


Abstract
In this study, a human carcinoembryonic antigen (CEA) transgenic (CEA.Tg) mouse model was evaluated for the preclinical assessment of agents directed against CEA. Methods: Cell-type and organ-specific expression of CEA was studied in CEA.Tg mice derived from a colony that carries the complete human CEA gene together with flanking regulatory sequences and also in wild-type controls. Biodistribution studies were performed on wild-type and CEA.Tg mice by intravenous injection of 125I-labeled anti-CEA (PR1A3) or isotype control (IC) murine monoclonal antibodies (mAbs). Studies were also performed on tumor-bearing CEA.Tg and nude mice. Results: As with humans, the CEA.Tg mice had low serum levels of CEA (mean, 8.8 ± 5.52 ng/mL), and cell-surface CEA expression was primarily localized in the gastrointestinal tract. Both mAbs showed similar biodistribution patterns in the wild-type mice, whereas in the CEA.Tg mice, PR1A3 specifically localized to the CEA-expressing tissues. In the gastrointestinal tract, the percentage injected dose for PR1A3 was significantly higher than that for IC mAb at all the time points sampled. In CEA.Tg mice bearing a murine tumor transfected with human CEA, PR1A3 targeted tissues with constitutive CEA expression and was retained at the tumor site at high levels, whereas in nude mice, PR1A3 localized only to the site of the transplanted tumor. Conclusion: These results demonstrate the targeting potential of the anti-CEA antibody, PR1A3, and emphasize the value of using a transgenic model in preclinical studies.
Several tumors express unique antigenic determinants, defined as tumor-associated antigens (TAAs), which distinguish them from normal cells. TAAs are often expressed ectopically or at high levels in tumors relative to normal tissue (1). Human carcinoembryonic antigen (CEA) is a TAA expressed on many epithelial tumors, including those of the gastrointestinal tract, breast, and lung (2,3). Colorectal cancer is one of the most common cancers in the developed world, accounting for 10% of cancer deaths in the United States (4). In colorectal cancer patients, elevated serum CEA levels have been associated with increased tumor size, disease progression, and poor prognosis (5,6). Significantly for CEA-directed immunotherapy, the pattern of CEA expression is altered in colon carcinomas such that the molecule is expressed over the entire cell surface rather than being confined to the luminal surface of columnar epithelial cells (2).
TAAs have been extensively explored as targets for radiolabeled antibodies in radioimmunoscintigraphy and radioimmunotherapy (7,8). To date, most preclinical in vivo studies have been performed using human tumor xenografts transplanted into immunodeficient (nude or severe combined immunodeficiency) mice (7,8). Although these models have been useful for the study of antibody activity in vivo, the effects of antibody cross-reactivity with normal TAA-expressing tissues cannot be assessed. In addition, using immunodeficient mice, it is difficult to determine the effect of targeted radiation on the immune system as a whole. Conversely, studies using immunocompetent, wild-type mice bearing syngeneic tumors transfected with human TAAs have been compromised because of the host’s intolerance to the xenoantigen (9). For these reasons, it can be argued that both model systems are of only limited value for the prediction of human responses to cancer radioimmunotherapy.
Although transgenic animals have been in existence for several decades (10), immunocompetent transgenic mice tolerant to human TAAs have only recently been developed to assess the efficacy of antitumor immunotherapies (11,12). Mice such as the human polymorphic epithelial mucin (MUC1) mouse have been used in anti-idiotypic antibody (13) and vaccine (14) studies. However, such transgenic models have not previously been extensively used for evaluation of radiopharmaceuticals. The transgenic mice used in this study were derived from a colony into which had been inserted the complete human CEA gene and flanking regulatory sequences allowing cell-type and organ-specific expression of CEA (11).
Elevated levels of CEA on colorectal carcinomas and its exposed nature on cancer cells make this TAA a potential candidate for targeted therapy (15). Using transgenic mice that are fully immunologically responsive and tolerant to human CEA (11,16) permits an assessment of CEA-directed therapies in an in vivo environment more closely resembling the human scenario. Presented here is a preclinical study using a high-affinity (1 nmol/L) murine IgG1k anti-CEA antibody called PR1A3 (17,18). The biodistribution of 125I-labeled PR1A3 and subsequent clearance within a fully immunotolerant transgenic model are described.
MATERIALS AND METHODS
Animals
A colony of heterozygous human CEA transgenic (CEA.Tg) mice (C57BL/6, H-2b) was obtained from John Thompson (University of Freiburg, Freiburg, Germany). The mice had been generated by microinjection of a 33-kb AatII DNA fragment containing the complete human CEA gene (11). The imported CEA.Tg mice were bred by backcrossing with a colony of wild-type C57BL/6 mice. Heterozygous CEA.Tg offspring were identified by polymerase chain reaction of tail snip-extracted DNA, as previously described (19). Female nu/nu mice of mixed genetic background were obtained from the breeding unit of the Imperial Cancer Research Fund (Clare Hall Laboratories, South Mimms, U.K.). All mice were housed and maintained in negative-pressure isolators. Experiments were conducted on 10- to 14-wk-old female mice in full accordance with the U.K. Home Office Animal (Scientific Procedures) Act of 1986.
Cell Lines and Tumors
The human CEA-transfected murine cell line C15 (C57BL/6, H-2b) used for tumor studies was kindly donated by F. James Primus (Vanderbilt University Medical Center, Nashville, TN). C15 is a subclone from the CEA-negative murine colon adenocarcinoma cell line MC38 (16). C15 and MC38 cell lines were maintained in Dulbecco’s modified Eagle’s culture medium, supplemented with 10% heat-inactivated fetal calf serum, 2 mmol/L l-glutamine, and 500 IU/mL penicillin/streptomycin (all from Cambridge Life Sciences, Ely, U.K.). The C15 medium was additionally supplemented with 500 μg/mL G418 (Cambridge Life Sciences). Solid tumors were established by subcutaneously injecting 1 × 106 tumor cells (either C15 or MC38) in 100 μL of phosphate-buffered saline into the mice. Tumors were measured every 3 or 4 d with vernier calipers. Tumor volumes were calculated as (a × b2)/2, where a represents the largest and b the smallest diameter. When tumors reached a mean size of approximately 0.05 cm3, the animals were used for radiolabeled antibody studies.
Antibodies
PR1A3, a murine IgG1k monoclonal antibody (mAb) against CEA (17), and HMFG1, a murine IgG1 mAb against MUC1 (20), were used for in vivo studies, with the latter serving as an isotype-matched control (IC). Humanized versions of PR1A3 (hPR1A3) and HMFG1 (hHMFG1) were used for immunohistochemistry studies. Humanized mAbs were constructed by transferring the complementarity-determining regions of the mouse antibodies onto human framework regions, as previously described (18). A purified immunoglobulin fraction of rabbit anti-human CEA antiserum (21), either unconjugated (code M0804; DAKO, Glostrup, Denmark) or conjugated with horseradish peroxidase (code P0167; DAKO), was used for immunohistochemistry.
Anti-CEA Antibody Binding
C15 and MC-38 cancer lines were used to assess mouse PR1A3 (mPR1A3) binding to membrane-bound CEA. Immunofluorescent staining of cells was conducted by sequential antibody incubation steps, as described in detail previously (18). Stained cells were fixed in 1% paraformaldehyde, and antibody labeling was assessed by flow cytometry on a FACScan (Becton, Dickinson and Co., Franklin Lakes, NJ). Data were analyzed using the Cell Quest software package (Becton, Dickinson) for Macintosh (Apple Computer, Inc., Cupertino, CA). Antibody binding to soluble CEA was studied by enzyme-linked immunosorbent assay (ELISA) using microtiter plates coated with 1 μg/mL of purified CEA (Sigma, Gillingham, U.K.). Primary antibody binding was detected with alkaline phosphatase-conjugated secondary antibodies and p-nitrophenylphosphate substrate (Sigma) (18).
Murine tissues were examined by immunohistochemistry for expression of human CEA. Cryostat sections (5–6 μm) were cut from frozen tissues, mounted on Superfrost slides (BDH Merck, Poole, U.K.), air dried, and fixed in acetone for 5 min. Sections were blocked in normal swine serum (1:5 with Tris-buffered saline) for 15 min, incubated for 1 h in primary antibody (either horseradish peroxidase-conjugated rabbit antihuman CEA, hPR1A3, or hHMFG1), and then incubated with a goat anti-human IgG horseradish peroxidase-conjugated polyclonal antibody (code A-0170; Sigma). All antibody dilutions were performed in normal swine serum. Labeling was detected with diaminobenzidine substrate solution (Sigma), counterstained with Mayer’s hematoxylin, and viewed and photographed with a light microscope (Leitz, Solms, Germany) attached to a digital camera (Sony Corp., Tokyo, Japan).
Radiolabeling
Murine antibodies were radiolabeled with 125I using the Bolton and Hunter reagent as follows. Antibodies were transferred into 0.1 mol/L N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) buffer, pH 8, and were concentrated by ultracentrifugation (Amicon; Millipore, Bedford, MA) to concentrations of 5–10 mg/mL. Twenty microliters (3.7 MBq) of N-succimidyl-3-(4-hydroxy-3-125I-iodophenyl) propionate (Nycomed-Amersham PLC, Buckinghamshire, U.K.) were dried in the bottom of a 1.5-mL polypropylene tube in a Speed Vac (Thermo Savant, Holbrook, NJ), and 50 μg of antibody were added. After mixing and incubation for 10 min, the reaction was quenched by the addition of 0.2 mL of 0.2 mol/L glycine in 0.1 mol/L HEPES and the labeled antibodies were purified by gel filtration on PD-10 Sephadex columns (Amersham-Pharmacia Biotech, Amersham, U.K.) using phosphate-buffered saline containing 0.5% polysorbate 20. The immunoreactivity of the radiolabeled PR1A3 antibody was confirmed to be greater than 90% in a cell-binding assay as previously described (22).
Biodistribution Studies
Through a tail vein, 70 kBq (50 μL) of radiolabeled murine antibody were injected into the mice. After injection, groups of mice (4 mice per group) were sacrificed at intervals (4, 24, and 48 h) and the entire intestine, kidney, liver, spleen, stomach, and tumor and samples of blood, bone, and muscle were removed. The tissue samples were counted on an automated γ-counter (Ultragamma; LKB-Wallac, Turku, Finland) together with a known dilution of the injected material, and the percentage injected dose (%ID) present was determined by dividing the counts in the tissue sample by an average of the total calculated radioactive counts injected. For incomplete tissue samples, that is, blood, bone, and muscle, as well as tumor, the %ID/g was determined by dividing the %ID by the weight of the tissue sample.
Group means were compared using a 2-tailed, unpaired Student t test. Probability values less than 0.05 were interpreted as statistically significant. All statistics were performed using Excel software (Microsoft, Redmond, WA).
CEA in mouse serum was quantitatively measured using a commercial solid-phase, 2-site, chemiluminescent enzyme immunometric assay (Immulite 2000 CEA; Diagnostic Products Corp., Los Angeles, CA).
RESULTS
mPR1A3 is a murine IgG1 mAb that has a high degree of specificity to cell-bound but not soluble CEA. In flow cytometric studies using C15, a murine colorectal line transfected with human CEA, mPR1A3 showed similar binding to a commercially available rabbit anti-CEA polyclonal antibody (Fig. 1A). mPR1A3 did not bind to the nontransfected parental line MC38. ELISAs indicate that unlike the anti-CEA rabbit polyclonal antibody, mPR1A3 has no specificity for plate-bound soluble CEA (Fig. 1B).
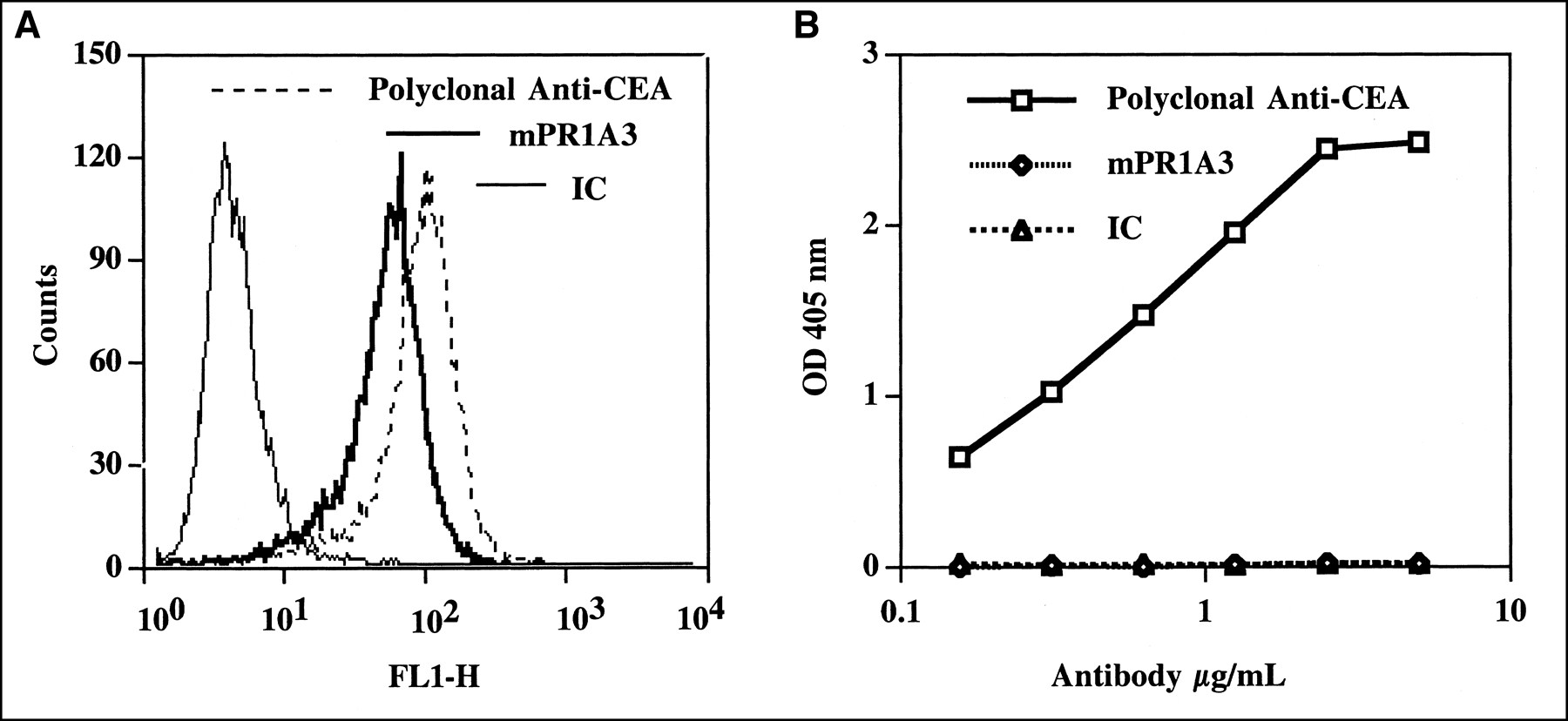
mPR1A3 binding to human CEA. (A) mPR1A3 binding to cell-surface CEA was determined by flow cytometry. C15 cells were stained with either mPR1A3, anti-CEA polyclonal serum, or IC mAb. (B) mPR1A3 binding to soluble CEA was determined by ELISA.
Expression of human CEA was assessed by histology on various tissues of the transgenic mice using both a rabbit anti-CEA polyclonal antibody and the humanized anti-CEA mAb, hPR1A3. Humanized PR1A3 was used in preference to murine PR1A3 because of the inability of most antimouse secondary antibodies to distinguish between the mouse primary antibody and endogenous mouse immunoglobulin in the tissue. Tissue distribution of CEA was similar to that reported previously (11), with CEA being localized in the gastrointestinal tract but not expressed in other organs examined, which included liver, kidney, and pancreas. Sites of high CEA expression in the gastrointestinal tract included the colon and cecum, where most luminal epithelial cells lining the crypts showed positive staining (Fig. 2). All tissue sections labeled with the IC mAb were negative, and tissues from wild-type littermates were negative for all anti-CEA antibodies (Fig. 2). Circulating human CEA was detected in the serum of all but 1 transgenic mouse tested (n = 10) (mean, 8.8 ± 5.52 ng/mL, with a range of 0–15.8 ng/mL), whereas CEA levels in the wild-type littermates (n = 10) were all below the sensitivity of the assay (<0.02 ng/mL).
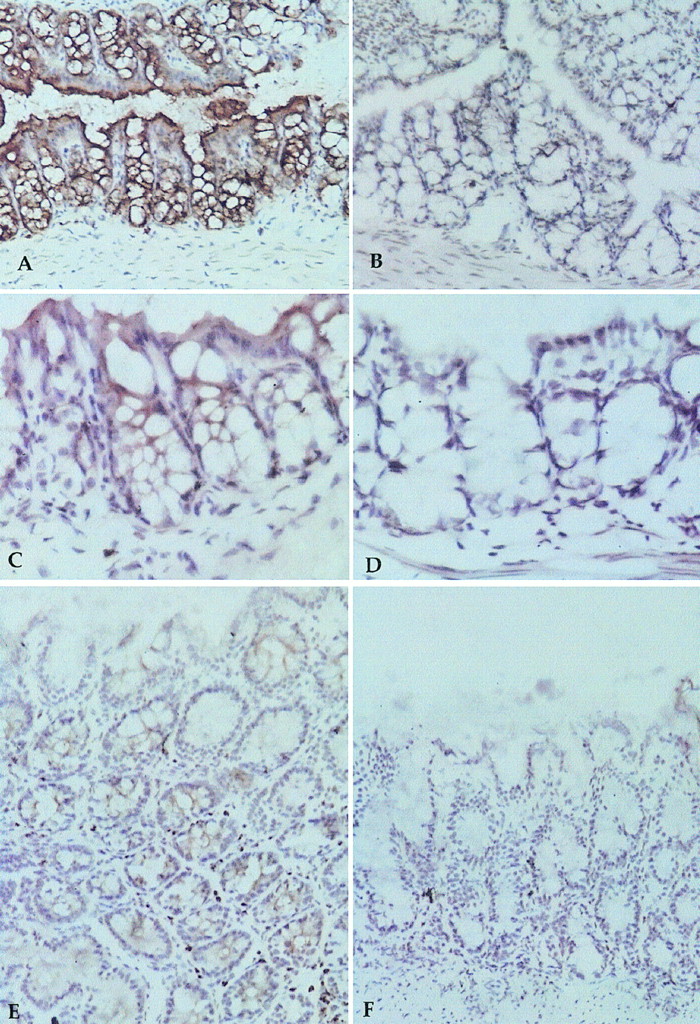
Human CEA expression in CEA.Tg mouse, shown by immunoperoxidase staining of gastrointestinal tract. Anti-CEA antisera stained CEA.Tg colon (A) and wild-type colon (B); hPR1A3 stained CEA.Tg colon (C) and wild-type colon (D); and hPR1A3 stained CEA.Tg cecum (E) and wild-type cecum (F).
CEA.Tg mice appear to have a CEA expression pattern comparable to that of humans (2,3). Whether this pattern affects the biodistribution of radiolabeled anti-CEA mAb was determined by giving wild-type and CEA.Tg mice an intravenous injection of either radiolabeled mPR1A3 or a radiolabeled murine IC. At 4, 24, and 48 h after injection, blood and tissues were removed and antibody uptake was measured. Uptake in the various tissues is shown in Tables 1 and 2.
Biodistribution of 125I-Radiolabeled mAbs in Wild-Type C57BL/6 Mice
Biodistribution of 125I-Radiolabeled mAbs in CEA Transgenic Mice
In wild-type animals, both antibodies demonstrated a similar pattern of biodistribution to that seen in previously published studies of labeled antibodies in mice (23). Clearance of antibody from blood was relatively slow, and the only tissues showing significant amounts of uptake were the excretory organs—liver, intestines, and kidney—all of which showed a pattern of declining activity with time. Statistical comparison (Student t test) of the biodistribution of the 2 antibodies in wild-type animals showed no significant differences in any tissue at any time point (Table 1).
In transgenic animals, however, clear differences were seen in the biodistribution of the 2 antibodies (Table 2). The most significant difference (P < 0.0001) was observed in the intestines (Fig. 3), in which uptake of mPR1A3 was up to 3 times greater than that of IC. There was also a difference in uptake in the stomach, with mPR1A3 uptake being significantly greater than IC uptake at later time points. Small differences were seen in kidney, liver, and blood, in which uptake of mPR1A3 was lower than that of IC (Table 2).
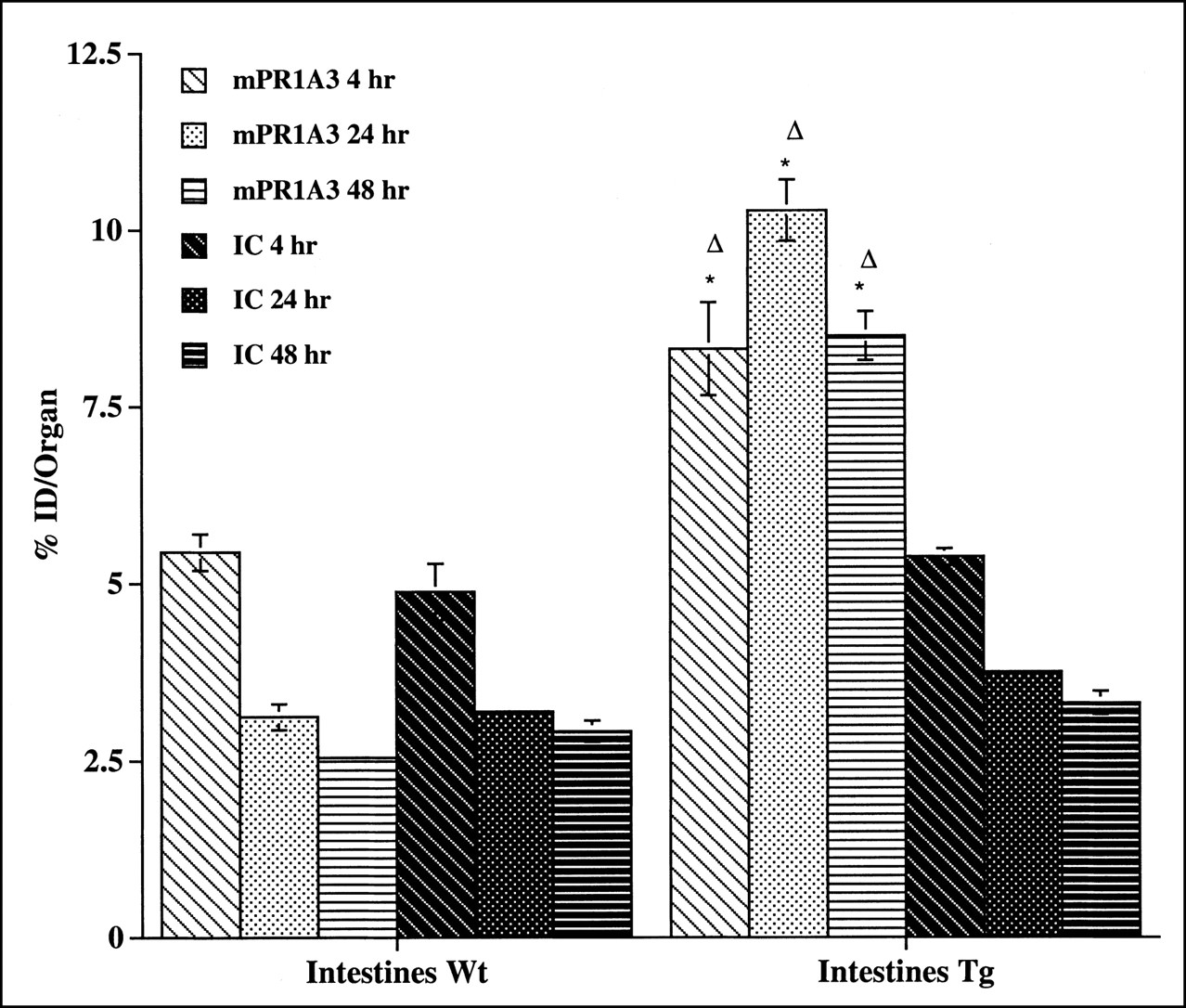
mPR1A3 binding to intestine of CEA.Tg (Tg) mice and wild-type (Wt) littermates. Mean %ID per organ and SEs are shown. Results are representative of 3 separate experiments. *Statistically significant difference (P < 0.05) between uptake of mPR1A3 in transgenic and wild-type animals. ▵Statistically significant difference (P < 0.05) between mPR1A3 and IC uptake in transgenic animals.
A statistical comparison of the biodistribution of mPR1A3 in wild-type and CEA.Tg mice also highlights significant differences, most notably in the intestine. At all time points, greater intestinal uptake of mPR1A3 was seen in CEA.Tg mice than in their nontransgenic littermates (Fig. 3), whereas no significant differences in tissue uptake of the IC antibody were observed between the 2 strains (Fig. 3).
Tumors were grown in CEA.Tg mice after a single subcutaneous injection of 1 × 106 C15 tumor cells. Two weeks later, the tumors were palpable and approximately 0.05 cm3 in size. Flow cytometric analysis of the C15 cells before injection indicated the presence of surface CEA, including the PR1A3 epitope (Fig. 1), and immunohistochemistry of 2-wk tumors with the anti-CEA polyclonal antibody confirmed the expression of CEA in vivo. Serologic analysis in transgenic mice indicated that the tumor-bearing mice had significantly higher levels of CEA (198 ± 156 ng/mL) than did non-tumor-bearing CEA.Tg mice (8.8 ± 5.5 ng/mL, P = 0.001).
In the normal tissues of transgenic mice with C15 tumors (Table 3), a similar biodistribution pattern of the radiolabeled antibodies was observed to that seen previously in the transgenic mice without tumors (Table 2). That is, compared with IC, mPR1A3 was preferentially taken up and retained in the intestine (Table 3). In addition, large differences between the tumor localization of the 2 antibodies were seen, with mice injected with radiolabeled mPR1A3 having significantly higher tumor uptake at all 3 time points studied.
Biodistribution of 125I-Radiolabeled mAbs in CEA Transgenic Mice Bearing CEA-Transfected Tumors
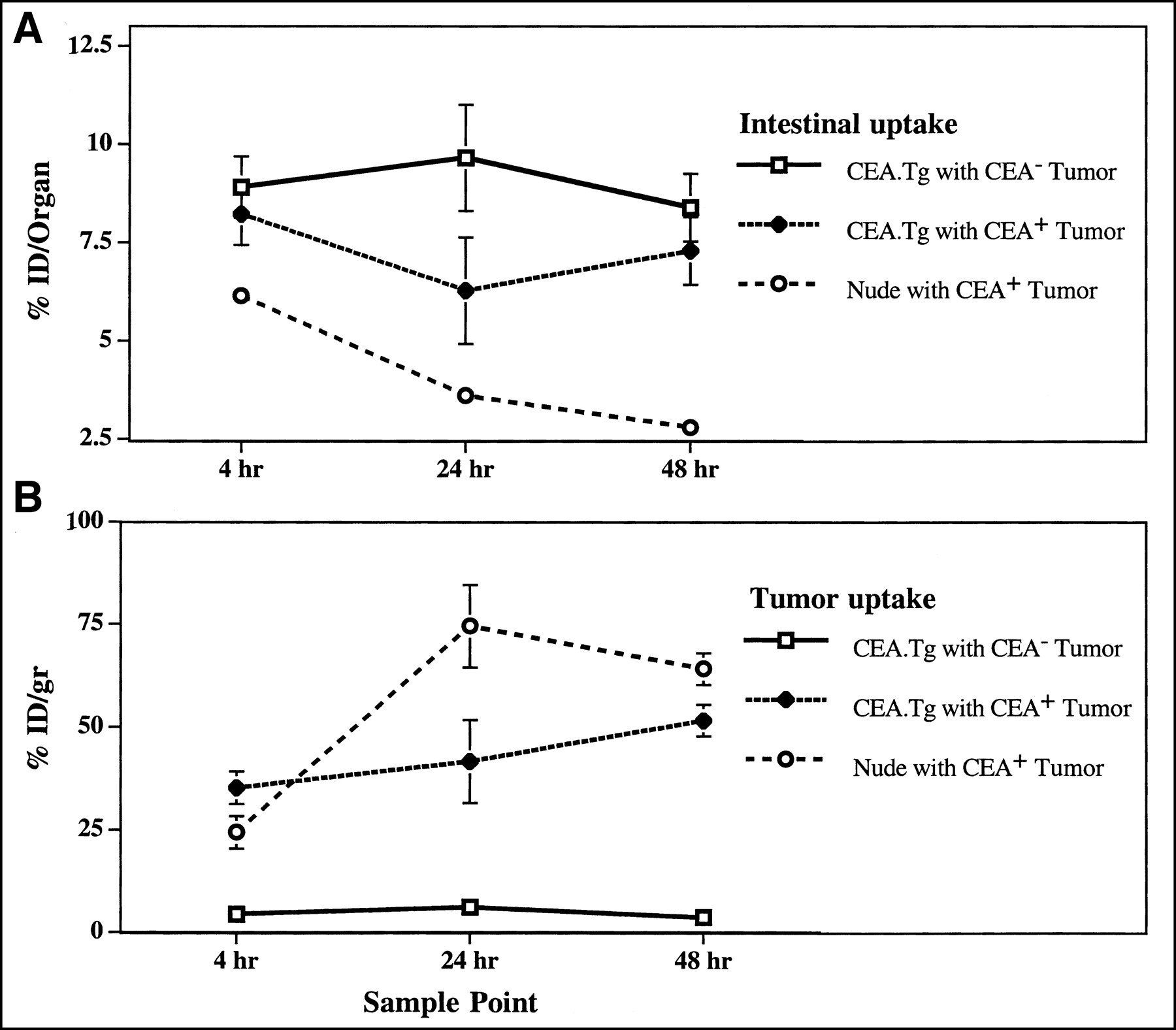
Biodistribution studies were also performed on nude mice bearing C15 (CEA-positive) tumors and CEA.Tg mice bearing MC38 (CEA-negative) tumors. The results were as expected, with significantly higher levels of mPR1A3 retention in the gastrointestinal tracts of the CEA.Tgs compared with nude mice (Fig. 4A) and significantly (8- to 10-fold) higher uptake in the CEA-positive than the CEA-negative tumors (Fig. 4B). These results confirm that mPR1A3 is specifically targeting the CEA expressed by the C15 tumors.

Comparisons of mPR1A3 uptake in tumor-bearing CEA.Tg and nude mice, including %ID in intestines of tumor-bearing mice (A) and %ID/g in tumor from same mice (B). Groups contained 4 mice, and SEs for mean %ID values are shown. Results are representative of 2 separate experiments.
DISCUSSION
The observation that CEA is overexpressed in gastrointestinal carcinomas has resulted in the development of diagnostic and therapeutic agents that target this molecule (2,15). Preclinical assessment of potential CEA-directed therapies has been hampered by the lack of appropriate animal model systems that accurately reflect target expression in humans. This communication has described the validation of the CEA.Tg mouse model for assessment of the anti-CEA antibody, PR1A3. Histologic and serologic analysis indicated that the CEA.Tg mouse exhibits CEA patterns analogous to those of humans. Thus, in CEA.Tg mice, the protein is primarily expressed on luminal epithelial cells of the gastrointestinal tissues and can be detected at low levels in blood serum.
PR1A3 has been shown to bind colorectal tumors regardless of their degree of differentiation (17). Radioimmunoscintigraphic studies have been performed with 99mTc-labeled PR1A3 in colorectal cancer patients. These have reported a 94% accuracy, with a positive predictive value for pelvic and abdominal recurrence of 92%. The “false-positive” uptake obtained was due in 3 cases to the presence of premalignant (antigen-positive) adenomas and in 3 cases to bladder or urinary activity incorrectly interpreted as tumor uptake. In no case was false-positive uptake observed in normal lymph nodes because of binding of the radiolabeled antibody to trapped shed antigen, as has been observed in other studies (24). It is likely that this is because the trapped soluble antigen does not present the epitope recognized by PR1A3. Previous studies have mapped the antibody epitope to the CEA B3 domain, proximal to the site of membrane attachment and distinct from the glycosyl-phospatidyl inositol anchor (25). PR1A3 differs from other anti-CEA antibodies in that it does not react in vitro with soluble serum CEA. Furthermore, it does not appear to cross-react with other CEA family members, including biliary glycoprotein (25) and meconium antigen (D. Snary, unpublished data, July 2000).
The performance of PR1A3 in tumor imaging taken in conjunction with the cell selectivity for antigen binding make it an excellent candidate for use as a tumor-targeting antibody in cancer therapy. Although the low doses of radioactivity and antibody used in human imaging studies were well tolerated, it is not clear what the effects would be of a higher therapeutic dose, especially in normal tissues that constitutively express CEA. Preclinical studies were therefore undertaken in the CEA.Tg mouse to obtain a more detailed insight into the biodistribution of mPR1A3 because these mice have a CEA expression pattern akin to that in humans and because animal work permits a more quantitative analysis of all organs and tissues.
The biodistribution of radiolabeled specific and nonspecific antibodies was studied in the CEA.Tg model and showed significant differences from that seen in the wild-type control mice. Most noticeable was a higher uptake of mPR1A3 in the (CEA-positive) gastrointestinal tracts of the CEA.Tg mice compared with their (CEA-negative) nontransgenic littermates. Detection of PR1A3 uptake in the gastrointestinal tract of the CEA.Tg mice is interesting because CEA is predominantly expressed intracellularly or on cells lining intestinal crypts. Uptake of mPR1A3 in the gastrointestinal tract may reflect the antibody homing via blood or after being secreted into the gut and targeting its epitope during excretion. During the sample period of 48 h, no obvious pathology was observed in the CEA.Tg mice, despite evidence of PR1A3 binding to CEA-positive tissues other than tumor. The dose used in these studies was about 2 μg, which corresponds to a dose of 7 mg in humans. A study involving further dose escalation would be required to determine the toxicity of higher doses, which might be required in clinical radioimmunotherapeutic studies.
When biodistribution studies were performed on animals implanted with CEA-expressing tumors (C15 cells), mPR1A3 specifically targeted CEA-positive tumors, reaching as high as 50 %ID/g, whereas uptake of the IC antibody was 3- to 6-fold less. This difference was highly statistically significant (P < 0.005). The intestines again showed a statistically significant difference (P < 0.05), but in terms of %ID/g, intestinal uptake was up to 10-fold less than uptake in the tumor. C15 tumor-bearing nude mice also showed PR1A3 tumor targeting, but this was greater than that seen in the CEA.Tg mice. Differences may reflect less mPR1A3 being available for binding in the tumors of CEA.Tg mice because of the “sink” effect of antibody-binding CEA-positive tissues, such as the gastrointestinal tract. This finding indicates the importance of using a preclinical model that expresses the target antigen in a similar manner to humans. No mPR1A3 uptake by CEA-negative tumors was seen, and no significant differences in the patterns of biodistribution of the nonspecific IC antibody were seen in the wild-type and transgenic strains. CEA.Tg mice appear to be fully tolerant to human CEA in that they did not develop a humoral response to the antigen despite its being expressed within the tumor microenvironment.
Szalai et al. recently reported the findings of a biodistribution study in a strain of CEA.Tg mice with an anti-CEA mAb, T84.66 (26). Unlike our study, no comparison was made between the binding of their anti-CEA mAb and a radioiodinated isotype control. However, insofar as one can compare results between 2 different studies, there were some points of similarity. Both showed high uptake of the anti-CEA antibody into the tumor, and both showed higher uptake of the antibody into the colon of nontumor transgenic mice than normal mice. That these 2 independent studies concurred in their 2 major findings despite the use of different antibodies and strains of mouse suggests that they can be considered to be representative of such models.
Future CEA.Tg studies will involve higher and repeated doses of labeled mPR1A3, and their effect on tumor regression will be studied. A further development of this model would be to induce malignant changes within both CEA-positive and CEA-negative tissues. The ability of therapies to treat such tumors in immunocompetent animals against the background of “normal” CEA expression would provide a real indication of the potential to treat gastrointestinal cancer in humans. Such preclinical models have been achieved by crossbreeding the CEA.Tg with a mutant strain prone to spontaneous cancer. For example, Thompson et al. (19) have back-crossed the CEA.Tg with the multiple-intestinal-neoplasia mouse, which is genetically predisposed to the development of intestinal polyps (27), and Mukherjee et al. (28) have reported the generation of a mouse in which pancreatic tumors positive for the human TAA MUC1 develop. Neither model as yet has been used to assess the potential of antibody-directed therapy.
CONCLUSION
The human CEA.Tg mouse represents a powerful model for preclinical assessment of CEA-directed reagents. Recently, PR1A3 has been humanized and in vitro analysis has indicated retention of its binding (18). The availability of this humanized antibody now allows therapeutic strategies that require repeated administration of antibody to be used in humans without the induction of strong human antimouse antibody responses. The CEA.Tg mouse model offers an excellent opportunity for preclinical refinement of such strategies before investigators embark on expensive and lengthy clinical trials of these products.
Acknowledgments
We thank Gillian Lewis (Hybridoma Development Unit, Imperial Cancer Research Fund, London, U.K.) for the antibody supply, Dr. Richard Poulson and George Elia (Histopathology Unit, Imperial Cancer Research Fund) for help with histology, Ruthline Laylor (Transplant Immunology Department, Hammersmith Hospital, London, U.K.) for help with ELISA and transgenic genotyping, and Hugh Mitchell (Charing Cross Hospital, London, U.K.) for serum CEA measurements. This study was supported financially by Antisoma PLC and the Imperial Cancer Research Fund.
Footnotes
Received Dec. 3, 2001; revision accepted Jun. 11, 2002.
For correspondence or reprints contact: Stephen J. Mather, PhD, Department of Nuclear Medicine, St. Bartholomew’s Hospital, London, EC1A 7BE, U.K.
E-mail: Stephen.Mather{at}cancer.org.uk





{kind=link}
{kind=link}
{kind=link}
{kind=link}