposure of anionic phospholipids on the cell surface as opposed to a specific phospholipid such as PS. These include mimics of annexin V such as those peptidic vectors found by phage display technologies (100), nonselective cationic liposomes (101), sensing of the phosphate moiety on PS with Zn(II)-dipicolylamine (DPA) complexes (102,103), and the development of radiolabeled and iron-labeled forms of the C2A domain of synaptotagmin I, a neural protein with a relatively weak PS binding capacity (10- to 100-fold less than that of wild-type annexin V) (104). A disadvantage of the nonselective anionic phospholipid and phosphate binding tracers is the potential for hemolysis and other forms of cytotoxicity found with other types of membrane-sensing agents such as Ro09-0198, a tetracyclic 19-amino-acid polypeptide that recognizes and forms a tight equimolar complex with phosphatidylethanolamine (PE) on biologic membranes (105).
Another set of tracers for imaging apoptosis target caspase 3 via specific binding peptides that rely not on membrane changes but on the activation of the caspase cascade. These agents include a small membrane–permeant, caspase-activable, far-red fluorescent peptide composed of a Tat-based permeation peptide fused to an l-amino acid effector caspase recognition sequence, DEVD (106), and WC-II-89 (a non–peptide-based isatin sulfonamide caspase-inhibiting analog), labeled with 18F for PET (107,108). Although caspase-based agents hold promise, previous attempts yielded relatively low target-to-background activities (109), and given that caspase activity is not unique to apoptosis, specificity for apoptosis will also be a problem.
The most recent class of tracers proposed for the imaging of apoptosis (110–113) are the ApoSense family of small molecules (such as N,N′-didansyl-l-cystine [DDC] and NST-732). These agents are quite small (MW = 300–700, depending on the formulation), with an amphipathic structure, having both specific hydrophobic and charged moieties. Although the small size of DDC and its entry into cells with scrambled membrane phospholipids are potentially attractive characteristics, but by no means unique, there are several unanswered questions.
For instance, the amount of material injected for imaging (70 mg/kg) (108) appears to be excessive (on both a weight and molar basis), unlike most radionuclide tracers (0.01–1 mg/kg) or fluorescent probes (1–5 mg/kg) currently used for small-animal imaging experiments. There also are no published data provided on the in vitro binding affinities for any of the ApoSense family of tracers for apoptotic cells. Furthermore, there is no explanation provided for the remarkably low uptake values (converted to percentage injected dose per gram [%ID/g]) observed at 2 h after injection of tracer in treated tumors (0.6 %ID/g vs. 0.05 %ID/g for control), severe unilateral ischemia (1.20% ± 0.13 %ID/g vs. 0.58% ± 0.12 %ID/g for control) (106), or cerebral ischemic-reperfusion injury (1.6 %ID/g for ischemia vs. 0.4 %ID/g for control) (105). There are also no clearance or conventional biodistribution data provided so that is not possible determine the potential imaging qualities of the ApoSense family of tracers, particularly with regard to target-to-background ratio and potential washin and washout problems. The rationale for the chosen imaging time of 2 h after injection of the tracer is also not given, but one article does provide a value for blood activity of 0.3 %ID/g (106).
Another concern is that the average uptake value of DDC within infarcted cerebral hemispheres was actually higher than what was reported for the severely injured kidney, an organ with no blood–brain barrier (BBB) (105). The authors also presented no control data for delineating BBB breakdown and nonspecific leakage of the tracer after cerebral infarction.
In summary, DDC and the ApoSense family of tracers will need far more detailed investigation than that provided in the literature to date.
LIPID PROTON MR SPECTROSCOPY
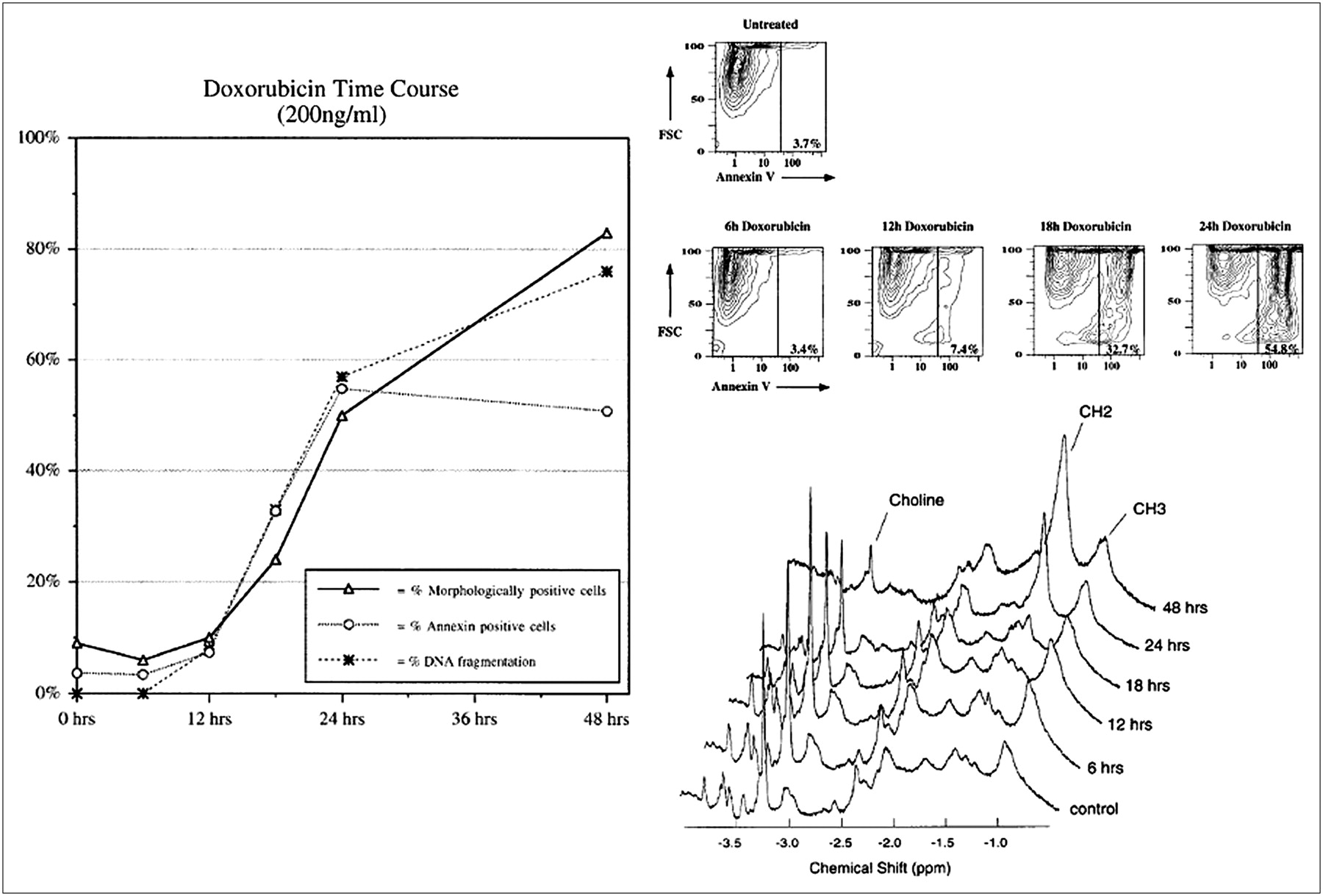
The first MRI technique applied to the detection of apoptosis was lipid proton magnetic resonance spectroscopy (114–116). These studies described apoptosis-specific changes, including a selective increase in CH2 (methylene) relative to CH3 (methyl) mobile lipid proton signal intensities at 1.3 and 0.9 ppm, respectively. The rise in CH2 resonance occurred with a wide range of apoptotic drugs as well as apoptosis associated with serum (growth factor) deprivation (Fig. 5) (114). The CH2/CH3 ratio also had a strong linear correlation with other markers of programmed cell death, including fluorescent annexin V cytometry and DNA ladder formation (Fig. 6) (115). Although there was an increase in the methylene resonance, there was no detectable change in total lipid composition or new lipid synthesis, suggesting an increase in membrane mobility as opposed to increased amounts of lipids within cells.
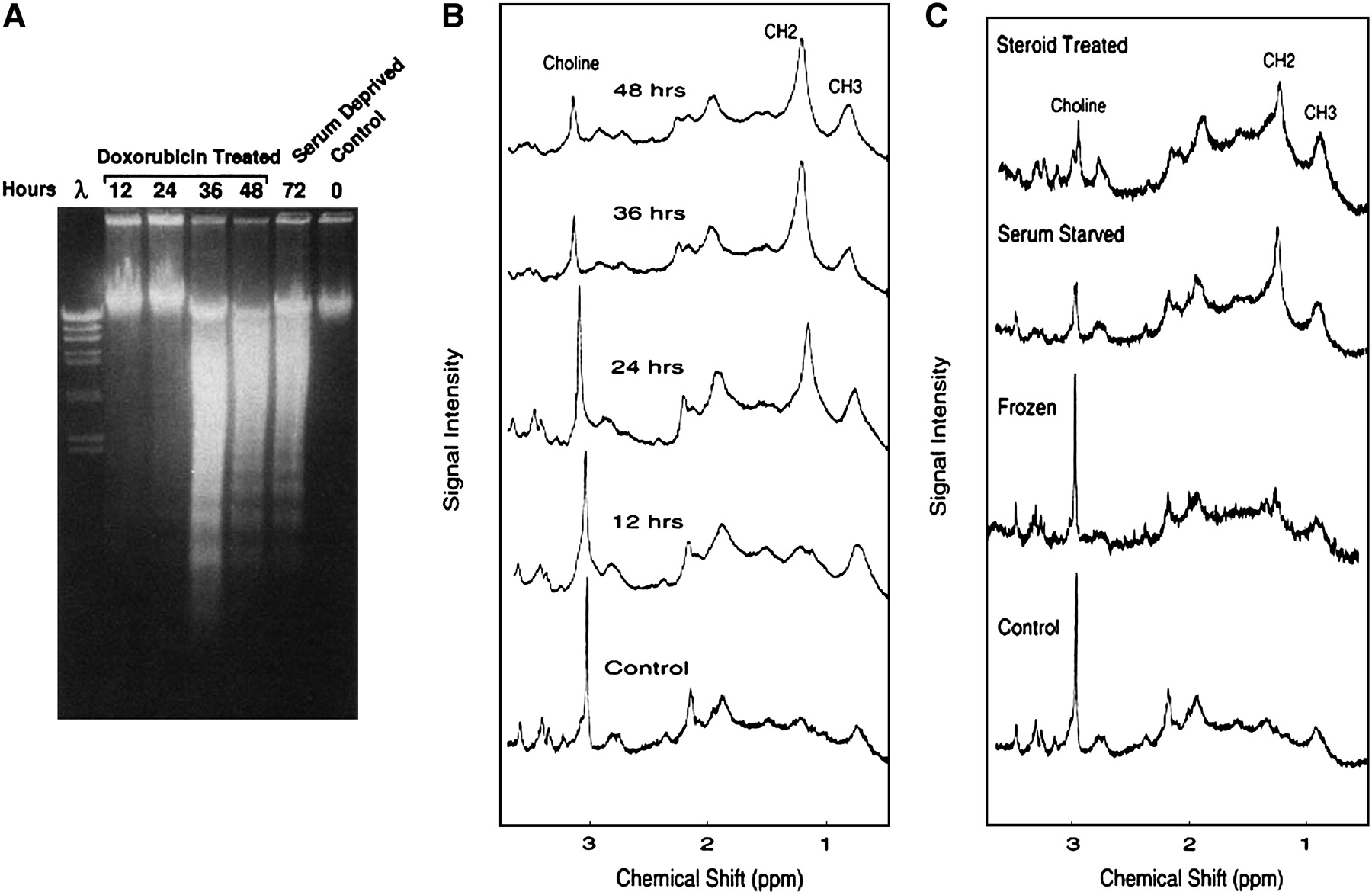
Relationship of proton lipid MR spectroscopy and apoptosis. Jurkat cells were incubated with doxorubicin (200 ng/mL) for 12, 24, 36, or 48 h. (A) DNA ladder formation (irreversible DNA cleavage). Lanes 0, 12, 24, 36, and 48 represent hours of incubation after addition of doxorubicin. DNA ladder formation from serum-deprived Jurkat cells is also shown (lane 72). Lane λ contained digest of bacteriophage λ DNA used as molecular weight standard. At least 50% of cells were apoptotic, as determined from morphologic criteria, in Jurkat cultures in which DNA laddering was present. Note that no laddering was identified until after 24 h. (B) 1H NMR spectra (obtained with 128 excitations). Spectral resonances of choline protons (-NH3) at 3.2 ppm, methylene protons (-CH2-) at 1.3 ppm, and methyl protons (-CH3) at 0.9 ppm are indicated. Note increase in CH2/CH3 ratio starting at 24 h before DNA ladder formation. (C) Jurkat cultures were treated with dexamethasone (25 pmol/L for 72 h), deprived of serum (for 72 h), and frozen (10 min on dry ice–ethanol). Dexamethasone treatment and serum deprivation induced apoptosis, as determined from morphologic criteria (such as DNA condensation and peripheral clumping, plasma membrane bleb formation, and cell shrinkage), whereas freezing resulted only in necrotic cell death (irreversible membrane damage resulting in staining of necrotic nuclei with propidium iodide). Note that there were no differences in lipid spectral signals between necrotic and apoptotic cell cultures. (Reprinted with permission of (114).)
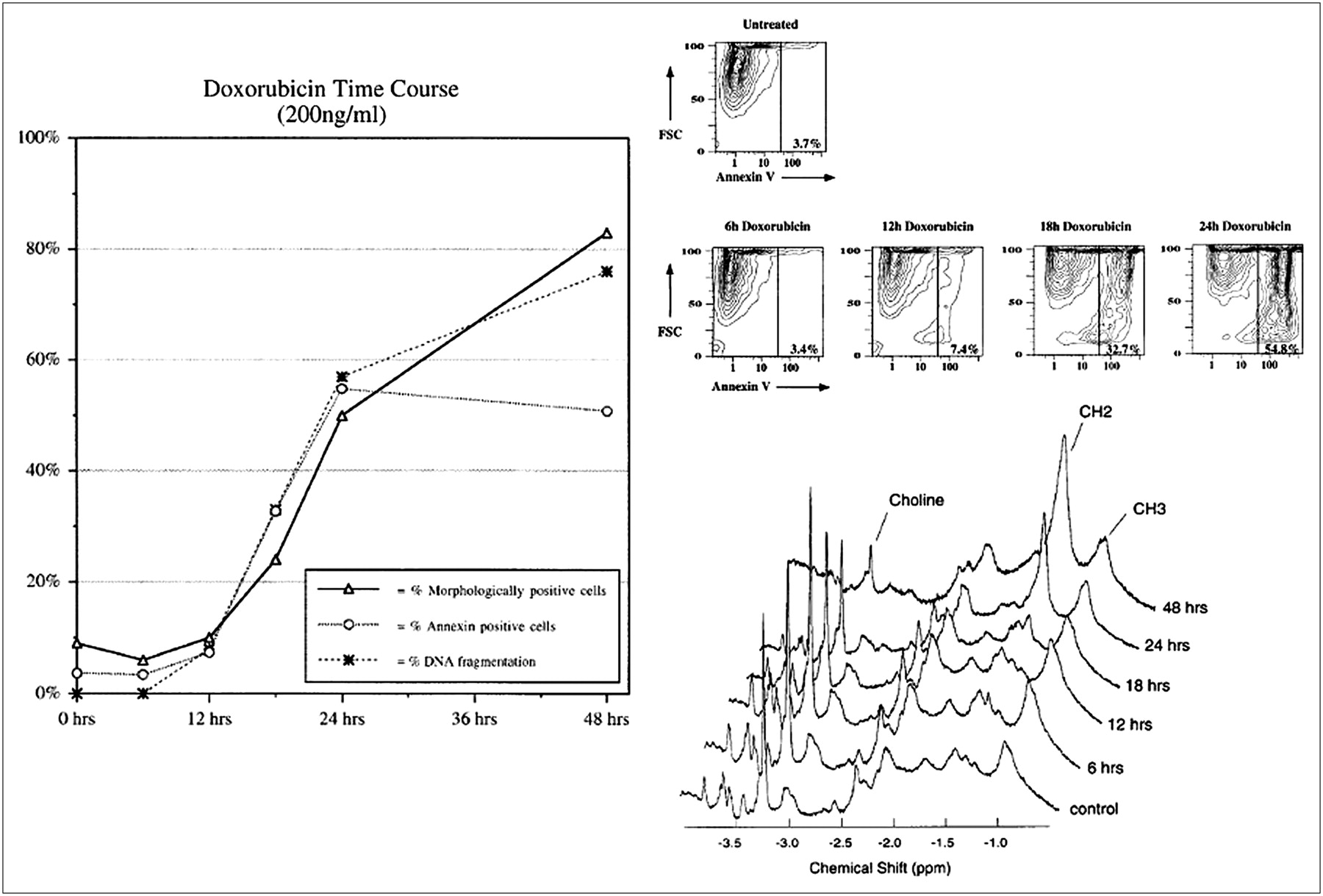
CH2/CH3 lipid proton signal ratios and traditional markers of apoptosis. Jurkat cultures were incubated with doxorubicin (200 ng/mL) for 0, 6, 12, 18, 24, and 48 h. (Left) Percentage of apoptotic cells in cultures, as observed by nuclear morphologic analysis, FITC-annexin V flow cytometry, and DNA gel electrophoresis (DNA laddering), from 0 to 48 h after doxorubicin treatment. (Top right) FITC-annexin V flow cytometric data from 0 to 24 h after doxorubicin treatment. FCS = forward scatter; FITC-annexin V = fluorescence intensity at 494 nm plotted on logarithmic scale. (Bottom right) 1H NMR spectra (obtained with 64 excitations) at 400 MHz. Spectral resonances of choline protons (-N(CH3)) at 3.2 ppm, methylene protons (-CH2-) at 1.3 ppm, and methyl protons (-CH3) at 0.9 ppm are indicated. CH2/CH3 signal intensity ratios were 0.28, 0.56, 0.33, 0.89, 2.41, and 4.42 at 0, 6, 12, 18, 24, and 48 h after doxorubicin treatment, respectively. (Reprinted with permission of (115).)
These observations have largely been confirmed in subsequent investigations, though the source of the increased methylene signal intensity seen with apoptosis has been determined to arise from the formation of osmophilic lipid (0.2−2.0 μm) droplets with the cytoplasm (117). These droplets contain variable amounts of polyunsaturated fatty acids associated largely with 18:1 and 18:2 lipid moieties and an accumulation of triacylglycerides. The accumulation of triacylglycerides is believed to be related to phospholipase A2 activation and the formation of ceramide (a regulatory molecule in the mediation of membrane-related apoptotic events with a long CH2 chain).
Despite the differences in the drugs used and their mechanisms of action and the different types of cells studied, the increase in the lipid signal, as evidenced by the increase in the 1.3-ppm intensity, seems to hold in all situations. Therefore, a selective increase in CH2 and CH3 mobile lipid protons, principally of CH2, permits the calculation of the CH2/CH3 ratio as a measure of the presence and degree of apoptosis within a sample or voxel in most situations. Decreases in other chemical species, such as glutamine and glutamate, choline-containing metabolites, taurine, and reduced glutathione, can also be seen with apoptotic cell death. In contrast, necrosis in general is characterized by a completely different 1H nuclear magnetic resonance (NMR) profile in which there is a significant increase in all of the metabolites examined, with the exception of CH2 mobile lipids that remain unchanged (coupled to a decrease in reduced glutathione).
This early spectroscopic work, however, was severely limited by the lack of a method that could permit high spectral resolution of excised whole tissue. Therefore, investigations were conducted with treated cell suspensions in deuterated (D2O) phosphate-buffered saline or lipid extracts of tissue, a process that necessarily introduces artifacts. These problems arise because tissues are semisolid in nature and contain highly heterogeneous microenvironments that lead to marked restriction of molecular motion and high magnetic susceptibility (such as dipole couplings and chemical shift anisotropy). The result is strong interactions between the spins of each proton leading to severe dephasing with T2 shortening (signal intensity loss) and relatively broad spectral lines. In addition, spin–spin interactions have an angular dependence with respect to the main magnetic field. In liquids (and lipid extracts of tissue), molecules can freely move at rates faster than these dipole interactions and are effectively averaged, giving sharp, well-defined spectral lines and long T2 relaxation times.
With the advent of magic angle spinning (MAS) proton spectroscopy in the early 1990s, the problems with obtaining high-resolution spectra from whole-tissue samples have largely been overcome (118–120). It is known that if a sample is spun mechanically, at a rate faster than the spectral broadening originating from these interactions (about 2.5 kHz) and at the “magic angle” of 54°44′ with respect to the main magnetic field, then the contribution from these interactions to the MR spectral broadening can be significantly reduced.
Ex vivo MAS and conventional in vivo MR spectroscopy of patients with cervical carcinomas before and after radiation therapy showed that the apoptotic activity could be predicted well from the lipid metabolites in high-resolution MAS MR spectra, whereas tumor cell fraction and density were predicted from cholines, creatine, taurine, glucose, and lactate (121,122). Clinical studies using lipid proton MR spectroscopy, however, will be, at least in the near term, limited to organs that are accessible to surface (or endovaginal or endorectal) coils, which are needed to detect the relatively small changes in lipid signals with scattered regions of apoptosis typically found within a voxel of tissue or in organs, such as the brain, that are mostly free of motion artifacts and far from subcutaneous fat; a very large nonspecific tissue signal can bleed as noise into the much smaller desired lipid signal of apoptotic cells.
DIFFUSION-WEIGHTED MRI
Diffusion-weighted MRI (DWI) is an alternative MRI modality that can image apoptosis in response to radiation and chemotherapy without the need for a contrast agent (123–126). DWI generates image contrast by using the diffusion properties of water within tissues. Diffusion can be predominantly unidirectional (anisotropic) or not (isotropic) and can be restricted or free depending on the amount of water in the extracellular (relatively unrestricted) or intracellular (restricted) compartments. Diffusion-sensitized (weighted) images can be acquired with magnetic gradients of different magnitudes, generating an apparent diffusion coefficient (ADC) map. As increases in cellularity are reflected as restricted motion, DWI has been used in cancer imaging to distinguish between tumor (restricted microenvironment) and peritumoral edema (unrestricted). DWI may also be valuable in monitoring treatment, where changes due to cell swelling and apoptosis are measurable as changes in ADC. The magnitude of changes, however, is small (i.e., <50% of control), and it may be difficult to separate tumor shrinkage, necrosis, and other processes that can occur with therapy. Therefore, more studies are needed to confirm the validity of DWI as a marker of therapeutic efficacy in the clinic.
DETECTION OF APOPTOSIS WITH ULTRASOUND
High-frequency ultrasound (40 MHz or greater) has been used to detect the unique specular reflections of apoptotic cells in vitro and in vivo (127). Backscatter from apoptotic nuclei is up to 6-fold greater than that from nonapoptotic cellular nuclei. The specific nuclear features resolved at 40 MHz include fragmentation of DNA and chromatin condensation, which occur relatively late in the apoptotic cascade. Unfortunately, the significant energy loss with the soft tissues at these higher frequencies currently limits high-frequency ultrasound to the study of the skin and other superficial structures. High-frequency ultrasound, however, could be quite useful for the study of apoptosis in the brain (and possibly other organs) of neonates and young infants. The open fontanels of neonates provide excellent sonographic windows for the high-frequency ultrasonographic study of apoptosis known to be associated with hypoxic ischemic injury (128). In fact, in the most recent study (2005), Tunis et al. (129) developed statistical methods at a frequency of 20 MHz that enable the monitoring of structural changes within a very low percentage of apoptotic cells in a tissue, raising the possibility of using this technique in vivo, particularly in the premature neonatal brain, which is at high risk for periventricular leukomalacia (PVL).
Contrast-enhanced ultrasound may also be applied to the study of apoptosis in a fashion similar to that of MRI with contrast agents. The development of novel microbubble-based contrast agents for ultrasound, however, has focused on the blood pool, macrophages, and the reticuloendothelial system for liver, spleen, lymph node, and atherosclerotic plaque imaging (130–132). Most of these blood-pool agents are composed of lipid, albumin, or perfluorocarbon shells encapsulating microbubbles of less than 5 μm in diameter, permitting easy access to all portions of the microcirculation. Because of their size and physical characteristics, these agents are confined to the imaging of the vascular space. There is, however, the potential of directing biotinylated-coated microbubbles with several avidin-labeled molecules, including anti-integrin αvβ3, anti–P-selectin, and anti–ICAM-1 antibodies (133,134). Indeed, it is possible to label microbubbles with apoptosis-specific agents such as annexin V for the imaging of myocardial infarction in mice (135).
CONCLUSION
As the field of apoptosis imaging is changing quite rapidly, it is difficult to make predictions about which set of tracers or imaging modalities will prove successful for clinical applications. For the time being, it appears that agents that bind to the surface of stressed or apoptotic cells, including annexin V and its derivatives, have the advantage with respect to sensitivity and specificity over metabolically directed tracers, such as caspase-related radiopharmaceuticals. MR spectroscopy may be useful inside the brain and requires no administration of contrast agent, but its low sensitivity for small absolute changes in the lipid signal relative to subcutaneous fat (1:100) and bleed-in from adipose tissue, as well as an inherent susceptibility to motion artifacts, limits its use in the clinic. DWI may be more robust outside the brain but, again, changes with cell death appear to be small and sensitive to many different types of artifacts.
It is also unclear which are the best imaging applications for any given apoptosis-directed agent (or modality) in the clinic. It is clear that assessment of the tumor response remains difficult because of the unknown time course of the tumor response to therapy for most malignancies, with non-Hodgkin's lymphoma being a notable exception. The imaging of inflammation-associated apoptosis, particularly chronic inflammation (such as that seen in Alzheimer's disease, multiple sclerosis, and rheumatoid arthritis) or postischemic inflammation (such as stroke or myocardial infarction), is an attractive area for further study with annexin and other PS membrane–directed agents.
Acknowledgments
This study was supported in part by NIH grant EB000898.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.
- 9.↵
- 10.↵
- 11.
- 12.
- 13.
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.
- 42.
- 43.
- 44.
- 45.
- 46.
- 47.
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.
- 58.
- 59.
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.
- 79.
- 80.
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.
- 112.
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.
- 125.
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- Received for publication August 14, 2007.
- Accepted for publication January 2, 2008.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- BASIC BIOCHEMICAL AND MORPHOLOGIC CHANGES OF APOPTOSIS
- PHYSIOLOGY OF ANNEXIN V BINDING AND INTERNALIZATION
- PET AND SPECT WITH RADIOLABELED ANNEXIN V
- RECENT DEVELOPMENTS IN ASSAYS OF MODIFIED FORMS OF ANNEXIN V
- OTHER POTENTIAL TRACERS
- LIPID PROTON MR SPECTROSCOPY
- DIFFUSION-WEIGHTED MRI
- DETECTION OF APOPTOSIS WITH ULTRASOUND
- CONCLUSION
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- Proteomic profiling of follicular and papillary thyroid tumors
- Specific biomarkers of receptors, pathways of inhibition and targeted therapies: pre-clinical developments
- Evaluation of Chemotherapy Response in VX2 Rabbit Lung Cancer with 18F-Labeled C2A Domain of Synaptotagmin I
- Apoptosis Imaging: Beyond Annexin V